

Abstract
Objective
To test the hypotheses that some thrombin-reactive anti-cardiolipin antibodies (aCL) may bind to protein C (PC) and/or activated PC (APC), and that some of the PC- and APC-reactive aCL may inhibit PC activation and/or the function of APC.
Methods
We studied the reactivity of patient-derived monoclonal aCL with PC and APC. Of the reactive antibodies, we examined their effects on PC activation and the activity of APC in plasma coagulation.
Results
Five of five patient-derived thrombin-reactive monoclonal aCL bind to PC and APC. In addition, one patient-derived monoclonal anti-prothrombin antibody (aPT) that displays aCL activity and reacts with thrombin also binds to PC and APC. Of these 6 PC- and APC-reactive aCL/aPT, all failed to inhibit PC activation, but one (CL15) shortened the plasma coagulation time in the presence of exogenous APC and thus inhibited the anticoagulant function of APC.
Conclusion
Most of the thrombin-reactive aCL in APS patients may bind to PC and APC. Of the APC-reactive aCL, some (like CL15) may inhibit the anticoagulant function of APC and thus are likely to be prothrombotic in the host.
INTRODUCTION
Immunological studies of antiphospholipid antibodies (aPL) in the antiphospholipid syndrome (APS) have shown that aPL represent a heterogeneous group of immunologically distinct antibodies (Ab) that recognize various phospholipids (PL), PL-binding plasma proteins and/or PL-protein complexes (1-6). The involved plasma proteins include β2 glycoprotein-1 (β2GPI), prothrombin (PT), protein C (PC), and protein S. Mechanistically, aPL of different binding specificities are thought to promote thrombosis via different mechanisms. For example, Ab against β2GPI and/or its complexes with cardiolipin (CL) were suggested to interact with endothelial cells (EC) and monocytes, and induce a tissue factor (TF)-dependent procoagulant state (7-10). On the other hand, Ab against PC, protein S, or PL in complexes with either PC or activated PC (APC) and protein S may inhibit activation of PC and/or function of APC (6, 11-15). Since APC proteolytically inactivates the activated factors V and VIII (denoted as Va an VIIIa, respectively), the reduced activation of PC and/or reduced APC function may lead to a procoagulant effect and thrombotic events. Of note, congenital, heterozygous PC deficiency increases the risk of venous thrombosis about five to ten fold (16).
Recently, during our studies of anti-PT Ab (aPT), we found that the IS6 monoclonal IgG aPT (derived from a primary APS patient) binds to CL and thrombin, and that 5/7 patient-derived monoclonal IgG aCL crossreact with PT and thrombin (17). Of these 6 thrombin-reactive IgG monoclonal Ab (mAb), CL24 could interfere with inactivation of thrombin by antithrombin (AT) (17). Therefore, CL24 and two other thrombin-reactive mAb were analyzed for their binding affinities to thrombin and PT by competitive inhibition. The results showed that soluble thrombin was more effective than PT in inhibiting all three mAb binding to either PT or thrombin on solid phase. Importantly, thrombin could inhibit all tested mAb from binding to thrombin and PT, while PT could only inhibit mAb from binding to PT but not thrombin. These results demonstrated that these three mAb are more specific for thrombin than PT. Based on these inhibition data, the relative Kd values of these “anti-thrombin antibodies” are around 1.7-7.5 × 10-6 M (17).
The discovery of thrombin-reactive aCL raised a possibility that such aCL may also react with PC, which contains a trypsin/thrombin-like serine protease domain. A comparison of PC and thrombin at the protein level shows these two proteins share a similarity of 50.5% with an identity of 40.8% (Figure 1). The homologous amino acid sequences reside mainly in the trypsin/thrombin-like serine protease domains of thrombin and PC, as well as APC, which is generated from PC after cleavage by the thrombin-thrombomodulin complex between residues R211 and L212 in the heavy chain of PC (based on the amino acid positions of the PC precursor, Figure 1). Similarly, thrombin and APC are homologous at the structural level. As can be seen in their 3-dimentional (3D) structures in a ribbon model (Figures 2 A and B), thrombin and APC share three α-helixes on the left side, two β-sheets at the bottom side, and a homologous active site at the center that consists of the identical catalytic triad residues (H406, D462, S568 for thrombin and H253, D300, S405 for APC) (18, 19). Moreover, the space-filling models of both molecules show that most of the amino acid residues in the highly homologous regions are on the surface (Figures 2 C and D). The two most homologous regions (colored in red and magenta, respectively) are in the active site cleft and contain His and Ser of the catalytic triad residues in both proteins (Figure 2). The third region (colored in orange) is in close proximity to the catalytic center. The fourth homologous region (colored in green) is a part of the domain that corresponds to the exosite I in thrombin and the loops 60-70 in APC, which are implicated in interaction with their macromolecular substrates (20, 21).
Figure 1.

Amino acid sequence comparison of human thrombin and human PC. The amino acid sequence of thrombin (residues 328-622 of PT, pir:tbhu) was compared with that of PC (residues 43-461 of the PC precursor, pir:kxhu) using the Gap program. PC is a two-chain molecule, consisted of a light chain (residues 43-197) and a heavy chain (residues 200-461). The dots (.) are introduced to maximize the similarity between the two sequences. The heavy chains (beginning with I364 in thrombin and D200 in PC, respectively) and trypsin/thrombin-like serine protease domains (encompassing residues 364-613 in thrombin and 212-445 in PC, respectively) in both proteins are marked. The pipe characters (∣) are put between amino acids that are the same; colons (:) between amino acids whose comparison values are greater or equal to 0.5; and periods (.) between amino acids whose comparison values are greater or equal to 0.1. Four regions of the most homologous amino acid sequence are underlined.
Figure 2.
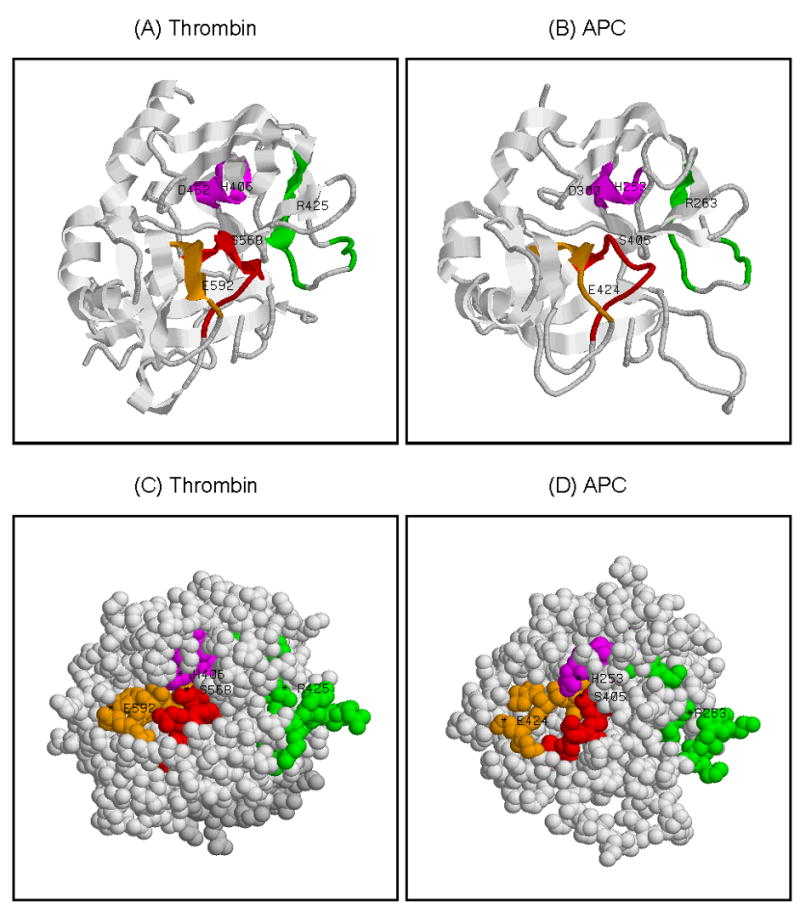
Human thrombin and APC share homologous structures and their surfaces contain regions of the highly homologous amino acid sequence. The 3D structures of thrombin and APC in the ribbon (panels A and B) and the space-filling (panels C and D) models are given. The homologous regions (in the order of regional similarity) are colored respectively in: red (D562-P571 for thrombin and D399-P408 for APC); magenta (W400-L408 for thrombin and W247-M255 for APC); orange (G586-C594 for thrombin and G418-C426 for APC); and green (L422-H429, R433-R436 for thrombin and L260-Y267, R271-K274 for APC). Thrombin consists of a light chain (residues 328-363, in light gray color, panels A and C) and a heavy chain (residues 364-622, in gray color, panels A and C). In panel A, the ribbon model of thrombin looking directly into the active site cleft, with the catalytic residues of thrombin (H406, D462, S568) at the center. In addition, residues implicated in fibrinogen recognition (R425) and antithrombin interaction (E592) (20, 36) are marked. In panel B, the ribbon model of APC is given in an orientation similar to thrombin in panel A. The catalytic residues of APC (H253, D300, S405) are at the center, and the residues R263 and E424 in APC that correspond respectively to R425 and E592 of thrombin are marked. In panel C, the space-filling model of thrombin looking directly into the active site cleft in the standard orientation (having the cleft running horizontally left to right) with the exosite I on the right side. In panel D, the space-filling model of APC is given in an orientation similar to thrombin in panel C.
This similarity of thrombin to PC and APC led us to hypothesize that some of the thrombin-reactive monoclonal aCL may bind to PC and/or APC and thus interfere with PC activation and/or APC function. We now report that 6/6 thrombin-reactive monoclonal IgG aCL/aPT bind to both PC and APC, and, of these, CL15 inhibits the anticoagulant activity of APC.
MATERIALS AND METHODS
Computer sequence analysis and 3-dimentional (3D) structure modeling
The amino acid sequence of human thrombin (residues 328-622 of PT, with the accession name of ‘tbhu’ in the Protein Information Resource/pir databases) was compared with that of human PC (residues 43-461 of the PC precursor, pir:kxhu) using the Gap program in the Genetics Computer Group software package (22). The Gap program uses a scoring matrix with matches scored as 1.5 and mismatches scored according to the evolutionary distance between the amino acids. The 3D structures of thrombin and APC in the ribbon and the space-filling models were generated based respectively on the coordinates of human thrombin (1A5G) (18) and human APC (1AUT) (19), using the RasMol software (V2.7; Bernstein & Sons, Bellport, NY).
Patient-derived monoclonal aCL and aPT
Seven IgG monoclonal aCL and one IgG monoclonal aPT were analyzed in the present study. The aCL included CL1, CL15, CL24, IS1, IS2, IS3 and IS4 (23), and the single aPT was IS6 (24). Their generation and characterization had been reported previously (23, 24).
Enzyme-linked immunosorbent assays (ELISA) for Ab against PC and APC
High-binding ELISA plates (Costar, Cambridge, MA) were coated with 5 μg/ml of human PC or human APC (both from Haematologic Technologies, Essex Junction, VT) in Tris-buffered saline (TBS, 20 mM Tris-HCl, 150 mM NaCl, pH 7.4) containing 2.5 mM CaCl2. After incubating overnight at 4 °C, plates were blocked with TBS/2.5 mM CaCl2 containing 0.3% gelatin. Then test mAb or control IgG (1 μg/ml) in TBS/2.5 mM CaCl2 containing 0.1% gelatin were distributed to wells in duplicate and incubated for 1 hour at room temperature. After washing with TBS/2.5 mM CaCl2, bound human IgG was detected with HRP-conjugated goat anti-human IgG (γ-chain specific; Biosource International, Camarillo, CA) and peroxidase substrate tetramethylbenzidine (Kirkegaard and Perry Laboratories, Gaithersburg, MD).
Preparation of PL liposomes
The PL 1, 2-dilinoleoyl-sn-glycero-3-phosphatidylethanolamine (PE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (PS) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (phosphatidylcholine) were purchased from Avanti Polar Lipids (Alabasta, AL). The PL liposomes were prepared by ultrasonication of a 1 mg/ml mixture of 40% PE, 20% PS and 40% phosphatidylcholine in TBS (15).
Effects of PC-reactive mAb on PC activation
This was done according to Xu et al. (25) with minor modifications. Briefly, the EAhy926 (endothelial hybridoma) cells were cultured at 2×104 cells/well in DMEM (Invitrogen, Carlsbad, CA) supplemented with 20% fetal calf serum in a 24-well culture plate. After incubation for 3 days, cells in wells were washed twice with a Hank’s balanced salt solution (HBSS) containing 3 mM Ca2+, 0.6 mM Mg2+ and 0.1% bovine serum albumin. Thereafter, 25 μl of PC (400 nM) was mixed with 150 μl of test mAb or control IgG (133.3 μg/ml) in the HBSS buffer, and the mixtures added to cells and incubated for 1 hour. The activation reactions were initiated by adding 25 μl of human thrombin (40 nM). The final concentrations of PC, thrombin and IgG were 50 nM, 5 nM and 100 μg/ml, respectively. After 15 minutes at 37 °C, the reactions were stopped by adding 50 μl of hirudin at 75 U/ml in a HEPES buffer (20 mM HEPES-HCl, 150 mM NaCl, pH 7.5). Then the amidolytic activities of APC were measured by transferring 200 μl of the supernatants from each well to a 96-well plate, and then adding 100 μl of APC chromogenic substrate S-2366 (0.6 mM; Chromogenix, Orangeburg, NY) in the HEPES buffer to each well. The rate of substrate hydrolysis (APC activity) was determined based on optical density (OD) measured at 405 nm using a microtiter plate reader. All rates were in a linear range as a function of time. The concentration of APC was determined by comparison with a standard curve of amidolytic activity versus APC concentration constructed with freshly prepared, fully activated PC. Under these conditions, less than 10% of PC was activated during the assay.
Effects of APC-reactive mAb on APC activity
The effects of APC-reactive mAb on APC function were studied initially on the amidolytic activity of APC. Briefly, 50 μl of APC (20 nM) was mixed separately with 50 μl of a test mAb, normal human IgG, or a monoclonal isotype control IgG3 (all at 200 μg/ml) in the HBSS buffer for 1 hour at room temperature. Then, to each reaction mixture was added 100 μl of S-2366 (0.4 mM). After 5 minutes, OD was measured and the APC activity determined as described above.
Subsequently the effects of APC-reactive mAb on APC activity were examined in plasma coagulation according to Smirnov et al. (15) with minor modifications. Experiments were conducted in TBS containing 0.1% gelatin at 25 °C in microtiter plates. The assay was initiated by incubating 10 μl of APC (170 nM) with 10 μl of a test mAb, normal human IgG or a monoclonal isotype control IgG3 (all at 850 μg/ml) for 5 minutes at room temperature. Then, to each reaction mixture was added 20 μl of human normal pooled plasma (Chromogenix), 10 μl of the PL liposomes (425 μg/ml) and 10 μl of a factor X activator (85 ng/ml; purified from Russell’s viper venom, Chromogenix). After one-minute incubation, clotting was initiated by adding 25 μl of 20 mM CaCl2, and OD at 405 nm was measured at every 3 or 5 seconds for real time kinetics in an iEMS microplate photometer (ThermoLab systems, Helsinki, Finland). The clotting time was determined as the time when the OD (above the plasma background) increased by 10% of the OD increase in fully clotted plasma, according to Smirnov et al. (26). The final concentrations of APC, PL liposomes, factor X activator and IgG were respectively 20 nM, 50 μg/ml, 10 ng/ml and 100 μg/ml, unless stated otherwise.
Characterization of the binding affinity of mAb to PC and APC
A competitive inhibition assay was used to study the binding affinity of selected mAb to PC and APC. Briefly, each mAb (1 or 2 μg/ml) was preincubated for 1.5 hours with various concentrations of either PC or APC. Then, the mixture was distributed to the PC- or APC-coated wells in duplicate. After incubation, bound IgG was measured. The inhibition data of each mAb were used to calculate its relative Kd toward PC and APC (27).
Statistical analysis
The effects of all mAb on APC activity in plasma were analyzed using ANOVA followed by the Dunnett’s multiple comparison test. When CL15 was analyzed alone with the control IgG, the student t test (two-tailed) was used. A p value of less than 0.05 was considered significant.
RESULTS
Reactivity of monoclonal aCL to PC and APC
To test our hypothesis that thrombin-reactive aCL may bind to PC and/or APC, we first analyzed our 7 IgG monoclonal aCL for their reactivity with PC. The results revealed that 5/5 thrombin-reactive monoclonal aCL bound to PC, while neither of two non-thrombin-reactive aCL (i.e., IS1 and IS2) bound to PC (Figure 3A). In addition, we analyzed the IS6 monoclonal aPT that also reacted with CL (24) and found that IS6 bound to PC. Of note, all 5 thrombin-reactive aCL also bind to PT, while neither of the two non-thrombin-reactive aCL bind to PT (17). Of these 6 PC-reactive monoclonal aCL/aPT, IS3 and CL1 at 1 μg/ml displayed the strongest binding activity.
Figure 3.
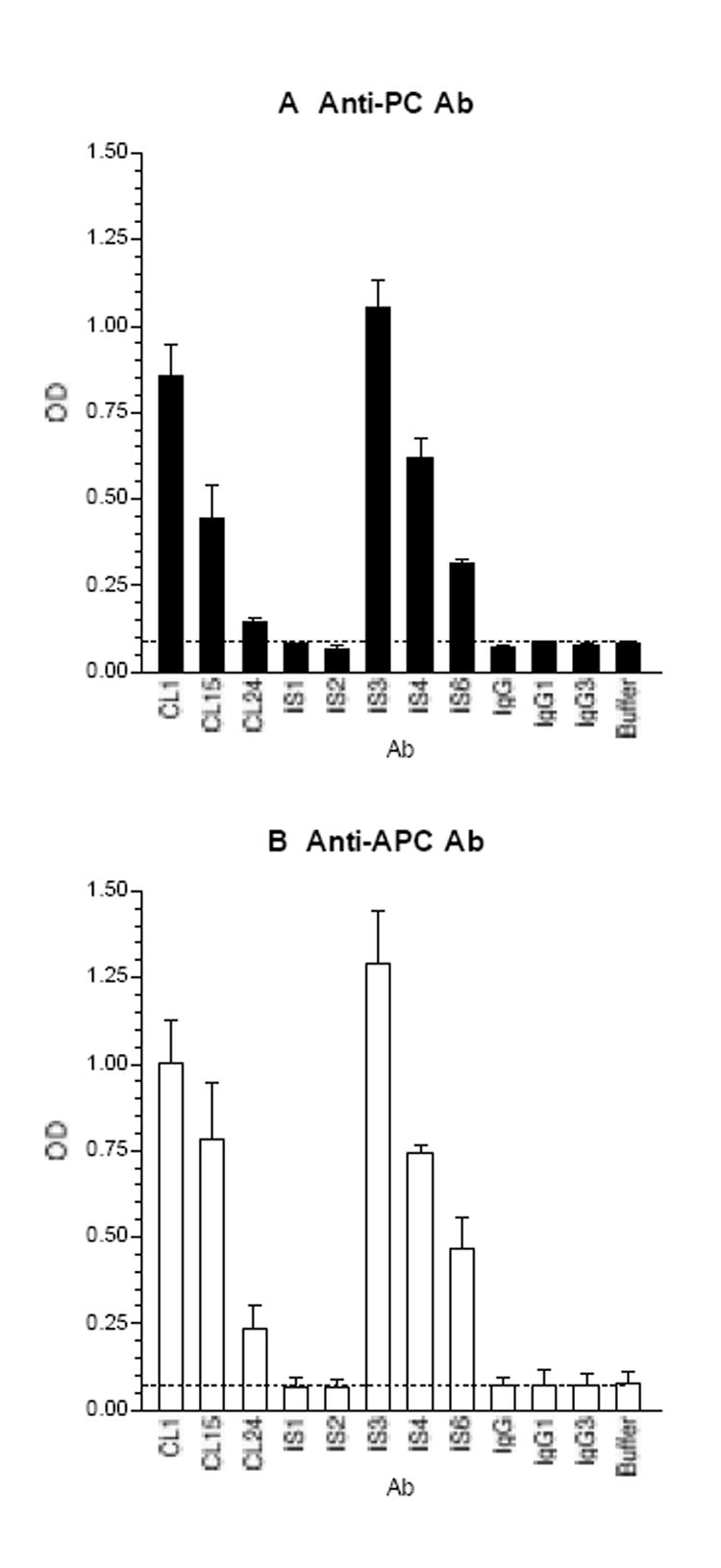
The thrombin-reactive monoclonal aCL/aPT bind to PC (panel A) and APC (panel B). Microtiter wells were coated with PC or APC, and the test mAb and control IgG were analyzed at 1 μg/ml. IS1 and IS2 are IgG1, and the other mAb are IgG3. Bound IgG was measured and expressed in OD; the means and the ranges are given (n = 2).
Subsequently, we analyzed all 8 mAb against APC. Figure 3B shows that 6/6 thrombin-reactive aCL/aPT bind to APC, but none of the non-thrombin-reactive aCL. The binding patterns of tested mAb to APC were similar to those to PC.
Effects of PC-reactive mAb on PC activation
Upon discovery of these 6 patient-derived, PC/APC-reactive IgG mAb, we studied the effects of these mAb on PC activation in an EC-based functional assay. None of these mAb affected the activation of PC on the EC surface (data not shown).
Effects of APC-reactive mAb on APC activity
We then examined these 6 mAb on the amidolytic activity of APC using a small chromogenic substrate of APC, S-2366. None of the 6 mAb affected the amidolytic activity of APC (data not shown). Since S-2366 is a small molecule, the assay might not reflect the inhibitory effects of some APC-reactive mAb on proteolytic inactivation of factors Va and VIIIa by APC. Accordingly, we examined effects of these APC-reactive mAb on APC in plasma coagulation.
To this end, the test mAb were first examined for their background activity (in the absence of APC) in plasma coagulation initiated by the addition of a factor X activator. As can be seen in Figure 4A, except for CL15, all test mAb, as well as the control human IgG (including polyclonal IgG and monoclonal IgG3) did not affect the baseline clotting time (i.e., in the presence of buffer only). CL15 at 100 μg/ml had a slightly extended clotting time of 123 seconds, as compared with 109 seconds (for buffer only) or 103 seconds (for the IgG control) in the presence of 50 μg/ml of PL liposomes. Of note, CL15 displayed a strong lupus anticoagulant activity in a kaolin clotting time test (28).
Figure 4.
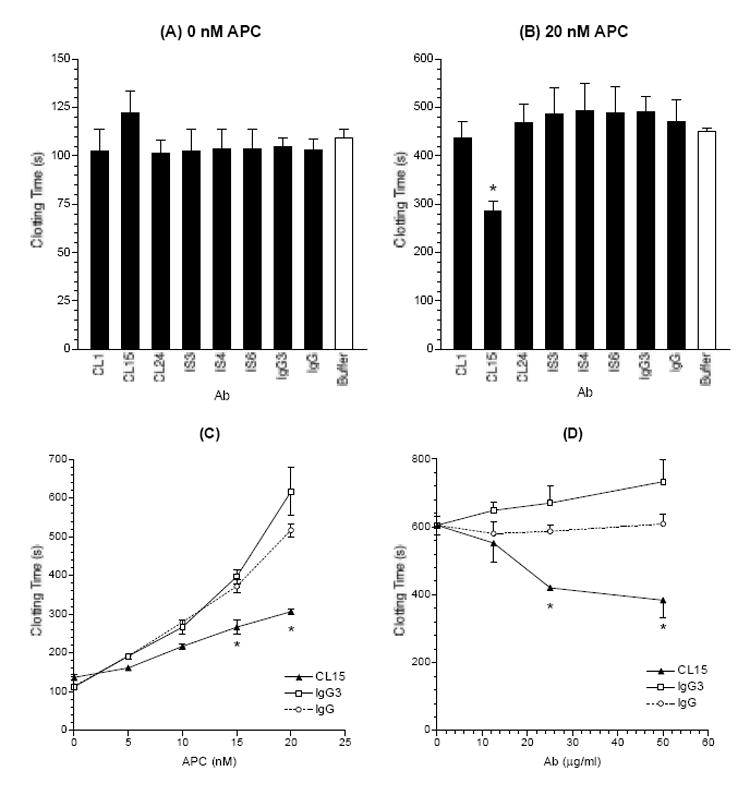
CL15 shortens the plasma coagulation times in the presence of exogenous APC. Test mAb and control IgG were first examined for their background activity on clotting times in the absence of APC (panel A) and then in the presence of 20 nM exogenous APC (panel B). APC was preincubated with test mAb; then to the mixture was added normal pooled plasma, the PL liposomes and the factor X activator. Clotting was initiated by CaCl2 and OD at 405 nm was measured every 3 or 5 seconds. Clotting times (in seconds) were determined as described in the Methods. The mean and standard error of mean (SEM) for each test sample are given (n = 2-3 for A, and 5-6 for B). C, The effects of CL15 were examined in the presence of APC at the indicated concentrations (0-20 nM). The means and the ranges are given (n = 2). D, CL15 was analyzed at the indicated concentrations (0 to 50 μg/ml). The mean and SEM are given (n = 3). * denotes a significant difference from both normal human IgG and the monoclonal isotype control (p < 0.05).
Then the above assays were repeated in the presence of 20 nM APC (final concentration), which increased the baseline clotting time (for buffer only) to 452 seconds. Under this condition, CL15 reduced the clotting time to 287 seconds (a 37% reduction from 452 seconds), while all other test mAb and control IgG did not affect significantly the clotting time (Figure 4B). These data show that CL15 inhibits the anticoagulant function of APC.
Subsequently we examined CL15 in the presence of APC from 0 to 20 nM. As can be seen in Figure 4C, CL15 caused a significant reduction in clotting times in the presence of APC at 15 and 20 nM, and the degree of reduction in clotting times was increased as the concentration of APC was increased. In addition, CL15-mediated inhibition of APC function was analyzed at various concentrations of CL15 (0 to 50 μg/ml). The results showed that CL15 at only 25 μg/ml reduced significantly the plasma coagulation time in the presence of exogenous APC (Figure 4D). Considering that the plasma IgG concentration is approximately 10 mg/ml, 25 μg/ml is equivalent to 0.25% of total plasma IgG, suggesting that the observed inhibition of APC function by CL15 is physiologically relevant.
The binding properties of two selected mAb to PC and APC
Why did only CL15 inhibit APC function? Was this because the binding affinity of CL15 to APC is higher than that of all other mAb? Besides, why did all mAb (including CL15) fail to interfere with PC activation? Was this because all mAb bind to PC with low affinities that are physiologically irrelevant? To test these hypotheses, we determined the binding affinities of CL15 and IS3 to PC and APC by competitive inhibition. We elected to study only two mAb because of the prohibitory cost of APC; and IS3 was chosen because it displayed the highest OD in binding to PC and APC on the plate (Figure 3). The results showed that PC and APC were comparable in inhibiting both mAb from binding to the corresponding antigens on the plate (Figure 5). Based on the inhibition data, the relative Kd values of these mAb to PC were calculated to be 1.6 × 10-6 and 3.6 × 10-6 M for CL15 and IS3, respectively; and the relative Kd values of CL15 and IS3 to APC were respectively 2.2 × 10-6 and 3.8 × 10-6 M.
Figure 5.
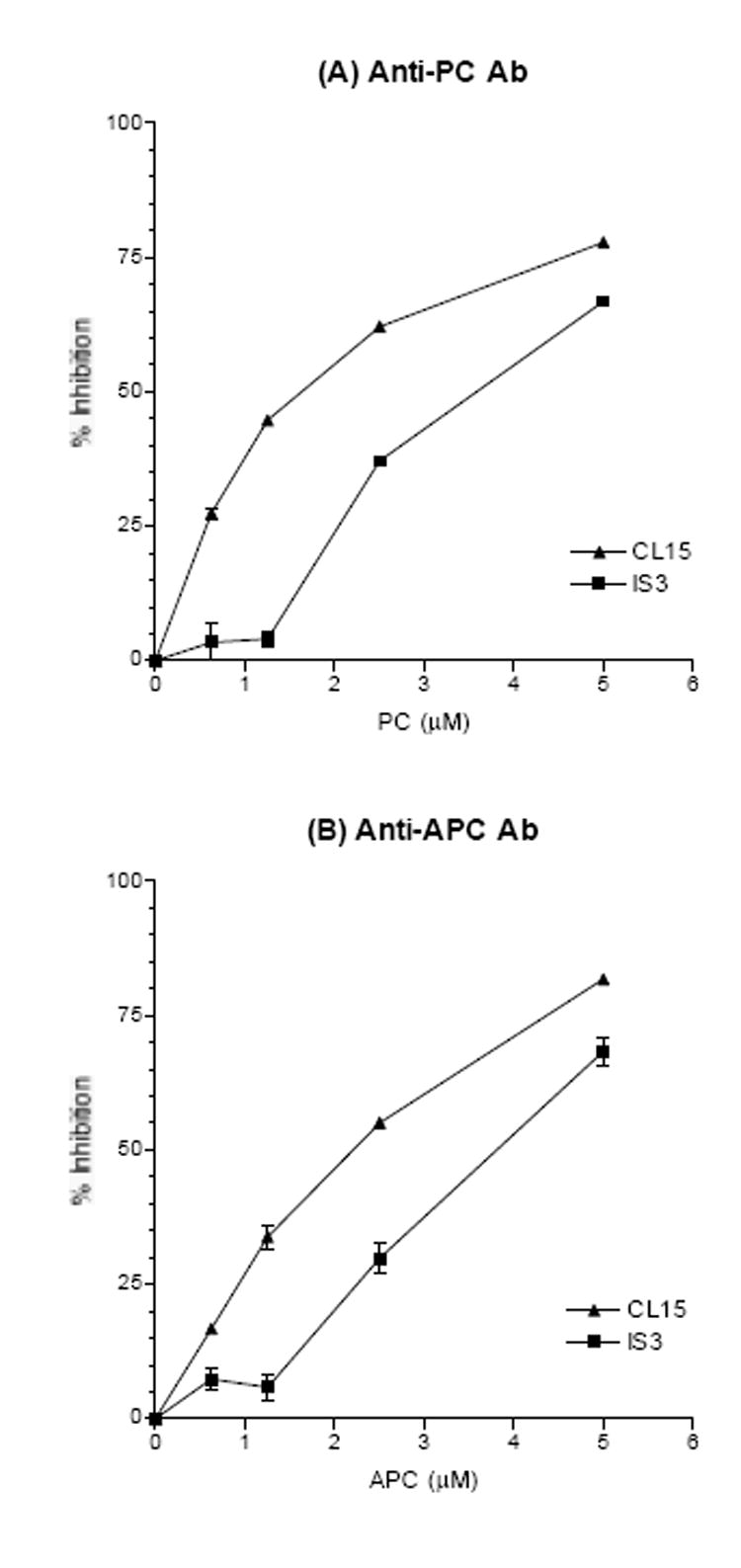
Competitive inhibition of CL15 and IS3 binding to PC (panel A) and APC (panel B). The results are expressed in % inhibition, and a representative result from two experiments is shown.
These data show that the binding affinity of CL15 to APC is only slightly higher than that of IS3 to APC, and thus suggest that the affinity difference between CL15 and IS3 probably does not account for their difference in affecting plasma coagulation time in the presence of exogenous APC. On the other hand, the binding affinity of CL15 to PC is even slightly higher than that of CL15 to APC.
DISCUSSION
To test the hypotheses that thrombin-reactive aCL may bind to PC and/or APC and consequently interfere with PC activation and/or anticoagulant function of APC, we found that 6/6 patient-derived thrombin-reactive IgG monoclonal aCL (including IS6) bind to both PC and APC, but not 2/2 non-thrombin-reactive monoclonal aCL. More importantly, of these 6 PC- and APC reactive mAb, CL15 at 25 μg/ml (equivalent to 0.25% of total plasma IgG) shortened plasma coagulation time in the presence of exogenous APC, indicating an inhibition of the APC activity by CL15. Of note, it is generally accepted that the diagnostic cutoff for IgG aCL in APS patients is 20 GPL, equivalent to 20 μg/ml of IgG aCL. CL15 binds to APC with a relative Kd value of 2.2 × 10-6 M. Combined, these data suggest that many thrombin-reactive aCL in APS bind to PC and APC and that a few of such Ab (like CL15) inhibit APC function.
The key coagulation cascade consists of: 1) expression of TF, which binds and activates factor VII (generating VIIa); 2) the TF-VIIa complexes activate factors IX and X (generating IXa and Xa, respectively); 3) factor Xa works with factor Va to convert PT to thrombin, while factor IXa works with factor VIIIa to generate more factor Xa; and 4) thrombin converts fibrinogen to a fibrin network, and feedback amplifies the cascade by activating more factors V and VIII (16). The coagulation cascade is subjected to three major feedback regulation mechanisms: 1) the TF-pathway inhibitor inhibits TF-VIIa complexes from activating factors IX and X; 2) AT binds to thrombin, IXa and Xa, and inactivates their enzyme activity; and 3) PC is activated by the thrombin-thrombomodulin complex on the EC surface, and then the APC forms a complex with protein S on PL surfaces and proteolytically inactivates factors Va and VIIIa (16, 29). In this context, CL15 may inhibit APC function by interfering with its binding to protein S and/or PL, or its subsequent cleavage of factors Va and/or VIIIa.
Studies have shown that familial thrombophilia and 20-40% of unrelated thrombophilic patients are associated with a single point mutation in the factor V gene (G1691A), resulting in an abnormal factor V (R506Q) that lacks one of the APC cleavage sites in factor V and thus is resistant to APC inactivation; the phenomena are termed APC resistance (APCR)(30-32). Subsequently, the aforementioned aPL-mediated inhibition of APC function has been frequently referred to as “acquired APCR.” In most studies of acquired APCR in APS, the target antigens of the involved aPL and the associated mechanisms are not well defined. Recently, it was reported that the EY2C9 and GR1D5 monoclonal IgM aCL bound to PC in the presence of both CL and β2GPI but not in the absence of either CL or β2GPI (33). Moreover, the study also showed that soluble PC could inhibit EY2C9 from binding to coated PC in the presence of both CL and β2GPI; and that EY2C9 shortened the clotting time of a normal plasma in the presence of APC in an activated partial thromboplastin time test (33). These findings led the authors to suggest that some aCL may bind to protein C via β2GPI. Taken together with the CL15 data, these studies suggest that there are at least two different species of aPL in APS, which could inhibit APC function; one binds directly to APC and the other binds indirectly to PC via β2GPI. In the future, it will be important to study these two species of aPL and determine the prevalence and the pathological significance of each aPL in APS.
In addition to aPL-mediated inhibition of APC function, acquired APCR was observed in two patients with thrombosis (34, 35). In the first case, the patient had two IgG paraproteins but the antigens recognized by the involved antibodies were not defined (34). In the second case, the patient’s IgG was shown to react with APC but not PC (35), and thus differs from CL15 that binds to both PC and APC. These data suggest that there are more than one epitopes recognized by anti-APC Ab that inhibit anticoagulant activity of APC and are associated with thrombosis.
It is noteworthy that only 1/6 APC-reactive mAb from APS patients inhibits the anticoagulant activity of APC. Therefore, it is likely to be fruitless to assess the clinical significance of all anti-APC Ab in APS by association studies of the presence of anti-APC Ab to APS, or to study the functional activities of affinity-purified polyclonal anti-APC Ab from patients. Instead, it will be first necessary to delineate the APC epitopes recognized by various anti-APC Ab with or without acquired APCR activities, such as CL15 (that inhibits APC activity) and IS3 (that does not inhibit APC activity but binds to APC with an affinity similar to that of CL15) (Figures 4 and 5). If the APC epitopes recognized by only prothrombotic anti-APC Ab are defined, then specific assays for the disease-relevant anti-APC Ab may be developed and used to study the roles of prothrombotic anti-APC Ab in thrombosis in APS patients.
Acknowledgments
We thank Mrs. Paifei Chen for technical assistance.
This work was supported by a grant from the Southern California Chapter of the Arthritis Foundation and a grant AR42506 from the National Institutes of Health. P. P. Chen is supported by a research award from the Alliance for Lupus Research.
Contributor Information
Kwan-Ki Hwang, Department of Medicine, Division of Rheumatology, University of California, Los Angeles, CA.
Cheng-De Yang, Department of Medicine, Division of Rheumatology, University of California, Los Angeles, CA.
Weihong Yan, Department of Chemistry and Biochemistry, University of California, Los Angeles, CA.
Jennifer M. Grossman, Department of Medicine, Division of Rheumatology, University of California, Los Angeles, CA.
Bevra H. Hahn, Department of Medicine, Division of Rheumatology, University of California, Los Angeles, CA.
Pojen P. Chen, Department of Medicine, Division of Rheumatology, University of California, Los Angeles, CA.
References
- 1.Matsuura E, Igarashi Y, Fujimoto M, Ichikawa K. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet. 1990;336:177–178. doi: 10.1016/0140-6736(90)91697-9. [DOI] [PubMed] [Google Scholar]
- 2.McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid binding inhibitor of coagulation: beta 2-glycoprotein I (apolipoprotein H) Proc Natl Acad Sci USA. 1990;87:4120–4124. doi: 10.1073/pnas.87.11.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galli M, Comfurius P, Maassen C, Hemker HC, de Baets MH, van Breda-Vriesman PJC, et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. 1990;335:1544–1547. doi: 10.1016/0140-6736(90)91374-j. [DOI] [PubMed] [Google Scholar]
- 4.Fleck RA, Rapaport SI, Rao LV. Anti-prothrombin antibodies and the lupus anticoagulant. Blood. 1988;72:512–519. [PubMed] [Google Scholar]
- 5.Bevers EM, Galli M, Barui T, Comfurius P, Zwaal RFA. Lupus anticoagulant IgG’s (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb Haemost. 1991;66:629–632. [PubMed] [Google Scholar]
- 6.Oosting JD, Derksen RHWM, Bobbink IWG, Hackeng TM, Bouma BN, de Groot PG. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: An explanation for their pathogenic mechanism? Blood. 1993;81:2618–2625. [PubMed] [Google Scholar]
- 7.Simantov R, LaSala JM, Lo SK, Gharavi AE, Sammaritano LR, Salmon JE, et al. Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest. 1995;96:2211–2219. doi: 10.1172/JCI118276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kornberg A, Blank M, Kaufman S, Shoenfeld Y. Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J Immunol. 1994;153:1328–1332. [PubMed] [Google Scholar]
- 9.Del Papa N, Guidali L, Sala A, Buccellati C, Khamashta MA, Ichikawa K, et al. Endothelial cells as target for antiphospholipid antibodies - Human polyclonal and monoclonal anti-β2-glycoprotein I antibodies react in vitro with endothelial cells through adherent β2-glycoprotein I and induce endothelial activation. Arthritis Rheum. 1997;40:551–561. doi: 10.1002/art.1780400322. [DOI] [PubMed] [Google Scholar]
- 10.Cuadrado MJ, López-Pedrera C, Khamashta MA, Camps MT, Tinahones F, Torres A, et al. Thrombosis in primary antiphospholipid syndrome - A pivotal role for monocyte tissue factor expression. Arthritis Rheum. 1997;40:834–841. doi: 10.1002/art.1780400509. [DOI] [PubMed] [Google Scholar]
- 11.Cariou R, Tobelem G, Bellucci S, Soria J, Soria C, Maclouf J, et al. Effect of lupus anticoagulant on antithrombogenic properties of endothelial cells--inhibition of thrombomodulin-dependent protein C activation. Thromb Haemost. 1988;60:54–58. [PubMed] [Google Scholar]
- 12.Marciniak E, Romond EH. Impaired catalytic function of activated protein C: A new in vitro manifestation of lupus anticoagulant. Blood. 1989;74:2426–2432. [PubMed] [Google Scholar]
- 13.Malia RG, Kitchen S, Greaves M, Preston FE. Inhibition of activated protein C and its cofactor protein S by antiphospholipid antibodies. Br J Haematol. 1990;76:101–107. doi: 10.1111/j.1365-2141.1990.tb07843.x. [DOI] [PubMed] [Google Scholar]
- 14.Borrell M, Sala N, de Castellanau C, Lopez S, Gari M, Foncuberta J. Immunoglobulin fractions isolated from patients with antiphospholipid antibodies prevent the inactivation of factor Va by activated protein C on human endothelial cells. Thromb Haemost. 1992;68:268–272. [PubMed] [Google Scholar]
- 15.Smirnov MD, Triplett DT, Comp PC, Esmon NL, Esmon CT. On the role of phosphatidylethanolamine in the inhibition of activated protein C activity by antiphospholipid antibodies. J Clin Invest. 1995;95:309–316. doi: 10.1172/JCI117657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahlback B. Blood Coagulation. Lancet. 2000;355:1627–1632. doi: 10.1016/S0140-6736(00)02225-X. [DOI] [PubMed] [Google Scholar]
- 17.Hwang K, Grossman J, Visvanathan S, Chukwuocha R, Woods V, Le D, et al. Identification of anti-thrombin antibodies in the antiphospholipid syndrome that interfere with the inactivation of thrombin by antithrombin. J Immunol. 2001;167:7192–7198. doi: 10.4049/jimmunol.167.12.7192. [DOI] [PubMed] [Google Scholar]
- 18.St Charles R, Matthews J, Zhang E, Tulinsky A. Bound structures of novel P3-P1’ betastrand mimetic inhibitors of thrombin. J Med Chem. 1999;42:1376–1383. doi: 10.1021/jm980052n. [DOI] [PubMed] [Google Scholar]
- 19.Mather T, Oganessyan V, Hof P, Huber R, Foundling S, Esmon C, et al. The 2.8 Å crystal structure of Gla-domainless activated protein C. EMBO J. 1996;15:6822–6831. [PMC free article] [PubMed] [Google Scholar]
- 20.Rose T, Di Cera E. Three-dimensional modeling of thrombin-fibrinogen interaction. J Biol Chem. 2002;277:18875–18880. doi: 10.1074/jbc.M110977200. [DOI] [PubMed] [Google Scholar]
- 21.Gale A, Tsavaler A, Griffin J. Molecular characterization of an extended binding site for coagulation factor Va in the positive exosite of activated protein C. J Biol Chem. 2002;277:28836–28840. doi: 10.1074/jbc.M204363200. [DOI] [PubMed] [Google Scholar]
- 22.Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu M, Olee T, Le DT, Roubey RAS, Hahn BH, Woods VL, Jr, et al. Characterization of IgG monoclonal anti-cardiolipin/antiβ2GP1 antibodies from two patients with the anti-phospholipid syndrome reveals three species of antibodies. Br J Haematol. 1999;105:102–109. [PubMed] [Google Scholar]
- 24.Zhao Y, Rumold R, Ahmed AE, Le DT, Hahn BH, Woods VL, Jr, et al. An IgG anti-prothrombin antibody enhances prothrombin binding to damaged endothelial cells and shortens plasma coagulation times. Arthritis Rheum. 1999;42:2132–2138. doi: 10.1002/1529-0131(199910)42:10<2132::AID-ANR13>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Esmon NL, Esmon CT. Reconstitution of the human endothelial cell protein C receptor with thrombomodulin in phospatidylcholine vesicles enhances protein C activation. J Biol Chem. 1999;274:6704–6710. doi: 10.1074/jbc.274.10.6704. [DOI] [PubMed] [Google Scholar]
- 26.Smirnov MD, Safa O, Esmon NL, Esmon CT. Inhibition of activated protein C anticoagulant activity by prothrombin. Blood. 1999;94:3839–3846. [PubMed] [Google Scholar]
- 27.Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods. 1985;77:305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 28.Pierangeli SS, Liu XW, Espinola R, Olee T, Zhu M, Harris NE, et al. Functional analyses of patient-derived IgG monoclonal anticardiolipin antibodies using in vivo thrombosis and in vivo microcirculation models. Thromb Haemost. 2000;84:388–395. [PubMed] [Google Scholar]
- 29.Esmon CT. Regulation of Blood Coagulation. Biochim Biophys Acta. 2000;1477:349–360. doi: 10.1016/s0167-4838(99)00266-6. [DOI] [PubMed] [Google Scholar]
- 30.Griffin JH, Evatt B, Wideman C, Fernandez JA. Anticoagulant protein C pathway defective in majority of thrombophilic patients. Blood. 1993;82:1989–1993. [PubMed] [Google Scholar]
- 31.Dahlback B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA. 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertina RM, Koeleman BPC, Koster T, Rosendaal FR, Dirven RJ, De Ronde H, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 33.Atsumi T, Khamashta MA, Amengual O, Donohoe S, Mackie I, Ichikawa K, et al. Binding of anticardiolipin antibodies to protein C via β2-glycoprotein I (β2-GPI): a possible mechanism in the inhibitory effect of antiphospholipid antibodies on the protein C system. Clin Exp Immunol. 1998;112:325–333. doi: 10.1046/j.1365-2249.1998.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitchell CA, Rowell JA, Hau L, Young JP, Salem HH. A fatal thrombotic disorder associated with an acquired inhibitor of protein C. N Engl J Med. 1987;317:1638–1642. doi: 10.1056/NEJM198712243172606. [DOI] [PubMed] [Google Scholar]
- 35.Zivelin A, Gitel S, Griffin J, Xu X, Fernandez J, Martinowitz U, et al. Extensive venous and arterial thrombosis associated with an inhibitor to activated protein C. Blood. 1999;94:895–901. [PubMed] [Google Scholar]
- 36.Tsiang M, Jain A, Gibbs C. Functional requirements for inhibition of thrombin by antithrombin III in the presence and absence of heparin. J Biol Chem. 1997;272:12024–12029. doi: 10.1074/jbc.272.18.12024. [DOI] [PubMed] [Google Scholar]