

Abstract
The aim of our study was to investigate the cellular mechanisms induced by hypercapnic stimulation of ventilation, during 6 weeks/30 min per day, in 10 mdx and 8 C57BL10 mice (10G0.2 months old). Ten mdx and eight C57BL10 mice served as control group. This respiratory training increases in vitro maximal tetanic tension of the diaphragm only in mdx mice. Western blot analysis of diaphragm showed: (1) an over-expression of a-dystrobrevin in mdx and C57BL10 training group compared to control group (8100G710 versus 6100G520 and 2800G400 versus 2200G250 arbitrary units); (2) a decrease in utrophin expression only in mdx training group compared to control group (2100G320 versus 3100G125 arbitrary units). Daily respiratory muscle training in mdx mice, induces a beneficial effect on diaphragm strength, with an over-expression of a-dystrobrevin. Further studies are needed to determine if, in absence of dystrophin, the over-expression of a-dystrobrevin could be interpreted as a possible pathway to improve function of dystrophic muscle.
Keywords: Ventilatory responses, Hypercapnia, alpha-dystrobrevin, Utrophin
Keywords: Adaptation, Physiological; Animals; Blotting, Western; Body Weight; Breathing Exercises; Carbon Dioxide; pharmacology; Citrate (si)-Synthase; metabolism; Diaphragm; cytology; physiology; Dystrophin; metabolism; Dystrophin-Associated Proteins; metabolism; Female; Hematoxylin; Hypercapnia; physiopathology; Hyperventilation; physiopathology; Isometric Contraction; physiology; Male; Mice; Mice, Inbred C57BL; Mice, Inbred mdx; Muscle Fibers; metabolism; Muscular Dystrophy, Animal; physiopathology; therapy; Organ Size; Respiratory Mechanics; physiology; Utrophin; metabolism
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked genetic disorder caused by gene mutations mapped to locus Xp21 [1], which encodes dystrophin. Dystrophin is a large, actin-binding protein that is connected to a multi-component complex known as the dystrophin protein complex (DPC). The DPC is localized at the cell membrane and interacts with the extracellular matrix [2]. Three sub-complexes form the DPC: the transmembrane dystroglycan complex, the membrane-spanning sarcoglycan complex, and the alpha-dystrobrevin-syntrophin cytoplasmic complex [3]. DMD is characterized by progressive weakness of all skeletal muscles including the respiratory muscles, which remains a major problem in the management of these patients.
Inspiratory muscle training has been proposed to delay the decline in respiratory muscle function. Although deleterious effects of exercise on dystrophin-deficient muscle fiber have been reported [4], several studies have shown that a daily increase in the contractile activities of the respiratory muscles appears to have a beneficial effect on respiratory muscle function as assessed by measurement of strength and endurance [5–8].
To investigate the cellular mechanisms of exercise in DMD patients, most authors have used a mouse model of DMD for ethical reasons. The mdx mouse lacks dystrophin due to a nonsense point mutation in exon 23 of the dystrophin gene [9]. However, in contrast to the situation in humans, most of the skeletal muscles of mdx mice show little fibrosis or functional alteration until late in life [10,11]. The one major exception is the diaphragm of these mice, which exhibits significant fibrosis as well as greatly impaired contractile function from an early age [10,12]. For these reasons, the mdx mouse diaphragm is widely considered to be the most clinically relevant murine model of DMD. The majority of authors have studied the effect of exercise only on limb muscles [13–15], however, and few reports to date have focused on the effects of exercise on mdx mouse diaphragm. Dupont-Versteegden et al. [13,16] showed that voluntary running exercise beginning at 3 weeks of age and ending at 12 months increased the maximal tetanic tension of the mdx diaphragm without changes in resistance to fatigue, myosin concentration or oxidative metabolism as measured by citrate synthase activity.
We hypothesized that the functional improvement induced by respiratory training could be due to several mechanisms: (i) increased oxidative metabolism, (ii) fiber-type modifications, and (iii) changes in the sarcolemmal expression of dystrophin-homologous proteins such as utrophin and alpha-dystrobrevin. Indeed, utrophin, which is located at the sarcolemma during muscle development before dystrophin replaces it, presents similarities of structure and binding partner with dystrophin, and many studies have shown that utrophin may have a cellular function similar to that of dystrophin [17,18]. In addition, other intracellular proteins such as alpha-dystrobrevin, a component of the DPC, are also highly homologous to the COOH-terminus of dystrophin and may thus participate in a common function [19]. Moreover, as alpha-dystrobrevin seems to be partly functional in the absence of dystrophin, previous authors have suggested that it is conceivable that the over-expression of alpha-dystrobrevin may be beneficial to dystrophin-deficient muscles [20,21].
To verify our hypothesis, (i) we conducted respiratory muscle training consisting of hyperventilation induced by hypercapnic stimulation of ventilation (8% CO2) in two groups of mice (mdx and C57BL10, the wild type of mdx), for 6 weeks/30 min per day, and (ii) we evaluated the effects of this training by in vitro assessment of diaphragm contractile properties in these two trained groups in comparison with mdx and C57BL10 controls.
Material and Methods
Animals
Breeding pairs of mdx mice were purchased from Alain Sebille Laboratory, France. The mice were randomly assigned to one of two groups: a training group of 10 male mdx mice (training mdx) that followed a respiratory training program induced by hypercapnic stimulation and a control group of 10 male mdx mice (control mdx). The wild-type C57BL10 mice were also included in our study to better assess the effect of training on the cellular adaptations of dystrophin-deficient respiratory muscles compared to normal muscles. Sixteen adult male C57BL10 mice were also randomly separated into two groups: one training (training C57BL10) and one control (control C57BL10). The mean age of our mice was 10G0.2 months.
Specific respiratory muscle training in mdx mice
For the respiratory muscle training program, we used a method of hypercapnic stimulation of ventilation. This consisted of placing each mouse in a hypercapnic chamber in order to increase the minute ventilation in relation to resting ventilation. The percentage of CO2 was regulated to significantly increase the resting minute ventilation.
Plethysmography
To quantify the increase in ventilation, we needed to know the hypercapnic ventilation sensitivity of the mdx mice compared to the normal mice. The ventilatory response to CO2 was thus measured before training using whole barometric body plethysmography, as previously used and adapted for mouse (Buxco, Troy, NY) based on Drorbaugh and Fenn’s principle [22]. The plethysmograph has been described previously [23]. Calibration was done before each test by injecting 150 ml of air into the measurement chamber with a syringe and by introducing the corresponding pressure into Drorbaugh and Fenn’s equation. A 1500 ml minK1 flow of dry air was delivered through the chamber to prevent CO2 and water accumulation and to maintain a constant temperature inside the plethysmograph, which allowed a long measurement period. Hypercapnic mixtures (8% CO2, 21% O2, and 71% N2) were obtained commercially.
One week before starting the respiratory training program, each mouse was weighed and placed in the plethysmograph chamber. The mice were introduced into the ventilated chamber at least 60 min before breath recording began. The respiratory stimuli were administered by replacing the air flow by the hypercapnic flow through the plethysmograph. Each test consisted of 15 min of resting air measurement followed by 15 min of hypercapnic stimulation and then by the measurement of the ventilatory response to hypercapnic gas for 15 min. In five mdx and five C57BL10 mice, we prolonged the hypercapnic stimulation to 30 min, to ensure that the increased ventilation remained constant during this period. The values of pressure changes were used to calculate the total time of the respiratory cycle (Ttot) and tidal volume (Vt) expressed in microliter divided by body weight in grams. Minute ventilation (ml minK1 gK1 ) was calculated from breathing frequency and tidal volume measurements: minute ventilationZ [breathing frequency!tidal volume)/body weight (body temperature, pressure and saturation (BTPS)].
Respiratory training protocol
After assessment of resting and hypercapnic minute ventilation of the mdx and C57BL10 mice, 6 weeks of daily 30 min sessions of respiratory muscle training began. During the study period, the control and training mice were housed in similar boxes. Each box was divided into small (9!9 cm), similar individual compartments to limit their daily movement. This ensured that they would not be able to increase the work of their respiratory muscles except during training, thus simulating the situation of DMD children. They had access to food and water ad libitum.
The training mdx mice were submitted to a hypercapnic atmosphere (8% CO2, 21% O2 and 71% N2) in their individual compartments. The entire box containing the training mice was hermetically sealed and the hypercapnic flow was delivered through a small opening (0.5 cm diameter). A second opening allowed the gas flow to exit. The flow rate was twice the mean minute ventilation of all the mice, as measured under hypercapnic stimulation. During respiratory muscle training, the gas from the cage was continuously sampled and analyzed through a third small opening using an O2 analyzer (Beckman OM11, France) and a CO2 analyzer (Cosma rubies 3000, France) to ensure constant gas level.
Measurement of isometric contractile properties
At the end of 6 weeks, each mouse was anesthetized with an intraperitoneal injection of 50 mg/kg pentobarbital. The diaphragms were surgically excised with ribs and central tendon attached for in vitro study of the isometric contractile properties as described previously [24]. Briefly, a muscle strip (2 mm wide) was anchored by the rib that was left attached to the base of an organ bath chamber filled with oxygenated Krebs solution maintained at 35 8C. A 4.0 silk thread was used to secure the central tendon of the opposite part of the strip to the isometric force transducer (Kent Scientific Instruments, Litchfield, CT). Supramaximal stimuli with a monophasic pulse duration of 3 ms were delivered using platinum plate electrodes connected to a square wave pulse stimulator (Model S48; Grass Instruments, West Warwick, RI) and muscle force data were acquired by computer. After adjusting each strip to optimal length (L0), the strips were then sequentially stimulated at 20, 30, 50, 60, 80, 100 and 120 Hz for 600 ms each, with 2 min between each contraction, to determine the maximal tetanic force at different stimulation frequencies. Values were determined after normalization for total muscle strip cross-sectional area and expressed as Newton per square centimeter (N/cm2). The total muscle strip cross-sectional area was determined by dividing muscle weight by its length and tissue density (1056 g/cm3). Force generation by the diaphragm during repeated contractions was measured by assessing the decline in peak muscle force during tetanic stimulation (100 Hz, 300 ms duration, 1 train/s) for 3 min. The changes in the diaphragm’s ability to generate force over time were quantified in terms of the time taken for the tension to decline to 50% of the initial value (T50%).
Tissue preparation for biochemical analysis
At the end of 6 weeks, the diaphragm of each mouse was partitioned and muscle samples were quickly flash-frozen in isopentane, cooled in liquid nitrogen and then stored at K80 8C.
Biochemical analysis
All subsequent steps were performed at 4 8C. Diaphragm samples were defrosted at room temperature and homogenized with a glass potter (10% weight/vol) in a 100 mM potassium phosphate buffer (pH 7.4) containing 10 mM EDTA, with 0.05% bovine serum albumin (BSA). Centrifugation of homogenates was performed at 300g at 3 8C for 10 min to remove cellular debris. Protein content was measured using the Bio-rad protein assay Sigma [25].
Citrate synthase (CS) activity was measured in diaphragm sample homogenate. A volume of homogenate containing 10 mg of protein was added to a reaction medium consisting of 100 mM dinitrothiobenzoic acid and 15 mM acetylcoenzyme A, according to the method of Srere [26]. The absorbance at 412 nm (DU 640 spectrophotometer, Beckman, CA) was checked for 3 min to determine the nonspecific activity. Reactions were then initiated by addition of 500 mM of oxaloacetate, and the change in absorbance was recorded for at least 3 min at 412 nm.
Histologic and protein analysis
Fiber type determination
Muscle samples were cut into 10 mm thick transverse sections in a cryostat at K20 8C. Each section was verified by light microscopy to ensure appropriate fiber orientation. For each mouse, two adjacent sections with no artefactual changes were selected and used to determine fiber type. These sections were stained for myofibrillar ATPase activity according to the single-step ethanol-modified technique. Depending on the staining intensity, the different fiber types were designated as type I (nonstained), type IIa (lightly stained), or type IIb (darkly stained). Muscle sections were then magnified and transmitted to an image analyzer (NIH image analysis software) to count and classify fibers.
Histologic process and immunostaining
The 10 mm cryostat sections of each mouse diaphragm were treated with specific antibodies: the utrophin C-terminal end (CPNVPSRPQAM) was used as a specific antigenic sequence against this molecule and antibodies were obtained by injecting the KLH-linked peptide as antigen, according to a previously described protocol [27]. Specific synthetic peptides C-KSPAKKLSNALS, which correspond to amino acids 293–304 of the mouse alpha-dystrobrevin, were used to produce antibody against different isoforms of alpha-dystrobrevin (alpha 1 and 2).
We then produced and obtained various polyclonal antibodies specific to all isoforms of utrophin (K7) and alpha-dystrobrevin (D124) in accordance with previously described works [27].
Cryostat sections were first labeled with each of the polyclonal antibodies, then washed and subsequently detected with Cy3-conjugated sheep anti-rabbit IgG (1/1000 from SIGMA BlO-Sciences Laboratory). Similarly, hematoxylin labeling was applied and the resulting images were comparatively analyzed on the control mdx and C57BL10 groups versus the training mdx and C57BL10 groups.
Immunoblot detection
Crude protein extracts from mdx diaphragm muscles were obtained as follows. Fifty cryostat sections (10 mm) of each muscle were homogenized in 100 ml of buffer containing 10% SDS, 5% beta-mercaptoethanol, 10% glycerol, 10 mM EDTA and 100 mM Tris–HCI buffer (pH 8.0). Each sample was immediately denatured by boiling for 4 min. Then, after centrifugation (5000 rpm) at 4 8C, 5 ml of supernatant were loaded on 3–12% gradient SDS polyacrylamide gels. After overnight electrotransfer (30 V, 100 mA) in transfer buffer (25 mM Tris–HCI, 192 mM glycine, 0.1% SDS and 20% methanol, pH 8.3), nitrocellulose membranes (0.2 mm) were blocked with 3% BSA dissolved in TEST buffer (10 mM Tris–HCI, 150 mM NACI, 0.05% Tween 20, pH 8) for 30 min at room temperature. Blots were incubated with the specific antibodies for 1 h at room temperature. We used a monoclonal antibody directed against alpha 1-and alpha 2-dystrobrevin that was commercially available from Transduction Laboratories (D63020). For utrophin, the same antibody described above (K7) was used.
A secondary antibody coupled to alkaline phosphatase (1/5000 dilution, Jackson) was used to visualize each specific protein band with p-nitroblue tetrazolium and 5-bromo-4 chloro-3 indolylphosphate substrates.
Scanning densitometry
Western blots were digitized with a 256 gray scale and images were quantitatively analyzed using the NIH 1.62 image program. Each lane was treated independently and corrected by estimation of the relative optical density found in the Coommasie blue gel corresponding to the amount of myosin present in each mouse protein extract. Each blot was obtained in triplicate to avoid errors due to differences in signal intensities. Values were normalized and averages for the three assays were compared between control and training mice. Their ratio was expressed as a percentage of utrophin (or alpha-dystrobrevin) found in control versus training mice.
Statistics
Statistical analysis was performed using SigmaStat statistical software. Data are reported as meansGstandard error of the mean (SEM). Differences in muscle contractility and T50% between training and control groups were compared by two-way analysis of variance for repeated measures. Any differences detected were evaluated by the Student-Neuman-Keuls test. Data concerning body and diaphragm weights, enzyme activity and scanning densitometry were analyzed using Student’s t-test. When data distribution was not normal, a Mann-Whitney test was used. A value of P!0.05 was considered significant.
Results
Ventilatory response to hypercapnic stimulation
In the mdx and C57 mice, we observed a large increase in the parameter of breathing pattern under hypercapnic stimulation compared to its value at rest: (1) breathing frequency: 323G63 versus 170G30 cycles per minute for mdx mice and 320G49 versus 174G40 cycles per minute for C57BL10 mice, (2) tidal volume: 15.9G3 versus 5.4G 3 ml/g for mdx mice and 15G5 versus 5.5G2 ml/g for C57BL10 mice, and (3) minute ventilation: 5.3G1.5 versus 0.87G0.21 ml/g per min for mdx mice and 4.8G1.8 versus 0.96G0.8 ml/g per min for C57BL10 mice. With hypercapnic stimulation we thus observed an increase of nearly two-fold for breathing frequency, three-fold for tidal volume and five-fold for minute ventilation. We observed no statistical difference between mdx and C57BL10 mice in terms of ventilatory response to hypercapnic stimulation.
Isometric contractile properties
The diaphragmatic force-frequency relationships in the training and control groups of mdx and C57BL10 mice are shown in Fig. 1. The respiratory muscle training in mdx mice induced a significant increase in diaphragmatic force over a frequency range of 30–120 Hz. The mean maximal isometric force was 9.50G0.70 N/cm2 in the training mdx group and 8G0.48 N/cm2 in the control mdx group. The same respiratory muscle training in C57BL10 mice, as shown in Fig. 1, did not induce any significant change in diaphragmatic force at any frequency (PZ0.9): the mean maximal isometric force was 17.1G1.3 N/cm2 in the training C57BL10 group and 17.5G0.7 N/cm2 in the control C57BL10 group. Concerning resistance to fatigue, we found no difference after repeated stimulations in T50% between the mdx training group (66.8G7.4 s) and the mdx control group (65.3G5.1 s). We also observed no difference (PZ0.24) in T50% between the training and control C57BL10 groups (49G4 versus 43G2 s).
Fig. 1.
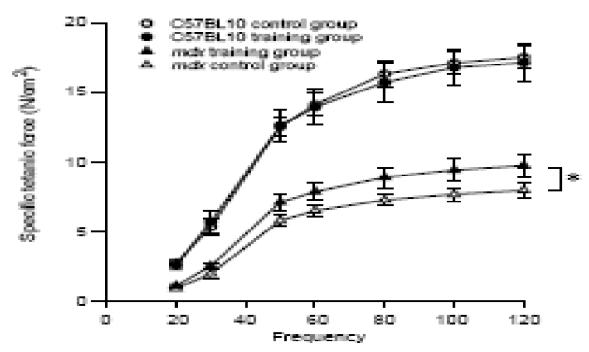
Effect of respiratory training induced by CO2 breathing on maximal specific tetanic force in adult mdx and C57BL10 mice as shown by the force-frequency relationship. Values are expressed as meansGSEM. *, P!0.05. White circle, control C57BL10 mice (nZ8); dark circle, trained C57BL10 mice (nZ8); white triangle, control mdx mice (nZ10); dark triangle, trained mdx mice (nZ10).
Biochemical analysis
Body and diaphragm weights
We found no significant difference in initial body weight between the training and control mdx groups (28.0G2.7 versus 29G3.7 g) and the training and control C57BL10 groups (33.3G4.9 versus 30.5G2.2 g).
After 6 weeks in individual cages coupled with 30 min of daily hypercapnic stimulation for the respiratory training groups, the body weights of each group did not significantly change. Moreover, after 6 weeks, we found no difference in diaphragm weight between the training and control mdx mice (78G16 versus 75.1G10 mg) or the training and control C57BL10 mice (75G7 versus 69G8 mg).
Diaphragm protein content and citrate synthase activity
After the 6 weeks of training, we found no significant difference in diaphragm protein content between the training and control mdx groups (3.6G0.4 versus 3.8G0.5 mg/g protein) or between the training and control C57BL10 groups (4.1G0.6 versus 4.4G0.4 mg/g protein). We also observed no significant difference in diaphragm CS activity between the training and control mdx mice (25.9G2.6 versus 27.8G3.6 mmol minK1gK1 protein) and between the training and control C57BL10 mice (33.3+/−4.9 versus 33.4+/−3.3 mol minK1gK1 protein). The results are shown in Fig. 2.
Fig. 2.
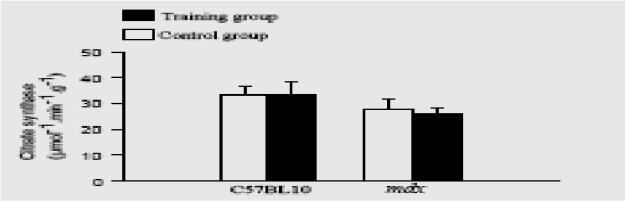
Effect of respiratory training induced by CO2 rebreathing on diaphragm citrate synthase activity in mdx and C57BL10 mice. White column, control groups; dark column, trained groups. Values are expressed as means +/− SEM.
Histologic and protein analysis
Fiber type determination
The proportions of the different fiber types in the training and control groups are summarized in Table 1. Respiratory muscle training did not induce any change in mdx fiber type. Moreover, we observed mostly type II fibers with only a few type I fibers in the training and control groups.
Table 1.
Percentage of type I, IIa, IIb fibers in diaphragm of control and training mdx mice
| Control group | Training group | |
|---|---|---|
| Type I | 11.7+/−1.9 | 10.4+/−2.0 |
| Type IIa | 60.3+/−5.1 | 56.6+/−5.0 |
| Type IIb | 28.0+/−4.1 | 33.0+/−4.3 |
Hematoxylin coloration and immunofluorescent detection of dystrophin
The status of each diaphragm muscle was controlled with both hematoxylin coloration and dystrophin detection on a 10 mm cryostat cross-section. As shown in Fig. 3, the mdx mouse diaphragm presented dystrophic patterns on hematoxylin coloration including variability in fiber size, central nuclei, and increased amounts of fat and connective tissue. Moreover, dystrophin detection was negative in mdx mouse diaphragm, as expected, and the dystrophic changes remained similar in the two mdx groups, without worsening or improvement, after respiratory training. For the C57BL10 mice, no significant histological change was observed in the training group compared with the control group.
Fig. 3.
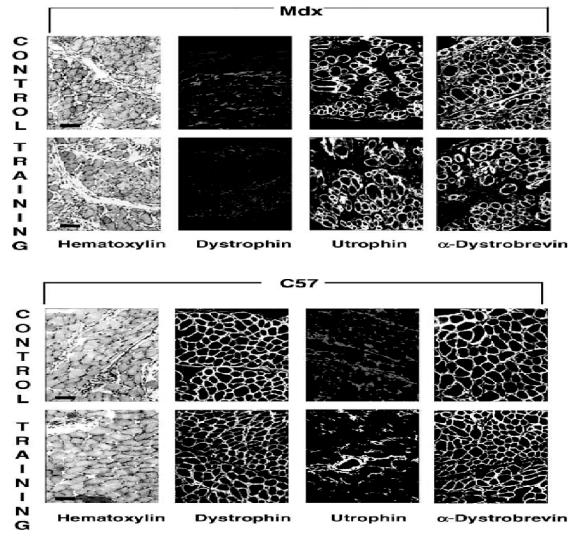
Comparative analysis on cryostat sections from (a) upper panel: trained and control mdx mice, (b) lower panel: trained and control C57BL10 mice. In mdx mice, compared to C57BL10, hematoxylin staining of diaphragm revealed infiltration by interstitial tissue and clear central nuclei distribution in some muscle cells. Immunofluorescence labeling was obtained using specific polyclonal antibodies directed specifically against dystrophin, utrophin and a-dystrobrevin. As expected, utrophin staining was clearly located in the plasma membrane from dystrophin-deficient muscles and dystrophin was absent from mdx muscles. α-Dystrobrevin was present in all muscles. There was no major difference revealed by immunofluorescence between control and training muscles in either mdx or C57BL10 mice.
Immunofluorescent study of diaphragm utrophin and alpha-dystrobrevin expression
As described previously [28], utrophin labeling was observed around each muscle fiber in mdx diaphragm and only at the synaptic junction in C57 diaphragm. In both the training and control groups, the presence of utrophin was detected with the same intensity.
All cell membranes were stained with the immunofluorescent labeling of alpha-dystrobrevin in the mdx and C57 mouse diaphragms, and we noted no difference between the training and control groups. The results are shown in Fig. 3.
Western blot analysis: utrophin and alpha-dystrobrevin expression
The different crude protein extracts obtained from the control and training groups in the mdx and C57 mice were comparatively analyzed on the same Western blot, shown in Fig. 4, with antibodies directed against utrophin and alpha-dystrobrevin. We observed a significant decrease (P<0.05) in utrophin quantity in extracts from training mdx mice (2100+/−320 arbitrary units) compared with control mdx mice (3100+/−125 arbitrary units) after scanning the utrophin and standardization of the results with myosin content. Indeed, we were able to use myosin as a reference because we found no difference on the same gel, after Coommassie staining, between the training and control groups in terms of myosin content (128+/−21 versus 116+/−29 arbitrary units). Moreover, in the C57BL10 mice, we observed no difference between the training and control groups in utrophin quantity (730+/−125 versus 592+/−100 arbitrary units). The results are shown in Fig. 5.
Fig. 4.
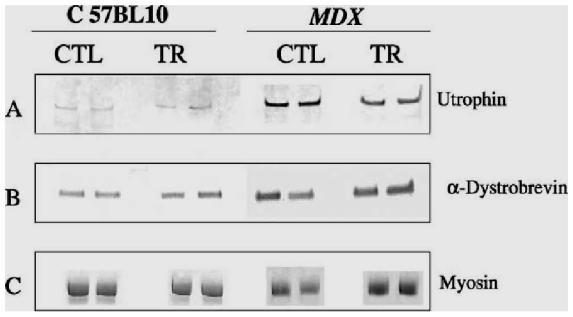
Comparative Western blot analyses of control and trained diaphragmatic muscle extracts from mdx and C57BL10 mice. Western blot analyses are presented in duplicate for protein extracts corresponding to control (CTL) and trained (TR) muscles from mdx and C57BL10 mice. A specific antibody was used and can be viewed after phosphatase alkaline revelation in panel (A) for utrophin; and in panel (B) for alpha-dystrobrevin. The protein extracts were analyzed on PAGE after Coommasie blue staining to estimate comparatively the content in myosin in each sample, as presented in panel (C).
Fig. 5.
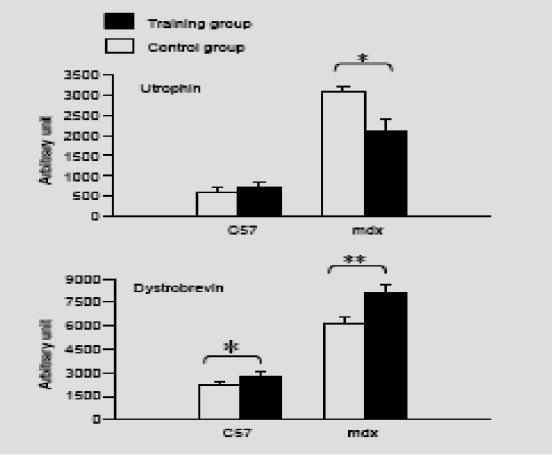
Effect of respiratory training on sarcolemmal expession of utrophin and alpha-dystrobrevin in diaphragm of mdx and C57BL10 mice. Numbers correspond to arbitrary units obtained after scanning the respective western blot analyses. White column, control groups; dark column, trained groups. Values are expressed in means +/− SEM. *, P<0.05; **, P<0.01.
Western blot analysis of the different diaphragm protein extracts from the mdx and C57BL10 mice were comparatively analyzed between the training and control groups with a specific monoclonal alpha-dystrobrevin antibody. As shown in Fig. 5, after scanning and normalization with myosin quantity, we observed a significant over-expression of alpha-dystrobrevin (P<0.01) in the training group of mdx mice (8100+/−710 arbitrary units) compared with the control group (6100+/−520 arbitrary units). In the C57BL10 mice, we also observed a significant increase in alpha dystrobrevin expression (P<0.05) in the training group compared with the control group (2800+/−400 versus 2200+/−250 arbitrary units).
Discussion
The aim of this study was to assess the cellular adaptations of mdx mouse diaphragm to respiratory muscle training. We showed that this 6-week trial induced an improvement in the maximal isometric force of the diaphragm, with an over-expression of alpha-dystrobrevin and a parallel decrease in utrophin expression in these mice. In trained C57BL10 mice, however, we found only an over-expression of alpha-dystrobrevin.
We developed a protocol for respiratory muscle training by hypercapnic stimulation of ventilation. Indeed, this training was as close as possible to one of the respiratory training programs followed by DMD children [5]. This program, in which the children voluntarily hyperventilate under air for 6 weeks/30 min per day, has been demonstrated to have a beneficial effect on respiratory muscle function [5]. To correctly follow a respiratory training program, DMD children generally must be at least 10-yearold to allow for sufficient learning ability, understanding of language, perceptual organization and motor coordination. Yet at this age, children already present an advanced stage of the disease with a decrease in diaphragmatic strength more than 50% of the control values [29]. To determine whether a dystrophic diaphragm with substantial functional alteration still retains adaptive capacity, we chose to study 10-month-old mdx mice. By this age, the entire mdx diaphragm is grossly pale owing to extensive myofiber loss and replacement fibrosis, and it thus presents physiopathologic properties similar to those found in the muscles from DMD patients undergoing respiratory training [10,30, 31]. Moreover, in our study, in vitro assessment revealed that mdx diaphragm strength reached approximately 50% of the strength of C57BL10 mouse diaphragm.
Plethysmography revealed a marked increase in minute ventilation under hypercapnic stimulation that remained constant for at least 30 min in both the mdx and C57BL10 mice. We found no difference in breathing frequency, tidal volume or minute ventilation under air and 8% CO2 between the mdx and C57BL10 mice. This result seems to be in contradiction with a previous study that found no increase in breathing frequency, tidal volume or minute ventilation in mdx mice under 8% CO2 compared to air [32]. This could be explained partly by differences in the measurement protocol we used. When the mouse was placed in the plethysmograph chamber, we waited at least 60 min, and the breath recording began when the mouse was nearly asleep, without movement. Respiratory stimuli with 8% of CO2 were then administered, with the mouse always in the same state of low vigilance. The 10-month-old mdx mice of our study presented normal sensitivity of their respiratory centers to CO2, with normal ventilatory output.
As the mice were quiet and without movement during the CO2 rebreathing, the respiratory training model we used allowed us to modulate the level of diaphragm activity to observe a potential dose effect, with a minimum of stress for the animal. The effect of hypercapnia alone on the respiratory muscles is currently not well understood. Kumagai et al. [33] found that constant hypercapnia in the rat for 19 weeks had no effect on fiber type in untrained quadriceps femoralis muscle. However, in response to induced hyperventilation, these authors showed an increase in the fatigue-resistant fibers in diaphragm.
An important result of our work was that specific respiratory muscle training in the mdx mice induced a significant increase in diaphragm strength without improving resistance to fatigue. Few authors have studied the effect of exercise on the diaphragm. Dupont-Versteedgen et al. [13,16] found that long-term exercise consisting of voluntary wheel-running from 3 weeks to about 13 months of age increased specific active tension by 30% in mdx diaphragm. But these authors did not find any strength improvement after shorter-term exercise. After a short term of 6 weeks of hypercapnic stimulation of ventilation, we found a significant increase of 20% in specific maximal tetanic tension. Thus, training of respiratory muscles with hypercapnic stimulation of ventilation seems to be more effective than voluntary wheel-running, given the positive effect of the former protocol after a short training period.
However, our training protocol did not affect the diaphragm’s resistance to fatigue. This may be related to the lack of effect on citrate synthase activity that we observed, which may indicate that 30 min per day of hyperventilation has no effect on the oxidative metabolism of mdx or C57BL10 diaphragm. Interestingly, a previous study [13] found that the diaphragms of mdx mice that had been exercised with voluntary wheel-running for 3 months were less susceptible to fatigue that those of sedentary control mdx mice, but the exercise had no effect on citrate synthase activity. Previous authors who showed an increase in resistance to fatigue after training in peripheral muscles [14,34] observed an increase in the proportion of type I fatigue-resistant oxidative muscle fibers only secondary to a long exercise training period. In our study, with hypercapnic stimulation of ventilation for 6 weeks/30 min per day, we did not observe a change in the percentage of fiber type. Indeed, we observed mostly type II fibers, in accordance with the results of Louboutin et al. [30], who found more than 90% of type II fibers at 9 months of age. Thus it seems that our short, specific respiratory training period, although more efficient than voluntary wheel-running to improve respiratory muscle strength, was not long enough to have an effect on in vitro resistance to fatigue, citrate synthase activity or the proportion of type I fatigue-resistant oxidative muscle fibers.
Another important result was that this respiratory muscle training with CO2 breathing did not induce any change in diaphragm strength or resistance in the C57BL10 mice. This result indicates that the mdx mouse diaphragm adapts differently to exercise induced by hypercapnic stimulation of ventilation than the C57BL10 diaphragm. Moreover, as we did not observe an effect on C57BL10 diaphragm, we may hypothesize that the mechanism of improved contractile function in mdx mouse diaphragm after respiratory training induced by CO2 rebreathing likely involves a functional replacement of the missing dystrophin or a decrease in the physiopathological processes of mdx mice, such as muscular fiber degeneration, fibrosis or inflammation.
We found similar levels of alpha-dystrobrevin expression in the diaphragms of our mdx and control mice. This result seems to contradict the finding of a previous study [35], which showed that alpha-dystrobrevin was dramatically reduced at the sarcolemma of mdx hindlimb muscle. This discrepancy may have several explanations. First, it is possible that alpha-dystrobrevin expression differs in mdx diaphragm and hindlimb muscle. Skeletal muscle groups do not respond in the same way to dystrophin deficiency, with distinct and differentially activated signaling pathways in the diaphragm and hindlimb muscles of dystrophic mice [36]. Second, whereas alpha-dystrobrevin 2 is mainly associated with dystrophin [37] and thus almost completely absent at the mdx mice sarcolemma [35,38], alphadystrobrevin 1 binds to dystrophin and utrophin [37]. In this study, we used a pan-dystrobrevin antibody that recognizes both isoforms of alpha-dystrobrevin. Our mdx mice were old (10G2 months) and thus likely to be presenting high levels of sarcolemmal utrophin expression, which would explain the relatively high level of alphadystrobrevin expression in our control mdx mice. Indeed, the utrophin level was clearly demonstrated to be over-expressed in an age-dependent manner [11,39]. Third, a previous study found that alpha-dystrobrevin 2 expression depended on the muscle region examined in mdx [37]. With immunostaining, these authors showed that alpha-dystrobrevin 2 labeling was similar to that of wild-type mice at the neuro-muscular junctions (NMJs) or on the perijunctional sarcolemma. They further demonstrated that it was dramatically reduced in regions far from NMJs. As the diaphragm is transversely crossed by multiple scattered NMJ bands [40], it is possible that we examined a region particularly rich in NMJs as compared to hindlimb muscle.
In association with the increase in specific tetanic force induced by our respiratory training in mdx mouse, we found an increase in the expression of alpha-dystrobrevin in both mdx and C57BL10 mouse diaphragm. This protein is of particular interest because its sequence is partly similar to the COOH terminus of dystrophin [41]. It appears to parallel dystrophin in its expression and was severely reduced in biopsy from Duchenne muscular dystrophy patient [42]. Although the fundamental role of alpha-dystrobrevin remains unclear, it is an element in a mechanical model that directly links the DPC with the Z-line of the contractile apparatus, and thus may play a role in maintaining muscle cell integrity [21]. Indeed, alpha-dystrobrevin-deficient mice develop mild muscular dystrophy, similar to that of the dystrophin-deficient mdx mouse [41]. Moreover, the function of alpha-dystrobrevin does not depend on tight connection to dystrophin [43], and previous studies have reported [19,20] that over-expression of alpha-dystropbrevin in Caenorhabditis elegans could partly compensate the absence of dystrophin in terms of functional recovery.
Thus, the increase in alpha-dystrobrevin that we observed after respiratory training in mdx mouse may partially explain the improvement in the specific tetanic strength of the mdx diaphragm. Moreover, we may also assume that the over-expression of alpha-dystrobrevin observed in C57BL10 mice, secondary to exercise induced by CO2 breathing, was inefficient to improve diaphragm contractility. This result does not seem surprising, given that over-expression of alpha-dystrobrevin in the presence of dystrophin would logically have little impact on normal diaphragm contractility.
The increased expression of alpha-dystrobrevin in mdx mouse diaphragm induced by specific training may help us to interpret the concomitant decrease in utrophin. We observed a 25% decrease in utrophin expression in the mdx training group compared with the control group. In the serial sections for immunostaining, the signal was too low to observe any difference between the training and control groups. Thus, it was not possible to determine if the muscle fibers expressing high levels of alpha-dystrobrevin expressed low levels of utrophin. This is an important issue that needs further investigation. The utrophin over-expression observed in the mdx mice was interpreted as a compensatory mechanism partially protecting the muscle fiber from further steps of degeneration and regeneration [44]. Since alpha-dystrobrevin is over-expressed when it is down-regulated, we suggest that the binding site of this protein does not have a specific affinity for utrophin and thus other protein candidates should be investigated in future studies. This result seems to be supported by a recent study that observed no correlation between utrophin levels and the expression of alpha-dystrobrevin [43]. The lower level of utrophin expression in mdx mouse diaphragm after hypercapnic stimulation of ventilation could thus be interpreted as a diminution of this compensatory mechanism correlated with a lessening in the severity of the muscular dystrophy. Concerning the C57BL10 mice, we observed no change in utrophin expression after ventilatory exercise. In the presence of dystrophin, utrophin is essentially present at the neuromuscular junction [28], and its level of expression, which compared to that in the mdx mice is down-regulated, seems to have been uninfluenced by our respiratory training induced by CO2 breathing.
Other mechanisms involved in CO2 rebreathing may explain the improved diaphragm function of our mdx mice. A previous study showed that hypercapnic acidosis or isocapnic acidosis and buffered hypercapnia has antiinflammatory effects by attenuating NF-B activation [45]. This is of particular interest since, when TNF alpha—which is mediated by activation of NF-B [46]—is knocked out of mdx mice, they present improved respiratory function in comparison with standard mdx mice [32]. Moreover, CO2 may also be involved in scavenging peroxynitrite, thereby preventing the protein nitration and oxidative damage [47] that is much implicated in DMD [48]. To evaluate whether these mechanisms are the predominant explanations for the functional improvement with CO2 rebreathing, rather than alpha-dystrobrevin expression, further studies with respiratory training induced by CO2 breathing should be performed in mdx mice with the alpha-dystrobrevin gene knocked out.
In conclusion, we developed a respiratory muscle training program in mdx and C57BL10 mice based on hypercapnic stimulation of ventilation. We found that 6 weeks/30 min per day of hyperventilation induced by CO2 breathing increased maximal tetanic force in mdx but not C57BL10 diaphragm without, however, improving endurance. The concomitant increase in the expression of alpha-dystrobrevin, a protein homologous to dystrophin, seemed to have a beneficial effect on the contractile function in mdx diaphragm. A better understanding of the potential benefits of the over-expression of alpha-dystrobrevin induced by short-term endurance training and the regulatory mechanism of its expression could thus be very useful for therapeutic management of the dystrophic muscle in DMD children.
Acknowledgments
This work was supported by the AFM: Association Française contre les Myopathies (Research Grant).
The authors are very grateful to Dr Alain Sebille for his constant help and for providing the mdx mice, and to Assistant Professor Alain Comtois and Dr Maurice Hayot for providing us counsel and the electrodes needed for functional evaluation of mdx diaphragm.
References
- 1.Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–17. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 2.Tinsley JM, Blake DJ, Roche A, et al. Primary structure of dystrophinrelated protein. Nature. 1992;360(6404):591–3. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- 3.Ervasti JM, Campbell KP. Membrane organization of the dystrophinglycoprotein complex. Cell. 1991;66(6):1121–31. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 4.Petrof BJ. The molecular basis of activity-induced muscle injury in Duchenne muscular dystrophy. Mol Cell Biochem. 1998;179(1–2):111–23. doi: 10.1023/a:1006812004945. [DOI] [PubMed] [Google Scholar]
- 5.Vilozni D, Bar-Yishay E, Gur I, Shapira Y, Meyer S, Godfrey S. Computerized respiratory muscle training in children with Duchenne muscular dystrophy. Neuromuscul Disord. 1994;4(3):249–55. doi: 10.1016/0960-8966(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 6.Wanke T, Toifl K, Merkle M, Formanek D, Lahrmann H, Zwick H. Inspiratory muscle training in patients with Duchenne muscular dystrophy. Chest. 1994;105(2):475–82. doi: 10.1378/chest.105.2.475. [DOI] [PubMed] [Google Scholar]
- 7.Winkler G, Zifko U, Nader A, et al. Dose-dependent effects of inspiratory muscle training in neuromuscular disorders. Muscle Nerve. 2000;23 (8):1257–60. doi: 10.1002/1097-4598(200008)23:8<1257::aid-mus15>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 8.Topin N, Matecki S, Le Bris S, et al. Dose-dependent effect of individualized respiratory muscle training in children with Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12(6):576–83. doi: 10.1016/s0960-8966(02)00005-6. [DOI] [PubMed] [Google Scholar]
- 9.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244(4912):1578–80. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 10.Stedman HH, Sweeney HL, Shrager JB, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352(6335):536–9. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 11.Pastoret C, Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci. 1995;129(2):97–105. doi: 10.1016/0022-510x(94)00276-t. [DOI] [PubMed] [Google Scholar]
- 12.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90(8):3710–4. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupont-Versteegden EE. Exercise and clenbuterol as strategies to decrease the progression of muscular dystrophy in mdx mice. J Appl Physiol. 1996;80(3):734–41. doi: 10.1152/jappl.1996.80.3.734. [DOI] [PubMed] [Google Scholar]
- 14.Hayes A, Williams DA. Beneficial effects of voluntary wheel running on the properties of dystrophic mouse muscle. J Appl Physiol. 1996;80(2):670–9. doi: 10.1152/jappl.1996.80.2.670. [DOI] [PubMed] [Google Scholar]
- 15.Lynn DJ, Woda RP, Mendell JR. Respiratory dysfunction in muscular dystrophy and other myopathies. Clin Chest Med. 1994;15(4):661–74. [PubMed] [Google Scholar]
- 16.Dupont-Versteegden EE, McCarter RJ, Katz MS. Voluntary exercise decreases progression of muscular dystrophy in diaphragm of mdx mice. J Appl Physiol. 1994;77(4):1736–41. doi: 10.1152/jappl.1994.77.4.1736. [DOI] [PubMed] [Google Scholar]
- 17.Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384(6607):349–53. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 18.Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin–dystrophin deficient mice. Nat Genet. 1998;19(1):79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- 19.Gieseler K, Mariol MC, Bessou C, et al. Molecular, genetic and physiological characterisation of dystrobrevin-like (dyb-1) mutants of Caenorhabditis elegans. J Mol Biol. 2001;307(1):107–17. doi: 10.1006/jmbi.2000.4480. [DOI] [PubMed] [Google Scholar]
- 20.Gieseler K, Grisoni K, Mariol MC, Segalat L. Overexpression of dystrobrevin delays locomotion defects and muscle degeneration in a dystrophin-deficient Caenorhabditis elegans. Neuromuscul Disord. 2002;12(4):371–7. doi: 10.1016/s0960-8966(01)00330-3. [DOI] [PubMed] [Google Scholar]
- 21.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82(2):291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 22.Epstein MA, Epstein RA. A theoretical analysis of the barometric method for measurement of tidal volume. Respir Physiol. 1978;32(1):105–20. doi: 10.1016/0034-5687(78)90103-2. [DOI] [PubMed] [Google Scholar]
- 23.Hamelmann E, Schwarze J, Takeda K, et al. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156(3 Pt 1):766–75. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 24.Petrof BJ, Acsadi G, Jani A, et al. Efficiency and functional consequences of adenovirus-mediated in vivo gene transfer to normal and dystrophic (mdx) mouse diaphragm. Am J Respir Cell Mol Biol. 1995;13(5):508–17. doi: 10.1165/ajrcmb.13.5.7576685. [DOI] [PubMed] [Google Scholar]
- 25.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Srere PA. The citrate enzymes: their structures, mechanisms, and biological functions. Curr Top Cell Regul. 1972;5:229–83. doi: 10.1016/b978-0-12-152805-8.50013-7. [DOI] [PubMed] [Google Scholar]
- 27.Pons F, Robert A, Fabbrizio E, et al. Utrophin localization in normal and dystrophin-deficient heart. Circulation. 1994;90(1):369–74. doi: 10.1161/01.cir.90.1.369. [DOI] [PubMed] [Google Scholar]
- 28.Blake DJ, Tinsley JM, Davies KE. Utrophin: a structural and functional comparison to dystrophin. Brain Pathol. 1996;6(1):37–47. doi: 10.1111/j.1750-3639.1996.tb00781.x. [DOI] [PubMed] [Google Scholar]
- 29.Smith PE, Calverley PM, Edwards RH, Evans GA, Campbell EJ. Practical problems in the respiratory care of patients with muscular dystrophy. N Engl J Med. 1987;316(19):1197–205. doi: 10.1056/NEJM198705073161906. [DOI] [PubMed] [Google Scholar]
- 30.Louboutin JP, Fichter-Gagnepain V, Thaon E, Fardeau M. Morphometric analysis of mdx diaphragm muscle fibres. Comparison with hindlimb muscles. Neuromuscul Disord. 1993;3(5–6):463–9. doi: 10.1016/0960-8966(93)90098-5. [DOI] [PubMed] [Google Scholar]
- 31.Pastoret C, Sebille A. Age-related differences in regeneration of dystrophic (mdx) and normal muscle in the mouse. Muscle Nerve. 1995;18(10):1147–54. doi: 10.1002/mus.880181011. [DOI] [PubMed] [Google Scholar]
- 32.Gosselin LE, Barkley JE, Spencer MJ, McCormick KM, Farkas GA. Ventilatory dysfunction in mdx mice: impact of tumor necrosis factor-alpha deletion. Muscle Nerve. 2003;28(3):336–43. doi: 10.1002/mus.10431. [DOI] [PubMed] [Google Scholar]
- 33.Kumagai M, Kondo T, Ohta Y, Ishihara T. Size and composition changes in diaphragmatic fibers in rats exposed to chronic hypercapnia. Chest. 2001;119(2):565–71. doi: 10.1378/chest.119.2.565. [DOI] [PubMed] [Google Scholar]
- 34.Hayes A, Lynch GS, Williams DA. The effects of endurance exercise on dystrophic mdx mice. I. Contractile and histochemical properties of intact muscles. Proc R Soc Lond B Biol Sci. 1993;253(1336):19–25. doi: 10.1098/rspb.1993.0077. [DOI] [PubMed] [Google Scholar]
- 35.Blake DJ. Dystrobrevin dynamics in muscle-cell signalling: a possible target for therapeutic intervention in Duchenne muscular dystrophy? Neuromuscul Disord. 2002;12(Suppl 1):S110–S7. doi: 10.1016/s0960-8966(02)00091-3. [DOI] [PubMed] [Google Scholar]
- 36.Lang JM, Esser KA, Dupont-Versteegden EE. Altered activity of signaling pathways in diaphragm and tibialis anterior muscle of dystrophic mice. Exp Biol Med (Maywood) 2004;229(6):503–11. doi: 10.1177/153537020422900608. [DOI] [PubMed] [Google Scholar]
- 37.Peters MF, Sadoulet-Puccio HM, Mark Grady R, et al. Differential membrane localization and intermolecular associations of alphadystrobrevin isoforms in skeletal muscle. J Cell Biol. 1998;142(5):1269–78. doi: 10.1083/jcb.142.5.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ge Y, Molloy MP, Chamberlain JS, Andrews PC. Proteomic analysis of mdx skeletal muscle: great reduction of adenylate kinase 1 expression and enzymatic activity. Proteomics. 2003;3(10):1895–903. doi: 10.1002/pmic.200300561. [DOI] [PubMed] [Google Scholar]
- 39.Taylor J, Muntoni F, Dubowitz V, Sewry CA. The abnormal expression of utrophin in Duchenne and Becker muscular dystrophy is age related. Neuropathol Appl Neurobiol. 1997;23(5):399–405. [PubMed] [Google Scholar]
- 40.Boriek AM, Miller CC, 3rd, Rodarte JR. Muscle fiber architecture of the dog diaphragm. J Appl Physiol. 1998;84(1):318–26. doi: 10.1152/jappl.1998.84.1.318. [DOI] [PubMed] [Google Scholar]
- 41.Grady RM, Grange RW, Lau KS, et al. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol. 1999;1(4):215–20. doi: 10.1038/12034. [DOI] [PubMed] [Google Scholar]
- 42.Metzinger L, Blake DJ, Squier MV, et al. Dystrobrevin deficiency at the sarcolemma of patients with muscular dystrophy. Hum Mol Genet. 1997;6(7):1185–91. doi: 10.1093/hmg/6.7.1185. [DOI] [PubMed] [Google Scholar]
- 43.Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol. 2000;150(6):1399–410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90(4):729–38. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 45.Takeshita K, Suzuki Y, Nishio K. Hypercapnic acidosis attenuates endotoxin-induced nuclear factor-[kappa]B activation. Am J Respir Cell Mol Biol. 2003;29(1):124–32. doi: 10.1165/rcmb.2002-0126OC. [DOI] [PubMed] [Google Scholar]
- 46.Li YP, Reid MB. NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am J Physiol Regul Integr Comp Physiol. 2000;279(4):R1165–R70. doi: 10.1152/ajpregu.2000.279.4.R1165. [DOI] [PubMed] [Google Scholar]
- 47.Vesela A, Wilhelm J. The role of carbon dioxide in free radical reactions of the organism. Physiol Res. 2002;51(4):335–9. [PubMed] [Google Scholar]
- 48.Rando TA, Disatnik MH, Yu Y, Franco A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul Disord. 1998;8(1):14–21. doi: 10.1016/s0960-8966(97)00124-7. [DOI] [PubMed] [Google Scholar]