

Abstract
Self-reactive T cells that escape negative selection in the thymus must be kept under control in the periphery. Mechanisms of peripheral tolerance include deletion or functional inactivation of self-reactive T cells and mechanisms of dominant tolerance mediated by regulatory T cells. In the absence of costimulation, TCR engagement results in unopposed calcium signaling that leads to the activation of a cell-intrinsic program of inactivation that makes T cells hyporesponsive to subsequent stimulations. The activation of this program in anergic T cells is a consequence of the induction of a NFAT-dependent program of gene expression. Recent studies have offered new insights into the mechanisms responsible for the implementation and maintenance of T cell anergy and have provided evidence that the increased levels of proteins encoded by the genes upregulated in anergic T cells are responsible for the implementation of anergy interfering with TCR signaling and directly inhibiting IL-2 gene transcription.
1. Introduction
T-cell mediated immunity relies on the ability of the T cell receptor (TCR) to recognize foreign antigens. To accomplish this goal, random rearrangements of the V(D)J segments at the TCR locus lead to the generation of an extraordinarily diverse TCR repertoire. However, this process may also generate dangerous self-reactive T cells bearing TCRs that can recognize self antigens. Many self-reactive T cells are eliminated in the thymus, however, thymocyte negative selection has limitations and additional mechanisms of tolerance are required in the periphery to limit autoimmunity. Peripheral tolerance is achieved through different mechanisms that include the induction of anergy, suppression by regulatory T cells (Tregs) and peripheral deletion of self-reactive T cells. Anergy is a mechanism of peripheral tolerance which results in functional inactivation of self-reactive T cells [1–3], which can be induced upon engagement of the TCR in the absence of costimulatory signals [4–6]. Although the first characterization of T cell anergy was described 20 years ago, it has only been recently that the molecular mechanisms responsible for the functional inactivation of T cells have been characterized.
In this review, we will discuss evidence that supports that specific mechanisms of transcriptional regulation play crucial roles in the induction and maintenance of T cell anergy. In response to an anergizing stimulus specific transcriptional complexes activate the expression of a distinct calcium-dependent program of gene expression that is intrinsically different from the set of genes that are upregulated in activated T cells [7, 8]. One of the most important pathways in the transduction of calcium signals in T cells is the calcineurin-mediated dephosphorylation and nuclear translocation of members of the nuclear factor of activated T cells (NFAT) family of transcription factors [9–11]. We will discuss evidence that supports that NFAT proteins play a key role in the induction of tolerance in T cells by driving the expression of anergy-inducing genes[7]. The specific expression of these genes is required to impose a state of functional unresponsiveness through different mechanisms that include, among others, downregulation of TCR signaling by inactivation or degradation of signaling molecules [12].
One of the hallmarks of anergic T cells is their inability to synthesize IL-2 in response to TCR engagement [2]. Although this defect can be explained by events that lead to the blockade of signaling pathways downstream of the TCR, evidence suggests that transcriptional mechanisms that directly repress IL-2 gene expression are also engaged and contribute to the general defect in IL-2 production in anergic T cells [13, 14]. The implementation and maintenance of anergy may also respond to the engagement of specific mechanisms of transcriptional repression that inhibit the expression of cytokine genes in anergic T cells [15–17].
2. Anergic T cells activate distinct programs of gene expression
Early studies on the mechanisms that control the establishment of an anergic state in T cells, supported the idea that new protein synthesis was necessary for T cells to become unresponsive, as suggested by the inability to anergize T cell clones in the presence of cycloheximide [6]. The existence of anergizing factors was also made evident by the possibility of transferring the anergic phenotype in somatic fusions between an anergic mouse T cell and a human T cell. These experiments indicated that anergic T cells were able to produce active repressor molecules [18]. Genome wide analysis of gene expression using DNA microarray technology has revealed that anergic T cells, and also B cells, express a specific set of genes. The proteins encoded by those genes are responsible for dampening TCR signaling in anergic T cells [7, 8, 19]. Differential analysis of gene expression has been performed in vitro, using T cell clones or primary Th1 cells [7], and also in vivo, analyzing gene expression in TCR transgenic mice crossed with mice that express the cognate antigen [8, 20], anergizing transgenic DO11.10 mice by oral administration of ovalbumin [7], or using systemic administration of a superantigen to induce T cell tolerance [21]. All these reports provide conclusive evidence that the induction of T cell anergy is an active process that results from the activation of a specific program of gene expression is response to anergizing stimuli. As we will discuss in this review and in other reviews in this issue, the analysis of the function of the proteins encoded in those genes has provided valuable information to understand the molecular mechanisms involved in the implementation and maintenance of an unresponsive status in anergic T cells.
3. Calcium/NFAT signaling in T cell anergy
Three of the four calcium-regulated members of the NFAT family of transcription factors (NFAT1, 2 and 4) are expressed in T cells [10, 11, 22]. In resting cells, these proteins are heavily phosphorylated and reside in the cytosol. TCR engagement results in the activation of phospholipase C-gamma (PLCγ) that mediates the production of inositol triphosphate, which induces depletion of intracellular calcium stores and the subsequent activation of store-operated calcium entry [23, 24]. Sustained increase of intracellular calcium induces the activation of the calmodulin-dependent phosphatase calcineurin, which docks at a specific motif (PxIxIT) in the N-terminus of NFAT and dephosphorylates multiple serine residues in the NFAT regulatory domain [25, 26]. Upon dephosphorylation, NFAT proteins undergo a conformational change that exposes their nuclear localization signals, leading to their translocation into the nucleus and the induction of NFAT-mediated gene transcription [27]. Several kinases, including GSK3, CK1, DYRK and several mitogen activated protein kinases (MAPK), have been reported to be required for the phosphorylation of NFAT transcription factors [28–33]. These kinases would control NFAT nuclear shuttling by specifically phosphorylating distinct serine-containing motifs on different NFAT family members in the cytosol, to prevent nuclear translocation, or in the nucleus, to induce nuclear export. During T cell activation, NFAT proteins cooperate with members of the activator protein 1 (AP-1) family of transcription factors to elicit the expression of activation-induced genes [34–36]. Ternary complexes containing Fos, Jun and NFAT proteins are formed on NFAT:AP-1 composite sites which are present in many genes induced during T cell activation [37, 38]. Cooperation of NFAT and AP-1 results from the integration of two of the major signaling pathways induced in response to T cell stimulation: calcium, which is responsible for NFAT activation, and Ras/MAP kinases, which induce expression and activation of Fos and Jun [36, 37]. In the classic two-signal model this will occur when both the TCR (signal 1) and costimulatory molecules, such as CD28 (signal 2), are engaged. Stimulation of T lymphocytes by engagement of their antigen receptor (Signal 1) in the absence of co-stimulation leads to clonal anergy [1, 2]. The absence of CD28:B7 interaction leads to unbalanced activation, where calcium-induced signaling pathways are fully activated but those other pathways that need CD28 engagement (e.g. Ras-MAPK, PKC, and IKK) are only partially induced. Under these circumstances, NFAT proteins are responsible for the activation of a very specific program of gene expression that is responsible for the implementation of a hyporesponsive state in anergic T cells [7]. Transcription of these genes is AP-1 independent, as their expression can be induced using the calcium ionophore ionomycin under conditions in which no AP-1 transcriptional complexes could be detected in the nucleus. The expression of those genes is, however, clearly dependent on NFAT, as it can be blocked with cyclosporine, an inhibitor of calcineurin, and is markedly reduced when T cells from an NFAT1−/− mouse are anergized. Both cases lead to ineffective anergy induction. More strikingly, introduction of a constitutively active NFAT1 protein in Th1 cells renders them unresponsive to TCR engagement in a state resembling anergy, which correlates with the upregulation of many anergy-associated genes [7]. The anergy-inducing program of gene expression includes, among others, several E3 ubiquitn ligases, tyrosine phosphatases, diacylglycerol kinase alpha (DGKα) and several transcriptional repressors [39–41]. Subsequent reports have confirmed that some of those genes such as Itch, Cbl-b, GRAIL and DGKα, are specifically upregulated in anergic T cells, and that their expression is calcium dependent and cyclosporine sensitive. Moreover, these studies have provided direct experimental evidence of the crucial role of these proteins in T cell anergy [42–46]. This data supports that T cell inactivation is achieved in anergic T cells through a calcium/NFAT-dependent transcriptional program that induces the expression of genes that mediate the simultaneous inhibition of several cellular processes involved at different levels in the cascade of events leading from TCR engagement to cytokine production and clonal expansion.
What are the NFAT-containing transcriptional complexes that upregulate the expression of anergy associated genes? Experiments in which introducing an active form of NFAT1 directed the expression of anergy associated genes suggested that, at least for those genes, complexes that contain only NFAT proteins can induce their transcription [7]. The nature of those complexes has not been characterized yet. Recently, it has been shown that on certain NFAT sites that bear homology to κB binding elements, NFAT1 can bind as a dimer. The DNA binding domain of NFAT is structurally homologous to the Rel domain of NF-κB proteins and two structures containing dimers of NFAT1 have been resolved on the κB-like sites of the IL-8 promoter and the HIV1 long terminal repeat [47, 48]. It might be possible, thus, that dimer complexes of NFAT proteins may be responsible for the transcriptional activation of those genes. Results from our laboratory suggest that direct transcriptional activation of anergy-associated genes by NFAT dimers may occur in T cells when stimulated under anergizing conditions (Soto-Nieves N et al., Manuscript in preparation). It is however, unclear whether all NFAT family members may be involved in the activation of an anergy-inducing transcriptional program. Regions involved in dimer interaction and DNA binding are highly conserved among different NFAT family members and it might be even possible that heterodimers between different NFAT family members can be formed. Although redundancy may be a factor in many of the T cell functions controlled by NFAT transcription factors, analysis of mice deficient in individual NFAT proteins suggest that several functions may be regulated by specific NFAT family members. In particular, programs that negatively regulate T cell activation seem to be dependent on NFAT1 and NFAT4 [49–54]. Further research will be needed to determine NFAT specificity in the regulation of T cell anergy.
Two sets of results support the possibility that NFAT may cooperate with other transcription factors to activate a full anergy-inducing program. First, NFAT alone is not able to activate the expression of all genes induced under anergizing conditions; and second, anergizing T cells using ionomycin, which is capable of fully activating NFAT proteins, causes a hyporesponsive state that, as opposed to clonal anergy induced by TCR engagement in the absence of costimulation, is short lived [2, 7]. It is quite likely then that other transcription factors, independently or cooperating with NFAT, may be involved in activating the expression of anergy-inducing genes. Experimental evidence has shown that NFAT proteins can cooperate not only with AP-1 transcription factors but also with a myriad of other proteins. These interactions are specific for certain conditions and tissues, and regulate different functions in T cells and other cell types [10, 11]. NFAT proteins act, thus, as integrators of calcium signaling with many other signaling pathways in T cells. The structure of monomeric NFAT1 bound to DNA has recently resolved and has demonstrated that the linker segment located between the N-terminal and the C-terminal regions of DNA binding domain shows a high degree of flexibility. Because of this flexibility, NFAT1:DNA complexes can acquire different structural conformations, which should allow interactions of NFAT with different transcriptional partners [55]. Protein-protein interactions have been described between NFAT and proteins belonging to several other families of transcription factors. Synergy between NFAT and cMaf occurs at the IL-4 promoter [56], and between NFAT and GATA3 at the 3′ IL4 enhancer [57]. Synergistic interactions have also been reported with IRF-4 in the IL-4 promoter [58] and with T-bet in an enhancer of the interferon gamma gene [57]. NFAT and MEF2 cooperation is involved in the expression of Nur77 in T cells and may also play a role in IL-2 transcription [59, 60]. Cooperation of NFAT proteins with any of those factors or new unidentified interactions may also be key to the regulation of the induction of T cell anergy.
The expression of Egr2 and Egr3 had previously been shown to be cyclosporine sensitive and to respond to NFAT binding to specific sites in the promoters of these transcription factors [61]. Cooperation between NFAT and Egr2 and Egr3 was also shown to be required for proper Fas ligand (FasL) expression in T cells, as cells lacking NFAT1 and NFAT4 showed defective activation of a FasL promoter reporter, which was restored by reconstitution with a vector expressing NFAT1 [61]. Recently, it has been shown that the expression of Egr2 and Egr3 is upregulated and persists for longer times in anergic T cells compared with fully activated cells [62, 63]. Expression of these genes is cyclosporine sensitive and can also be detected in an in vivo model of T cell anergy [63]. Moreover, Egr3 deficient cells are resistant to anergy induction. The role of Egr proteins in T cell anergy seems to be to induce the expression of anergy-inducing genes, as Egr3−/− T cells fail to upregulate the expression of Cbl-b in response to ionomycin [63]. Egr2 and Egr3 would therefore be induced by NFAT in response to anergizing stimuli and would then activate transcription of other anergy-inducing genes, such as Cbl-b. Whether Egr2 and Egr3 can activate the expression of Cbl-b as independent complexes or in cooperation with NFAT remains to be determined. The fact that Egr2 and Egr3 expression seems to be dependent on NFAT1 and NFAT4 but not on NFAT2 suggests, as discussed before, a specific role for these two NFAT family members in the induction of clonal T cell anergy [61].
The involvement of NFAT proteins in the regulation of peripheral tolerance is not only limited to T cell anergy. Studies carried out in a well established model of B cell tolerance using transgenic mice that express anti HEL-Ig and a soluble form of this lysozyme have shown that NFAT1 also plays a role in B cell anergy, as transgenic B cells from mice deficient in NFAT1 are inefficiently anergized in the presence of soluble HEL [64].
The function of regulatory T cells (Treg) has also been shown to be regulated by NFAT. Dominant tolerance mediated by CD4+CD25+ Tregs constitutes another key mechanism of peripheral tolerance. Tregs have the ability to suppress effector functions, such as IL-2 production and proliferation, in CD4+ T cells [65]. The mechanisms underlying this form of suppression are still poorly understood. Natural CD4+CD25+Tregs are generated in the thymus, although recent evidence supports the extrathymic generation of bona fide Tregs in response to suboptimal stimulation [66]. The forkhead transcription factor Foxp3 has been identified as a hallmark of this population of suppressor cells. It is expressed almost exclusively in CD4+CD25+ Tregs and mice deficient in this transcription factor fail to produce a normal population of Tregs and develop a multiorgan autoimmune disorder [67, 68]. In addition to Foxp3, Tregs also show upregulated expression of other markers such as CD25, CTLA-4 and GITR, although the role of these surface proteins in the suppressor function of Tregs is not yet known [65]. Although FoxP3 was originally thought to be the factor responsible for the pattern of gene expression that defined Tregs, its mechanisms of action has not been characterized yet. Recent experiments suggested that this protein may interact with NFAT and NF-κB and inhibit their transcriptional activity [69]. A very elegant report has recently resolved the crystal structure of a complex containing the forkhead domain of FoxP2 and the DNA binding domain of NFAT1, demonstrating that Foxp proteins can associate in transcriptional complexes with NFAT transcription factors [70]. These experiments have also shown that these complexes bind to NFAT:AP1 composite sites on the IL-2 promoter and likely displace NFAT:AP-1 complexes, directly repressing IL-2 transcription. Foxp3 interaction with NFAT is crucial for Treg function, since the expression of a mutant Foxp3 unable to bind NFAT1 results in defective IL-2 repression and suppressor function in transfected T cells [70]. The IL-2 promoter region is not properly remodeled in Tregs and remains in an inactive closed state [71]. As Foxp3 has recently been shown to bind the promoters of the IL-2 and IFNγ genes in a cyclosporine A sensitive way and actively induce histone deacetylation [72], it is possible that NFAT:Foxp3 complexes might be able to induce the epigenetic changes that result in IL-2 silencing in Tregs. Interestingly NFAT:FoxP3 complexes can also activate transcription of CD25 and CTLA-4 [70]. The mechanisms that determine whether these complexes behave as a repressors or activators of transcription and the possible role of NFAT:Foxp3 interactions during Treg generation in the thymus or in secondary lymphoid organs remain yet to be elucidated.
On the other side of the suppressor reaction, it has been reported that cells defective in NFAT1 and NFAT4 are resistant to suppression by Tregs [73]. Although it is not clear how these two NFAT proteins may be involved in regulating T cell suppression, interaction with other transcription factors, similar to what has been shown with Foxp3 in Tregs, may be taking place. NFAT proteins have already been shown to cooperate with several transcriptional repressors, including ICER, a member of the CREB/CREM family of transcription factors, PPRγ and p21SNFT [74–76]. As we have seen during anergy induction, the choice of NFAT transcriptional partners may underlie the different patterns of gene expression that can be detected in CD4+ T cells when they are stimulated in the presence or the absence of CD4+CD25+ Tregs [77]. These results suggest that NFAT may not only control T cell inactivation due to anergy induction, but also suppression caused by regulatory T cells. Common pathways may therefore be shared between these two mechanisms of peripheral T cell tolerance
4. Mechanisms of transcriptional repression in the implementation and maintenance of T cell anergy
Initial molecular characterizations of T cell anergy identified a defect in the activation of the Ras/MAP kinase signaling pathway that could account for the defective activation of AP-1 in anergic T cells [78, 79]. Several mechanisms have been proposed to explain this block of TCR signaling. Proteins with ubiquitin ligase activity, including Itch, Grail and Cbl-b, have been implicated in negatively regulating TCR signals in anergic T cells [44–46, 80]. These enzymes can specifically target proteins such as PLCγ and PKCθ or RhoGDI, leading to their functional inactivation or degradation [45, 81]. Upregulation of DGKα has also been recently shown to prevent MAPK activation in anergic T cells by increasing the conversion of diacylglycerol into phosphatidic acid [42, 43].
Although all those proteins could cause severe signaling defects that would lead to defective TCR engagement-mediated upregulation of IL-2 expression, evidence suggested that an active mechanism of transcriptional repression was also activated in anergic T cells that directly inhibited IL-2 transcription. Early studies using reporter vectors containing different fragments of the IL-2 promoter, identified several regions that were required to detect a decrease in expression when transfected into anergic T cell clones [13, 14]. A site located at the -180 position in the IL-2 promoter was later shown to specifically bind CREB/CREM complexes in anergic T cells, suggesting that transcriptional inactive isoforms of CREM, such as ICER, could be forming repressor complexes with CREB bound to the IL-2 promoter [17, 82]. Specific complexes formed by Bcl-3 and NF-κB p50 homodimers have also been reported to bind the IL-2 promoter in T cells anergized with superantigen and possibly exert an inhibitory effect on IL-2 transcription [83].
In an in vivo model of T cell tolerance induced by immunization in the presence of antiCD40L and a CTLA-4-Ig fusion protein, it has been recently shown that tolerant T cells fail to inactivate Smad3 due to defective phosphorylation by cyclin dependent kinases [15]. In tolerized T cells these kinases are not properly activated likely due to the increased levels of p27kip [15, 84]. Smad3 would remain then active, secondary to this increase in p27kip, and could inhibit IL-2 expression and proliferation in tolerized T cells, as adoptively transferred Smad3 knocked-down T cells are resistant to tolerization in this model [15]. Smad3 had been previously shown to mediate TGF-β-induced inhibition of IL-2 expression in T cells [85], it is possible then that it plays a similar role in anergic T cells directly or indirectly inhibiting IL-2 transcription. cAMP-dependent signaling seems to be responsible for p27kip upregulation in tolerant T cells [84]. These results together with the possible inhibition of IL-2 expression by CREB/CREM complexes suggest that cAMP-mediated signaling may also be an important regulator of the transcriptional mechanisms that control T cell tolerance.
Our laboratory has shown that an active mechanism of transcriptional repression is engaged in anergic Th1 cells. Anergic cells undergo epigenetic changes in the IL-2 promoter locus, leading to a stable inhibition of IL-2 expression [16]. T cells that have been anergized upregulate the expression of Ikaros [7, 16], a transcription factor with key functions during lymphoid lineage development [86, 87]. Our results indicate that Ikaros may also control mature T cell function. In anergic T cells, Ikaros binds to the IL-2 promoter and recruits histone deacetylases (HDAC), inducing chromatin deacetylation at this locus [16]. Chromatin remodeling has already been suggested to control IL-2 gene expression in T cells. The distal region of the IL-2 promoter shows a partially open conformation in naïve T cells [88], which is further remodeled, as indicated by increased susceptibility of this locus to nucleases and increased histone acetylation, when IL-2 expression is induced following TCR and CD28 engagement [88, 89]. Increased histone acetylation at the IL-2 promoter seems to require signals derived from CD28 engagement [90]. Active histone deacetylation of the IL-2 promoter occurs in Th1 cells following partial stimulation which causes stable inhibition of IL-2 transcription. The induction of anergy would thus engage an active program of differentiation that would result in the generation of a population of T cells in which a specific cytokine expression pattern in response to stimulation has been epigenetically imprinted. Regulation of locus accessibility plays also key role in programs of T cell differentiation such as T helper cell differentiation [57, 91]. Changes in histone acetylation of the IL-4 and IFNγ genes define stable T cell populations and determine whether those genes will become silent or will be ready to be activated upon stimulation in Th1 or Th2 subpopulations. Whether other chromatin modifications occur concomitantly or are facilitated by histone deacetylation of the IL-2 promoter in anergic T cells remains to be determined. Specific Ikaros binding could also account for some degree of selectivity in the inhibition of cytokine expression in anergic T cells. Further research would be needed to determine this point. Therefore, epigenetic changes may contribute to induce a stable inactive status on the IL-2 locus, and to make inhibition of the IL-2 gene expression long-lasting in anergic T cells.
5. Conclusions
Transcriptional mechanisms regulate the two different phases of T cell anergy. First, the induction phase of T cell anergy results from an unbalanced activation of NFAT in the absence of full AP-1 activation caused by suboptimal or partial stimulation. Tolerizing stimuli induce the expression of a Ca2+/Cn/NAFT-dependent AP-1-independent program of gene expression. Anergy is then implemented and maintained by proteins encoded by those genes. Many of theses proteins, such as the E3 ubiquitin ligases or DGKα inhibit T cell function by blocking signaling initiated from the TCR; some others, like Ikaros, actively cause repression of IL-2 transcription of and possibly other cytokines. Keeping self-reactive T cells in check is crucial to prevent autoimmunity and it may explain the existence of multiple complementary mechanisms designed to inhibit self-reactive T cell activation. The characterization not only of the mechanisms that result in the inhibition of TCR signaling and cytokine transcription, but also of the transcriptional complexes that are responsible for the expression of anergy-inducing genes, may offer new therapeutic targets to modulate T cell function and promote tolerance in the treatment of autoimmune disease and in the prevention of graft rejection.
Figure 1. NFAT induces a specific program of gene expression in anergic T cells.
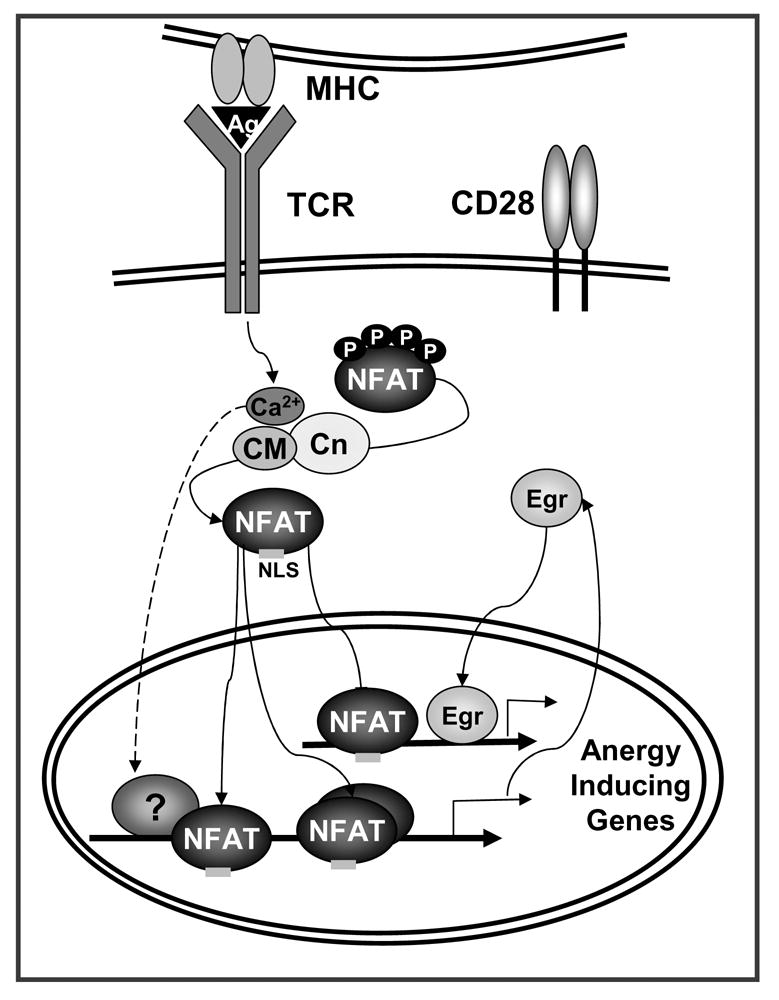
TCR engagement without concomitant costimulation results in the unbalance activation of calcium signaling in the absence of full activation of other pathways (i.e. Ras/MAPK, PKC, IKK). Sustained increased intracellular calcium activates the calmodulin (CM) dependent phosphatase calcineurin (Cn) which dephosphorylates NFAT, exposing a nuclear localization signal (NLS) and inducing NFAT nuclear translocation. In the absence of AP-1 proteins, NFAT induces an anergy-inducing program of gene expression. NFAT also upregulates the expression of Egr2 and Egr3 (Egr) that contribute in cooperation with NFAT or in an independent manner, to activate the expression of anergy-inducing genes. The proteins encoded by those genes (e.g. Itch, Grail, Cbl-b, DGKα, Ikaros) will cause dampening of TCR signaling and IL-2 silencing in anergic T cells.
Figure 2. Active mechanisms of transcriptional repression contribute to the implementation and maintenance of anergy in T cells.
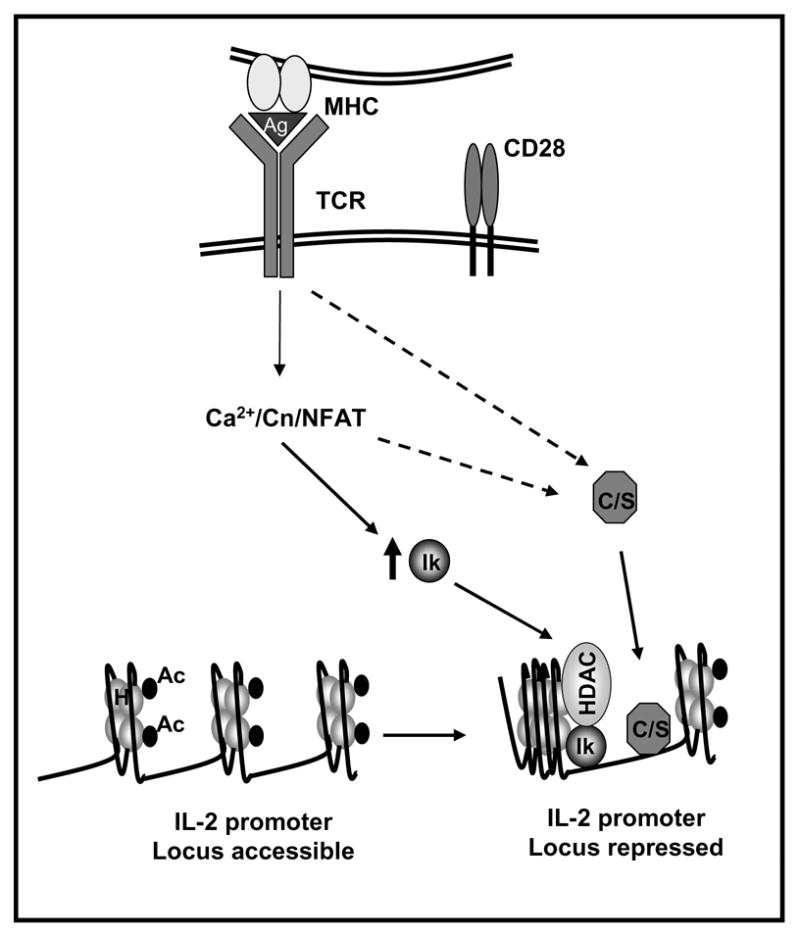
Anergizing stimuli induce the calcium/NFAT-dependent upregulation of the expression of Ikaros (Ik). In anergic T cells Ikaros binds to the IL-2 promoter and recruits histone deacetylases (HDAC). These enzymes would remove acetyl groups (Ac) from core nucleosome histones (H) leading to the establishment of epigenetic changes that result in stable silencing of the IL-2 gene expression. Other transcriptional repressors (i.e. CREM proteins and Smad3 (C/S)) may be activated by calcium or other signals and bind to the IL-2 promoter also inhibiting IL-2 expression.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM007288 and AI059738) and the Irene Diamond Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Macian F, Im SH, Garcia-Cozar FJ, Rao A. T-cell anergy. Curr Opin Immunol. 2004;16:209–16. doi: 10.1016/j.coi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 3.Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol. 2002;2:11–9. doi: 10.1038/nri701. [DOI] [PubMed] [Google Scholar]
- 4.Jenkins MK, Chen CA, Jung G, Mueller DL, Schwartz RH. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J Immunol. 1990;144:16–22. [PubMed] [Google Scholar]
- 5.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165:302–19. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quill H, Schwartz RH. Stimulation of normal inducer T cell clones with antigen presented by purified Ia molecules in planar lipid membranes: specific induction of a long-lived state of proliferative nonresponsiveness. J Immunol. 1987;138:3704–12. [PubMed] [Google Scholar]
- 7.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 8.Lechner O, Lauber J, Franzke A, Sarukhan A, von Boehmer H, Buer J. Fingerprints of anergic T cells. Curr Biol. 2001;11:587–95. doi: 10.1016/s0960-9822(01)00160-9. [DOI] [PubMed] [Google Scholar]
- 9.Crabtree GR. Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611–4. doi: 10.1016/s0092-8674(00)80571-1. [DOI] [PubMed] [Google Scholar]
- 10.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–32. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 11.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 12.Mueller DL. E3 ubiquitin ligases as T cell anergy factors. Nat Immunol. 2004;5:883–890. doi: 10.1038/ni1106. [DOI] [PubMed] [Google Scholar]
- 13.Kitagawa-Sakakida S, Schwartz RH. Multifactor cis-dominant negative regulation of IL-2 gene expression in anergized T cells. J Immunol. 1996;157:2328–39. [PubMed] [Google Scholar]
- 14.Becker JC, Brabletz T, Kirchner T, Conrad CT, Brocker EB, Reisfeld RA. Negative transcriptional regulation in anergic T cells. Proc Natl Acad Sci U S A. 1995;92:2375–8. doi: 10.1073/pnas.92.6.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Iwamoto Y, Berezovskaya A, Boussiotis VA. A pathway regulated by cell cycle inhibitor p27(Kip1) and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat Immunol. 2006;7:1157–65. doi: 10.1038/ni1398. [DOI] [PubMed] [Google Scholar]
- 16.Bandyopadhyay S, Dure M, Paroder M, Soto-Nieves N, Puga I, Macian F. Interleukin 2 gene transcription is regulated by Ikaros-induced changes in histone acetylation in anergic T cells. Blood. 2006 doi: 10.1182/blood-2006-07-037754. Epub 2006 Dec 05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Powell JD, Lerner CG, Ewoldt GR, Schwartz RH. The -180 site of the IL-2 promoter is the target of CREB/CREM binding in T cell anergy. J Immunol. 1999;163:6631–9. [PubMed] [Google Scholar]
- 18.Telander DG, Malvey EN, Mueller DL. Evidence for repression of IL-2 gene activation in anergic T cells. J Immunol. 1999;162:1460–5. [PubMed] [Google Scholar]
- 19.Glynne R, Akkaraju S, Healy JI, Rayner J, Goodnow CC, Mack DH. How self-tolerance and the immunosuppressive drug FK506 prevent B-cell mitogenesis. Nature. 2000;403:672–6. doi: 10.1038/35001102. [DOI] [PubMed] [Google Scholar]
- 20.Knoechel B, Lohr J, Zhu S, Wong L, Hu D, Ausubel L, et al. Functional and molecular comparison of anergic and regulatory T lymphocytes. J Immunol. 2006;176:6473–83. doi: 10.4049/jimmunol.176.11.6473. [DOI] [PubMed] [Google Scholar]
- 21.Kurella S, Yaciuk JC, Dozmorov I, Frank MB, Centola M, Farris AD. Transcriptional modulation of TCR, Notch and Wnt signaling pathways in SEB-anergized CD4+ T cells. Genes Immun. 2005;6:596–608. doi: 10.1038/sj.gene.6364245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 23.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–85. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 24.Feske S, Okamura H, Hogan PG, Rao A. Ca2+/calcineurin signalling in cells of the immune system. Biochem Biophys Res Commun. 2003;311:1117–32. doi: 10.1016/j.bbrc.2003.09.174. [DOI] [PubMed] [Google Scholar]
- 25.Aramburu J, Heitman J, Crabtree GR. Calcineurin: a central controller of signalling in eukaryotes. EMBO Rep. 2004;5:343–8. doi: 10.1038/sj.embor.7400133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aramburu J, Garcia-Cozar F, Raghavan A, Okamura H, Rao A, Hogan PG. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol Cell. 1998;1:627–37. doi: 10.1016/s1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- 27.Okamura H, Aramburu J, Garcia-Rodriguez C, Viola JPB, Raghavan A, Tahiliani M, et al. Concerted Dephosphorylation of the Transcription Factor NFAT1 Induces a Conformational Switch that Regulates Transcriptional Activity. Mol Cell. 2000;6:539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 28.Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, et al. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature. 2006;441:646–50. doi: 10.1038/nature04631. [DOI] [PubMed] [Google Scholar]
- 29.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–4. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 30.Chow CW, Dong C, Flavell RA, Davis RJ. c-Jun NH(2)-terminal kinase inhibits targeting of the protein phosphatase calcineurin to NFATc1. Mol Cell Biol. 2000;20:5227–34. doi: 10.1128/mcb.20.14.5227-5234.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez del Arco P, Martinez-Martinez S, Maldonado JL, Ortega-Perez I, Redondo JM. A role for the p38 MAP kinase pathway in the nuclear shuttling of NFATp. J Biol Chem. 2000;275:13872–8. doi: 10.1074/jbc.275.18.13872. [DOI] [PubMed] [Google Scholar]
- 32.Zhu J, Shibasaki F, Price R, Guillemot JC, Yano T, Dotsch V, et al. Intramolecular masking of nuclear import signal on NF-AT4 by casein kinase I and MEKK1. Cell. 1998;93:851–61. doi: 10.1016/s0092-8674(00)81445-2. [DOI] [PubMed] [Google Scholar]
- 33.Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A. A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Mol Cell Biol. 2004;24:4184–95. doi: 10.1128/MCB.24.10.4184-4195.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, et al. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature. 1993;365:352–5. doi: 10.1038/365352a0. [DOI] [PubMed] [Google Scholar]
- 35.Jain J, McCaffrey PG, Valge-Archer VE, Rao A. Nuclear factor of activated T cells contains Fos and Jun. Nature. 1992;356:801–4. doi: 10.1038/356801a0. [DOI] [PubMed] [Google Scholar]
- 36.Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–89. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 37.Macian F, Garcia-Rodriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 2000;19:4783–95. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–8. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- 39.Heissmeyer V, Macian F, Varma R, Im SH, Garcia-Cozar F, Horton HF, et al. A molecular dissection of lymphocyte unresponsiveness induced by sustained calcium signalling. Novartis Found Symp. 2005;267:165–74. [PubMed] [Google Scholar]
- 40.Serfling E, Klein-Hessling S, Palmetshofer A, Bopp T, Stassen M, Schmitt E. NFAT transcription factors in control of peripheral T cell tolerance. Eur J Immunol. 2006;36:2837–43. doi: 10.1002/eji.200536618. [DOI] [PubMed] [Google Scholar]
- 41.Borde M, Barrington RA, Heissmeyer V, Carroll MC, Rao A. Transcriptional basis of lymphocyte tolerance. Immunol Rev. 2006;210:105–19. doi: 10.1111/j.0105-2896.2006.00370.x. [DOI] [PubMed] [Google Scholar]
- 42.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, et al. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7:1174–81. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 43.Zha Y, Marks R, Ho AW, Peterson AC, Janardhan S, Brown I, et al. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat Immunol. 2006;7:1166–73. doi: 10.1038/ni1394. [DOI] [PubMed] [Google Scholar]
- 44.Anandasabapathy N, Ford GS, Bloom D, Holness C, Paragas V, Seroogy C, et al. GRAIL: an E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity. 2003;18:535–47. doi: 10.1016/s1074-7613(03)00084-0. [DOI] [PubMed] [Google Scholar]
- 45.Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–65. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- 46.Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21:167–77. doi: 10.1016/j.immuni.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 47.Giffin MJ, Stroud JC, Bates DL, von Koenig KD, Hardin J, Chen L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR kappa B element. Nat Struct Biol. 2003;10:800–6. doi: 10.1038/nsb981. [DOI] [PubMed] [Google Scholar]
- 48.Jin L, Sliz P, Chen L, Macian F, Rao A, Hogan PG, et al. An asymmetric NFAT1 dimer on a pseudo-palindromic kappa B-like DNA site. Nat Struct Biol. 2003;10:807–11. doi: 10.1038/nsb975. [DOI] [PubMed] [Google Scholar]
- 49.Heyer J, Kneitz B, Schuh K, Jankevics E, Siebelt F, Schimpl A, et al. Inefficient termination of antigen responses in NF-ATp-deficient mice. Immunobiology. 1997;198:162–9. doi: 10.1016/S0171-2985(97)80037-X. [DOI] [PubMed] [Google Scholar]
- 50.Hodge MR, Ranger AM, Charles de la Brousse F, Hoey T, Grusby MJ, Glimcher LH. Hyperproliferation and dysregulation of IL-4 expression in NF-ATp-deficient mice. Immunity. 1996;4:397–405. doi: 10.1016/s1074-7613(00)80253-8. [DOI] [PubMed] [Google Scholar]
- 51.Ranger AM, Oukka M, Rengarajan J, Glimcher LH. Inhibitory function of two NFAT family members in lymphoid homeostasis and Th2 development. Immunity. 1998;9:627–35. doi: 10.1016/s1074-7613(00)80660-3. [DOI] [PubMed] [Google Scholar]
- 52.Rengarajan J, Tang B, Glimcher LH. NFATc2 and NFATc3 regulate T(H)2 differentiation and modulate TCR-responsiveness of naive T(H)cells. Nat Immunol. 2002;3:48–54. doi: 10.1038/ni744. [DOI] [PubMed] [Google Scholar]
- 53.Schuh K, Kneitz B, Heyer J, Bommhardt U, Jankevics E, Berberich-Siebelt F, et al. Retarded thymic involution and massive germinal center formation in NF-ATp-deficient mice. Eur J Immunol. 1998;28:2456–66. doi: 10.1002/(SICI)1521-4141(199808)28:08<2456::AID-IMMU2456>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 54.Xanthoudakis S, Viola JP, Shaw KT, Luo C, Wallace JD, Bozza PT, et al. An enhanced immune response in mice lacking the transcription factor NFAT1. Science. 1996;272:892–5. doi: 10.1126/science.272.5263.892. [DOI] [PubMed] [Google Scholar]
- 55.Stroud JC, Chen L. Structure of NFAT bound to DNA as a monomer. J Mol Biol. 2003;334:1009–22. doi: 10.1016/j.jmb.2003.09.065. [DOI] [PubMed] [Google Scholar]
- 56.Ho IC, Hodge MR, Rooney JW, Glimcher LH. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996;85:973–83. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- 57.Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat Immunol. 2002;3:643–51. doi: 10.1038/ni808. [DOI] [PubMed] [Google Scholar]
- 58.Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002;195:1003–12. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Youn HD, Chatila TA, Liu JO. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO J. 2000;19:4323–31. doi: 10.1093/emboj/19.16.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan F, Ye Z, Cheng L, Liu JO. Myocyte enhancer factor 2 mediates calcium-dependent transcription of the interleukin-2 gene in T lymphocytes: a calcium signaling module that is distinct from but collaborates with the nuclear factor of activated T cells (NFAT) J Biol Chem. 2004;279:14477–80. doi: 10.1074/jbc.C300487200. [DOI] [PubMed] [Google Scholar]
- 61.Rengarajan J, Mittelstadt PR, Mages HW, Gerth AJ, Kroczek RA, Ashwell JD, et al. Sequential involvement of NFAT and Egr transcription factors in FasL regulation. Immunity. 2000;12:293–300. doi: 10.1016/s1074-7613(00)80182-x. [DOI] [PubMed] [Google Scholar]
- 62.Harris JE, Bishop KD, Phillips NE, Mordes JP, Greiner DL, Rossini AA, et al. Early growth response gene-2, a zinc-finger transcription factor, is required for full induction of clonal anergy in CD4+ T cells. J Immunol. 2004;173:7331–8. doi: 10.4049/jimmunol.173.12.7331. [DOI] [PubMed] [Google Scholar]
- 63.Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. 2005;6:472–80. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 64.Barrington RA, Borde M, Rao A, Carroll MC. Involvement of NFAT1 in B cell self-tolerance. J Immunol. 2006;177:1510–5. doi: 10.4049/jimmunol.177.3.1510. [DOI] [PubMed] [Google Scholar]
- 65.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 66.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–27. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 67.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 68.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 69.Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A. 2005;102:5138–43. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 71.Su L, Creusot RJ, Gallo EM, Chan SM, Utz PJ, Fathman CG, et al. Murine CD4+CD25+ regulatory T cells fail to undergo chromatin remodeling across the proximal promoter region of the IL-2 gene. J Immunol. 2004;173:4994–5001. doi: 10.4049/jimmunol.173.8.4994. [DOI] [PubMed] [Google Scholar]
- 72.Chen C, Rowell EA, Thomas RM, Hancock WW, Wells AD. Transcriptional regulation by foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J Biol Chem. 2006;281:36828–34. doi: 10.1074/jbc.M608848200. [DOI] [PubMed] [Google Scholar]
- 73.Bopp T, Palmetshofer A, Serfling E, Heib V, Schmitt S, Richter C, et al. NFATc2 and NFATc3 transcription factors play a crucial role in suppression of CD4+ T lymphocytes by CD4+ CD25+ regulatory T cells. J Exp Med. 2005;201:181–7. doi: 10.1084/jem.20041538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bodor J, Habener JF. Role of transcriptional repressor ICER in cyclic AMP-mediated attenuation of cytokine gene expression in human thymocytes. J Biol Chem. 1998;273:9544–51. doi: 10.1074/jbc.273.16.9544. [DOI] [PubMed] [Google Scholar]
- 75.Iacobelli M, Wachsman W, McGuire KL. Repression of IL-2 promoter activity by the novel basic leucine zipper p21SNFT protein. J Immunol. 2000;165:860–8. doi: 10.4049/jimmunol.165.2.860. [DOI] [PubMed] [Google Scholar]
- 76.Yang XY, Wang LH, Chen T, Hodge DR, Resau JH, DaSilva L, et al. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J Biol Chem. 2000;275:4541–4. doi: 10.1074/jbc.275.7.4541. [DOI] [PubMed] [Google Scholar]
- 77.Sukiennicki TL, Fowell DJ. Distinct molecular program imposed on CD4+ T cell targets by CD4+CD25+ regulatory T cells. J Immunol. 2006;177:6952–61. doi: 10.4049/jimmunol.177.10.6952. [DOI] [PubMed] [Google Scholar]
- 78.Fields PE, Gajewski TF, Fitch FW. Blocked Ras activation in anergic CD4+ T cells. Science. 1996;271:1276–8. doi: 10.1126/science.271.5253.1276. [DOI] [PubMed] [Google Scholar]
- 79.Li W, Whaley CD, Mondino A, Mueller DL. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science. 1996;271:1272–6. doi: 10.1126/science.271.5253.1272. [DOI] [PubMed] [Google Scholar]
- 80.Soares L, Seroogy C, Skrenta H, Anandasabapathy N, Lovelace P, Chung CD, et al. Two isoforms of otubain 1 regulate T cell anergy via GRAIL. Nat Immunol. 2004;5:45–54. doi: 10.1038/ni1017. [DOI] [PubMed] [Google Scholar]
- 81.Su L, Lineberry N, Huh Y, Soares L, Fathman C. A Novel E3 Ubiquitin Ligase Substrate Screen Identifies Rho Guanine Dissociation Inhibitor as a Substrate of Gene Related to Anergy in Lymphocytes. J Immunol. 2006;177:7559–7566. doi: 10.4049/jimmunol.177.11.7559. [DOI] [PubMed] [Google Scholar]
- 82.Bodor J, Feigenbaum L, Bodorova J, Bare C, Reitz MS, Jr, Gress RE. Suppression of T-cell responsiveness by inducible cAMP early repressor (ICER) J Leukoc Biol. 2001;69:1053–9. [PubMed] [Google Scholar]
- 83.Grundstrom S, Anderson P, Scheipers P, Sundstedt A. Bcl-3 and NFkappaB p50-p50 homodimers act as transcriptional repressors in tolerant CD4+ T cells. J Biol Chem. 2004;279:8460–8. doi: 10.1074/jbc.M312398200. [DOI] [PubMed] [Google Scholar]
- 84.Boussiotis VA, Freeman GJ, Taylor PA, Berezovskaya A, Grass I, Blazar BR, et al. p27kip1 functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat Med. 2000;6:290–7. doi: 10.1038/73144. [DOI] [PubMed] [Google Scholar]
- 85.McKarns SC, Schwartz RH, Kaminski NE. Smad3 is essential for TGF-beta 1 to suppress IL-2 production and TCR-induced proliferation, but not IL-2-induced proliferation. J Immunol. 2004;172:4275–84. doi: 10.4049/jimmunol.172.7.4275. [DOI] [PubMed] [Google Scholar]
- 86.Georgopoulos K. Haematopoietic cell-fate decisions, chromatin regulation and ikaros. Nat Rev Immunol. 2002;2:162–74. doi: 10.1038/nri747. [DOI] [PubMed] [Google Scholar]
- 87.Su RC, Sridharan R, Smale ST. Assembly of silent chromatin during thymocyte development. Semin Immunol. 2005;17:129–40. doi: 10.1016/j.smim.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 88.Ward SB, Hernandez-Hoyos G, Chen F, Waterman M, Reeves R, Rothenberg EV. Chromatin remodeling of the interleukin-2 gene: distinct alterations in the proximal versus distal enhancer regions. Nucleic Acids Res. 1998;26:2923–34. doi: 10.1093/nar/26.12.2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rao S, Gerondakis S, Woltring D, Shannon MF. c-Rel is required for chromatin remodeling across the IL-2 gene promoter. J Immunol. 2003;170:3724–31. doi: 10.4049/jimmunol.170.7.3724. [DOI] [PubMed] [Google Scholar]
- 90.Thomas RM, Gao L, Wells AD. Signals from CD28 induce stable epigenetic modification of the IL-2 promoter. J Immunol. 2005;174:4639–46. doi: 10.4049/jimmunol.174.8.4639. [DOI] [PubMed] [Google Scholar]
- 91.Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity. 1998;9:765–75. doi: 10.1016/s1074-7613(00)80642-1. [DOI] [PubMed] [Google Scholar]