

Abstract
Narcolepsy is characterized by excessive daytime sleepiness (EDS), cataplexy and/or other dissociated manifestations of rapid eye movement (REM) sleep (hypnagogic hallucinations and sleep paralysis). Narcolepsy is currently treated with amphetamine-like central nervous system (CNS) stimulants (for EDS) and antidepressants (for cataplexy). Some other classes of compounds such as modafinil (a non-amphetamine wake-promoting compound for EDS) and gamma-hydroxybutyrate (GHB, a short-acting sedative for EDS/fragmented nighttime sleep and cataplexy) given at night are also employed. The major pathophysiology of human narcolepsy has been recently elucidated based on the discovery of narcolepsy genes in animals. Using forward (i.e., positional cloning in canine narcolepsy) and reverse (i.e., mouse gene knockout) genetics, the genes involved in the pathogenesis of narcolepsy (hypocretin/orexin ligand and its receptor) in animals have been identified. Hypocretins/orexins are novel hypothalamic neuropeptides also involved in various hypothalamic functions such as energy homeostasis and neuroendocrine functions. Mutations in hypocretin-related genes are rare in humans, but hypocretin-ligand deficiency is found in many narcolepsy-cataplexy cases. In this review, the clinical, pathophysiological and pharmacological aspects of narcolepsy are discussed.
1. Introduction
Gélineáu first coined the term “narcolepsy” in 1880 with the complete description of a patient with excessive daytime sleepiness (EDS), sleep attacks and episodes of muscle weakness triggered by emotions (1). In the current international classification, narcolepsy is characterized by “excessive daytime sleepiness that is typically associated with cataplexy (i.e., narcolepsy with cataplexy) and/or with abnormal rapid eye movement (REM) sleep phenomena such as sleep paralysis and hypnagogic hallucinations”. Narcolepsy is a chronic neurological condition but is not a progressive disorder (2, 3).
Narcolepsy is an under-diagnosed sleep disorder that affects 0.03 to 0.16% of the general population in various ethnic groups (4-6). Most cases of human narcolepsy are sporadic; however, genetic predisposition and environmental factors are important for the development of narcolepsy, and a few familial cases (up to 5%) of human narcolepsy have also been reported (7).
Narcolepsy is mainly treated with pharmacological compounds. EDS is typically treated using central nervous system (CNS) stimulants and/or modafinil, and these compounds are effective in reducing daytime sleepiness but have little effect on cataplexy, hypnagogic hallucinations and sleep paralysis (8). Antidepressants (one of the most commonly used anticataplectic treatments) alleviate cataplexy and REM sleep abnormalities but have little effect on EDS (8). Sodium oxybate, a newly approved hypnotic (or novel compound), effectively controls cataplexy while also helping relieve daytime sleepiness.
The major pathophysiology of human narcolepsy has been recently elucidated based on the discovery of narcolepsy genes in animals. Using forward (i.e., positional cloning in canine narcolepsy) and reverse (i.e., mouse gene knockout) genetics, the genes involved in the pathogenesis of narcolepsy (hypocretin/orexin ligand and its receptor) in animals have been identified (9, 10). Hypocretins/orexins are novel hypothalamic neuropetides also involved in various hypothalamic functions such as energy homeostasis and neuroendocrine functions (11, 12). Mutations in hypocretin-related genes are rare in humans, but hypocretin-ligand deficiency is found in many cases (13-15), and this is manifested clinically as undetectable or low cerebrospinal fluid (CSF) hypocretin levels (14, 16). This discovery is likely to lead to the development of new diagnostic tests and targeted treatments. Indeed, low CSF hypocretin levels (less 30% of mean control value) were included for the diagnostic criteria for narcolepsy in the second revision of the International Classification of Sleep Disorders (ICSD) (17). Since hypocretins are involved in various hypothalamic functions, hypocretin-deficient narcolepsy now appears to be a more complex condition than just a simple sleep disorder (see (18)). This review begins with an overview of the clinical aspects of human narcolepsy, followed by those in the canine model of narcolepsy, an update on the pathophysiology of narcolepsy (with emphasis on the role of the hypocretins), and pharmacological treatments of narcoleptic symptoms and their mechanisms. Finally, the expectations from future narcolepsy research will also be discussed.
2. Epidemiology
2.1. Prevalence of narcolepsy
The prevalence of narcolepsy has been investigated in several ethnic groups and countries. One of the most sophisticated prevalence studies was a Finnish cohort study consisting of 11,354 twin individuals (5). All subjects who responded to a questionnaire with answers suggestive of narcolepsy were contacted by telephone. Clinical interviews were performed and polysomnographic recordings were then conducted in five subjects considered to be narcoleptic. Sleep-monitoring finally identified three narcoleptic subjects with cataplexy, thus leading to a prevalence of 0.026% (5). All three subjects were dizygotic twins and the co-twins were not affected (5). Other prevalence studies have led to similar prevalence values (0.02 to 0.067%) in Great Britain (19), France (20), the Czech Republic (21), five European countries (22) and the United States (23, 24).
A study performed in 1945 in African American navy recruits also led to 0.02% in this ethnic group for narcolepsy-cataplexy (25). Narcolepsy-cataplexy may be more frequent in Japan and less frequent in Israel. Two population-based prevalence studies led to prevalence figures of 0.16% and 0.18% in Japan (6, 26). However, these studies used only questionnaires and interviews but not polysomnography to confirm the diagnosis. In Israel, only a few narcoleptic patients have been identified when compared to the large population of subjects recruited into sleep clinics (27). This has led to the suggestion that the prevalence of narcolepsy could be as low as 0.002% in this ethnic group.
The age of onset varies from early childhood to the fifties, with two peaks, a larger one that occurs at around 15 years of age and a smaller peak at approximately 36 years of age (28). Similar results were found in two different populations but the reasons for the bimodal distribution remains obscure. Incidence of the disease was reported to be 1.37/100.000 per year (1.72 for men and 1.05 for women) in Olmsted County in Minnesota (24). The incidence rate was highest in the second decade, followed in descending order by the third, fourth and first decade.
2.2. Genetic vs. environmental factors
A familial tendency for narcolepsy has long been recognized since its description in the late 19th century (29). Starting in the 1940s, several studies were published, which investigated the familial history of small cohorts of narcoleptic probands (30-35) (Table 2). Using standard diagnostic criteria, more recent studies have shown rates of familial cases as 4.3% in Japan (36), 6% in the United States (7), 7.6% in France and 9.9% in Canada (28). In addition to subjects who fulfill all diagnostic criteria for narcolepsy, other relatives may report only recurrent sleep episodes; they may suffer from an incomplete and milder form of the disease (37). Studies also revealed that the risk of a first-degree relative developing narcolepsy-cataplexy is 1-2.0%, a risk 10 to 40 times higher than in the general population (37-39).
Table 2.
Current pharmacological treatment for human narcolepsy and its related disorders.
| Compound | Mode of action | Usual Daily Doses | Half life (hrs) | Side effects/Notes |
|---|---|---|---|---|
| Wake-promoting Compounds for EDS: | ||||
| Sympathomimetic stimulants: | ||||
| D-amphetamine sulfate | Dopamine enhancer (Dopamine release/ Dopamine uptake inhibition) | 5-60 mg | 16-30 | Irritability, mood changes, headaches, palpitations, tremors, excessive sweating, insomnia |
| Methylphenidate HCl | Dopamine enhancer | 10-60 mg | ∼3 | Same as amphetamines, less reduction of appetite or increase in blood pressure. |
| Pemoline* | Dopamine enhancer | 20-115 mg | 11-13 | less sympathomimetic effect, milder stimulant slower onset of action, occasionally produces liver toxicity |
| Non-amphetamine wake-promoting compounds: | ||||
| Modafinil | Unknown, inhibits dopamine uptake inhibition | 100-400 mg | 11-14 | No peripheral sympathomimetic action, headaches, nausea. |
| Short acting hypnotics: | ||||
| Gamma hydroxybutyric acid | Unknown, may act GABA-B or specific GHB receptors, reduces dopamine release | 6-9 g (divided nightly) | ∼2 | Sedation, nausea |
Potentially hepatotoxic - frequent liver function monitoring required.
On the other hand, 16 monozygotic (MZ) twin pairs with at least one affected twin have been reported in literature, and only four (or five, depending on the criteria) of these pairs were concordant for narcolepsy (38). Although genetic predisposition is likely to be involved in the development of narcolepsy, the relatively low rate of concordance in narcoleptic MZ twins indicates that environmental factors also play a role in the development of the disease. The nature of the possible environmental factors involved is not yet fully understood. Frequently cited factors are head trauma (40), sudden change in sleep/wake habits (41) or various infections (21). While these factors may be involved, there are no documented studies demonstrating increased frequency when compared to control groups.
3. Clinical characteristics of narcolepsy
3.1. Excessive daytime sleepiness (EDS) and related symptoms
EDS and cataplexy are considered to be the two primary symptoms of narcolepsy, with EDS often being the most disabling symptom. EDS most typically mimics the feeling that people experience when they are severely sleep-deprived but may also manifest itself as chronic tiredness or fatigue. Narcoleptic subjects generally experience a permanent background of baseline sleepiness that easily leads to actual sleep episodes in monotonous sedentary situations. This feeling is most often relieved by short naps (15-30 min), but in most cases the refreshed sensation only lasts a short time after waking. The refreshing value of short naps is of considerable diagnostic value. Sleepiness also occurs in irresistible waves in these patients, a phenomenon best described as “sleep attacks”. Sleep attacks may occur in very unusual circumstances, such as in the middle of a meal, a conversation or bicycle ride. These attacks are often accompanied by microsleep episodes (42) where the patient “blanks out”. The patient may then continue his or her activity in a semi-conscious manner (writing or speaking incoherently, etc.), a phenomenon called automatic behavior (42-44). Learning problems and impaired concentration are frequently associated with narcolepsy (42-46), but psychophysiological testing is generally normal.
Sleepiness is usually the first symptom to appear, followed by cataplexy, sleep paralysis and hypnagogic hallucinations (2, 21, 36, 47, 48). Cataplexy onset occurs within five years after the occurrence of daytime somnolence in approximately two-thirds of the cases (21, 36). Less frequently, cataplexy appears many years after the onset of sleepiness. The mean age of onset of sleep paralysis and hypnagogic hallucinations is also two to seven years later than that of sleepiness (2, 49).
In most cases, EDS and irresistible sleep episodes persist throughout the patient's lifetime, although symptoms often improve after retirement (possibly due to better management of activities), daytime napping and adjustment of nighttime sleep.
3.2. Cataplexy
Cataplexy is distinct from EDS and is pathognomonic of the disease (50). The importance of cataplexy for the diagnosis of narcolepsy has been recognized since its description (51, 52) and in subsequent reviews on narcolepsy (53, 54). Most authors now recognize patients with recurring sleepiness and cataplectic attacks as a homogeneous clinical entity, and this is now shown to be associated with hypocretin-deficiency (see the section on the pathophysiology of the disease). Cataplexy is defined as a sudden episode of muscle weakness triggered by emotional factors, most often in the context of positive emotions (such as laughter), and less frequently by negative emotions (most typically anger or frustration). All antigravity muscles can be affected, leading to a progressive collapse of the subject, but respiratory and eye muscles are not affected. The patient is typically awake at the onset of the attack but may experience blurred vision or ptosis. The attack is almost always bilateral and usually lasts a few seconds. Neurological examination performed at the time of an attack shows a suppression of the patellar reflex and sometimes Babinski's sign.
Cataplexy is an extremely variable clinical symptom (55). Most often, it is mild and occurs as a simple buckling of the knees, head dropping, facial muscle flickering, sagging of the jaw or weakness in the arms. Slurred speech or mutism is also frequently associated. It is often imperceptible to the observer and may even be only a subjective feeling difficult to describe, such as a feeling of warmth or that somehow time is suspended (54, 55). In other cases, it escalates to actual episodes of muscle paralysis that may last up to a few minutes. Falls and injury are rare and most often the patient will have time to find support or will sit down while the attack is occurring. Long episodes occasionally blend into sleep and may be associated with hypnagogic hallucinations.
Patients may also experience “status cataplecticus”. This rare manifestation of narcolepsy is characterized by subintrant cataplexy that lasts several hours per day, and confines the subject to bed. It can occur spontaneously or more often upon withdrawal from anticataplectic drugs (47, 56, 57).
Cataplexy often improves with advancing age. In rare cases, it disappears completely, but in most patients it becomes better controlled (probably after the patient has learned to control his emotions) (2, 58).
3.3. Sleep Paralysis
Sleep paralysis is present in 20-50% of all narcoleptic subjects (21, 31, 59, 60), and is often associated with hypnagogic hallucinations. Sleep paralysis is best described as a brief inability to perform voluntary movements at the onset of sleep, upon awakening during the night or in the morning. Contrary to simple fatigue or locomotion inhibition, the patient is unable to perform even a small movement, such as lifting a finger. Sleep paralysis may last a few minutes and is often finally interrupted by noise or other external stimuli. The symptom is occasionally bothersome in narcoleptic subjects, especially when associated with frightening hallucinations (61).
Whereas EDS and cataplexy are the cardinal symptoms of narcolepsy, sleep paralysis occurs frequently as an isolated phenomenon affecting 5-40% of the general population (62-64). Occasional episodes of sleep paralysis are often seen in adolescence and after sleep deprivation; thus, prevalence is high for single episodes.
3.4. Hypnagogic and hypnopompic hallucinations
Abnormal visual (most often) or auditory perceptions that occur while falling asleep (hypnagogic) or upon waking up (hypnopompic) are frequently observed in narcoleptic subjects (65). These hallucinations are often unpleasant and are typically associated with a feeling of fear or threat (59, 61). Polygraphic studies indicate that these hallucinations occur most often during REM sleep (59, 66). These episodes are often difficult to distinguish from nightmares or unpleasant dreams, which also occur frequently in narcolepsy.
Hypnagogic hallucinations are most often associated with sleep attacks, and the patient can criticize the content of the hallucinations. The hallucinations are most often complex, vivid and dream-like experiences (“half sleep” hallucinations), and may follow episodes of cataplexy or sleep paralysis (a feature that is not uncommon in severely affected patients). These hallucinations are usually easy to distinguish from hallucinations observed in schizophrenia or related psychotic conditions.
3.5. Other important symptoms
One of the most frequently associated symptoms is insomnia, best characterized as a difficulty to maintain nighttime sleep. Typically, narcoleptic patients fall asleep easily, only to wake up after a short nap and unable to fall asleep again for an hour or so. Narcoleptic patients do not usually sleep more than normal individuals over the 24-hour cycle (67-69) but frequently have a very disrupted nighttime sleep (67-69). This symptom often develops later in life and can be very disabling.
Frequently associated problems are periodic leg movements (70, 71), REM behavior disorder, other parasomnias (72, 73) and obstructive sleep apnea (71, 74, 75).
Narcolepsy was reported to be associated with changes in energy homeostasis several decades ago. Narcolepsy patients frequently (1) are obese (76, 77), (2) have insulin-resistant diabetes mellitus (76), (3) exhibit reduced food intake (78) and (4) have lower blood pressure and temperature (79, 80). These findings, however, did not receive much attention since they were believed to be secondary to sleepiness or inactivity during the daytime. More recently, however, it was shown that these metabolic changes might be found more specifically in hypocretin deficient patients (14), suggesting a direct pathophysiological link. Additional research in this area is warranted in order to clarify this association.
Narcolepsy is a very incapacitating disease. It interferes with every aspect of life. Additionally, the negative social impact of narcolepsy has been extensively studied, and patients experience impairments in driving and a high prevalence of either car- or machine-related accidents. Narcolepsy also interferes with professional performance, leading to unemployment, frequent changes of employment, worker's disability or early retirement (81-83). Several subjects also develop symptoms of depression, although these symptoms are often masked by anticataplectic medications (43, 81, 84).
4. Symptomatic narcolepsy/EDS
The symptoms of narcolepsy can also occur during the course of other neurological conditions (i.e., symptomatic narcolepsy). In a recent meta analysis, 116 symptomatic cases of narcolepsy were reported in the literature (cases meet with the ICSD criteria for narcolepsy and are also associated with a significant underlying neurological disorder that accounts for EDS and temporal associations) (85). As several authors previously reported, inherited disorders (n=38), tumors (n=33), and head trauma (n=19) are the three most frequent causes for symptomatic narcolepsy. Of the 116 cases, 10 are associated with multiple sclerosis, one case of acute disseminated encephalomyelitis, and relatively rare cases were reported with vascular disorders (n=6), encephalitis (n=4), degeneration (n=1), and hererodegenerative disorder (autosomal dominant cerebrospinal ataxia with deafness, three cases in a family). EDS without cataplexy or any REM sleep abnormalities is also often associated with these neurological conditions, and defined as symptomatic cases of EDS.
Although it is difficult to rule out the comorbidity of idiopathic narcolepsy in some cases, a review of the literature reveals numerous unquestionable cases of symptomatic narcolepsy (85). These include cases with human leukocyte antigen (HLA) negative and/or late onset, and cases in which the occurrences of the narcoleptic symptoms are parallel with the rise and fall of the causative disease. Interestingly, a review of these cases (especially those with brain tumors), illustrates clearly that the hypothalamus is most often involved (see (85)).
5. Idiopathic hypersomnia and other primary EDS
Less common forms of hypersomnia include the idiopathic and recurrent hypersomnias (86). Idiopathic hypersomnia is marked by excessive nocturnal sleep of good quality, and by EDS which is not as severe as in the narcoleptic patients (but not refreshing after naps) though not REM sleep–related (Table 1). The best characterized recurrent hypersomnia is the Kleine-Levin syndrome, a pervasive functional disorder of the hypothalamus characterized by hypersexuality, binge eating, and irritability associated with periods of EDS and sleep periods as long as 18-20 hours (87, 88). For details of clinical symptoms and treatments of these primary EDS, refer to Black et al. (86).
Table 1.
Clinical characteristics of narcolepsy-cataplexy, narcolepsy without cataplexy and idiopathic hypersomnia
| Daytime sleepiness | Other symptoms | MSLT | HLADQB1*0602 positivity | Low CSF hypocretin levels (<110 pg/ml) | |||
|---|---|---|---|---|---|---|---|
| Duration | Awaken-refreshed | Sleep latency | SOREMPS | ||||
| Narcolepsy-cataplexy | short (< 30 minutes) | (+) | Cataplexy REM sleep-related symptoms | <8 min* | ≥ 2 | >90% | 85-90% >90% in HLA positive |
| Narcolepsy without cataplexy | short (< 30 minutes) | (+) | Cataplexy (−) REM sleep-related symptoms | <8 min* | ≥ 2 | 40-50% | 10-20% (almost all HLA positive) |
| Idiopathic hypersomnia with long sleep time | long (> 30 minutes) | (−) | Cataplexy (−) prolonged nighttime sleep (>10hrs) autonomic nervous dysfunction | <8 min* | ≤1 | no consistent results | normal |
| Idiopathic hypersomnia without long sleep time | varied | (−) | Cataplexy (−) no prolonged nighttime sleep (<10hrs) autonomic nervous dysfunction | <8 min* | ≤1 | no consistent results | normal |
Less than 8 minutes is considered for both narcolepsy and idiopathic hypersomnia in the 2nd revision of ICSD.
6. Diagnosis
6.1. Polysomnography, nocturnal and daytime sleep evaluations
The clinical diagnosis is based on the presence of EDS and/or sudden onsets of sleep occurring almost daily during a period of at least six months and on the presence of a clear clinical history of cataplexy (89). In the current International Classification of Sleep Disorders (17), diagnosis (i.e., narcolepsy without cataplexy) can be made in the presence of EDS without cataplexy, if one associated symptom (including sleep paralysis, hypnagogic hallucinations, automatic behaviors and/or disrupted sleep) is present together with the following polygraphic abnormalities: average sleep latency < 8 min, or presence of two sleep-onset REM periods (SOREMPs) during the multiple sleep latency test (MSLT). Sleep prior to the MSLT must be at least six hours. (see Table 1). The regular MSLT consists of five naps, scheduled at two-hour intervals starting between 9 and 10 am (90-92). The test is terminated after a sleep period of 15 minutes or after 20 minutes if the patient did not fall asleep. SOREMPs are defined as the occurrence of REM sleep within 15 minutes after sleep onset.
A nocturnal polysomnogram is useful for eliminating other possible causes of EDS such as periodic leg movements and obstructive sleep apnea. The diagnosis of upper airway resistance syndrome must also be very carefully considered. An MSLT is generally performed the following day. Sleep efficiency during nocturnal polysomnography may be normal or low.
The American Sleep Disorders Association recommends that an MSLT be performed for all patients suspected of narcolepsy (93). The diagnostic value of the MSLT for narcolepsy has, however, been recently questioned by some authors (94, 95). First, approximately 15% of narcoleptic subjects with clear-cut cataplexy do not have a short sleep latency and/or more than two SOREMPs during a single MSLT (16, 95). Conversely, a small number of patients with abnormal breathing may display typical narcolepsy-like MSLT results. As the prevalence of upper airway resistance syndrome and sleep apnea is 100 times greater than those with narcolepsy-cataplexy, false positives may be frequent if the test is interpreted without carefully excluding all other causes of EDS(96).
In a number of countries, the MSLT is not commonly performed, especially if clear-cut cataplexy is present. Some investigators rely on the presence of SOREMPs during a single-night recording, an abnormality present in less than half of narcoleptic patients with cataplexy. A single daytime nap study is also used by some to analyze for the presence of a SOREMP or short sleep latency (97-99). Other groups have advocated the use of continuous 24-hour or 36-hour polysomnographic recordings (100), or ambulatory electroencephalographic (EEG) polygraphic recordings (101), but most investigators have found that the MSLT is more predictive than all of the above-mentioned tests for diagnosing narcolepsy.
Other polygraphic methods have been developed to measure EDS in narcolepsy, such as the sleep latency on the maintenance of wakefulness test (MWT) (102). The major difference with the MSLT is the instruction given to the subject. In a MWT the subject is told to attempt to remain awake. Generally, this testing method is not used for the diagnosis but rather to assess the effect of treatment with psychostimulants (103).
6.2. Biological/diagnostic markers
In addition to these clinical and polysomnographic criteria, HLA typing showing the association with HLA DQB1*0602 is supportive of the diagnosis, but the specificity of DQB1*0602 positivity is low (38). Today, CSF hypocretin-1 measurement has become a major positive diagnostic tool in the diagnosis of narcolepsy-cataplexy (16, 104), and low CSF hypocretin levels (110 pg/ml, one-third of mean control value) are included in the diagnostic criteria of narcolepsy in the second revision of ICSD (see the section of pathophysiology) (17).
6.3. Differential diagnosis
Narcolepsy is often misdiagnosed as a psychiatric condition or as an epilepsy variant. It may also be confounded with other forms of hypersomnia, such as sleep apnea syndrome, idiopathic hypersomnia or hypersomnia associated with depression. The presence of cataplexy is the key factor to single out narcolepsy from the other forms of hypersomnia. However, when cataplexy is predominant, narcolepsy can be misdiagnosed as syncopes, drop attacks, atonic attacks or attacks of a histrionic nature (some well-informed individuals may mimic the symptoms of narcolepsy in order to benefit from a disability leave from work or to obtain a prescription of psychostimulants). Narcolepsy without cataplexy may overlap with idiopathic hypersomnia, a heterogeneous disorder of chronic sleepiness (105). By definition, patients with idiopathic hypersomnia lack cataplexy and have less than two SOREMPs on the MSLT (Table 1). Some of these individuals have deep, excessively long periods of sleep, difficulty waking from sleep, and long unrefreshing naps, but many have symptoms similar to narcolepsy (105, 106).
7. Canine narcolepsy
In 1972, the Stanford Sleep Research Center (directed by Dr. William C. Dement) hosted an educational exhibit about human narcolepsy at the annual convention of the American Medical Association in San Francisco. As part of the exhibit, a film clip of narcoleptic patients having cataplexy was shown, and one member of the audience (a faculty member from the school of veterinary medicine at UC-Davis) mentioned that he took care of a dog that had the same attacks. Unfortunately, the dog had been euthanized since it was believed to be affected with refractory epilepsy. However, film clips of cataplectic attacks from this dog remained. Dr. Dement started to use those film clips along with those of human cataplexy for his lectures, including the one at the annual meetings of American Academy of Neology in Boston in 1973. After that lecture, a neurologist talked to Dr. Dement and mentioned that one of his friends had a dog that showed similar attacks. The dog was a miniature French Poodle named Monique, and was donated to Stanford. Monique revealed that the emotion of eating triggered multiple flaccid paralyses, and using surface electrodes, the loss of electromyogram (EMG) activities was observed during the attacks. An EEG was found to be normal, without any signs of an epileptic seizure, but showing patterns of typical of REM sleep and/or wakefulness (107). This together with an independent report of a narcoleptic dachshund dog by Knecht et al. (108) confirmed the existence of narcolepsy in dogs. In 1976, four narcoleptic Dobermans were obtained, and a breeding program for these dogs was initiated (109). Subsequently, it was demonstrated that narcolepsy in Dobermans (and Labradors) was transmitted with single autosomal recessive gene, named canarc-1, while the disease in other breeds was sporadic (109). With these multiple narcoleptic dogs, the Stanford Canine Narcolepsy Colony was established, and a breeding program for narcoleptic dogs was initiated (109) (see the section of inheritance of narcolepsy in canines) (Table 2).
7.1. Symptoms of canine narcolepsy
Affected dogs exhibit very pronounced attacks of cataplexy (which are mainly triggered by positive emotional experiences), such as being fed a favorite food or engaging in play. Cataplectic attacks in dogs often begin as a buckling of both hind legs, and this is often accompanied by a drooping of the neck (see Fig. 1). The dog may collapse to the floor and remain motionless for a few seconds or several minutes. In contrast to the some forms of epilepsy, excess salivation or incontinence are not observed during cataplectic attacks. During long cataplectic attacks, rapid eye movements, muscle twitching and/or slow repetitive movement of the fore and hind limbs may occur. These phenomena are related to the active phase of REM sleep. The muscle is always flaccid and never stiff during cataplectic attacks, and this is also different from most forms of seizure attacks. Dogs usually remain conscious (especially at the beginning of attacks) with eyes open and are capable of following moving objects with their eyes. If the attack lasts for an appreciable length of time (usually longer than 1 to 2 minutes), the dog may transit into sleep (often REM sleep). Dogs are often easily aroused out of an attack either by a loud noise or by being physically touched. Like narcoleptic humans, narcoleptic dogs are sleepier (fall asleep much more quickly) during the day. However, this was not noticeable at in usual circumstances, because even normal dogs take multiple naps during the daytime. While hypnagogic hallucinations and sleep paralysis may also occur in these dogs, there is no objective way to determine this at the present time.
Fig. 1. Cataplectic attacks in Doberman pinschers.

Emotional excitations, appetizing food or playing, easily elicit multiple cataplectic attacks in these animals. Most cataplexy attacks are bilateral (97.9%). Atonia initiated partially in the hind legs (79.8%), front legs (7.8%) neck/face (6.2%), or whole body/complete attacks (6.2%) Progression of attacks was also seen (49% of all attacks) (242).
A series of polygraphic studies clearly demonstrated that the dog's sleep/wake pattern is very fragmented and its wake/sleep bouts much shorter than age- and breed-matched dogs, and the narcoleptic dogs change their sleep states more frequently than they change their vigilance states (Fig. 2) (110-112). In other words, narcoleptic subjects could not maintain long bouts of wakefulness and sleep. By systematic polygraph assessments with multiple daytime nap tests, it was demonstrated that narcoleptic Dobermans showed shortened sleep latency and reduced latency to REM sleep (Fig. 2) (110) suggesting that these dogs have very similar phenotype to those in human narcolepsy.
Fig. 2. Percent of Time Spent in, Mean Frequency of, and Mean Duration for Each Vigilance State of Narcoleptic and Control Canines during Daytime 6-Hour Recordings (10:00 to 16:00).

(a, b) No significant difference was found in the percentage of time spent in each vigilance state between narcoleptic and control dogs. However, the mean duration of wake, drowsy, and deep sleep episodes were significantly shorter in the narcoleptics, suggesting a fragmentation of the vigilance states (wake and sleep) in these animals. To compensate for the influence of cataplectic episodes on wake and drowsiness, those episodes interrupted by the occurrence of cataplexy were excluded. (c) Mean latency (min) to each sleep stage and occurrences (number/total sessions) of cataplexy and sleep onset REM periods (SOREMPs) during the multiple sleep latency test (MSLT) in narcoleptic and control Dobermans. Drowsy and light sleep occurred in all sessions. Deep sleep, REM sleep or cataplexy (for narcoleptic dogs) occurred in some sessions, and the number of sessions where each state occurred/total number of sessions is shown in parentheses. Narcoleptic dogs exhibited cataplexy in 9 out of 100 sessions, and these events were differentiated from REM sleep episodes. Narcoleptic dogs show a significantly shorter latency to drowsy and light sleep in overall sessions. Note that narcoleptic dogs exhibited SOREMPs (i.e., REM sleep occurring within 15 min of sleep onset) significantly more often than control animals, although both narcoleptic (36.0 % of total session) and control dogs (21.7 %) showed REM sleep during the MSLT.
7.2. Inheritance of narcolepsy in canines
Unrelated narcoleptic Poodles, Beagles and Dachshunds donated to Stanford were bred, but none of the offspring from two primary crosses or a backcross were affected (113). Thus, it appears that in those breeds narcolepsy seems to be sporadic and involvement of a high-penetrant single major gene is unlikely. In humans, about 95% of narcoleptic subjects are sporadic, but familial occurrences were noted in about 5% of narcoleptic subjects (38) (Table 2). In 1976, the Stanford Sleep Research Center received four affected Dobermans (including two littermates) (109). These dogs were also bred, and it was discovered that all offspring from the two affected parents developed cataplexy around two months old. In 1978, familial narcolepsy in a Labrador retriever was also discovered (109). Thereafter, more sporadic cases of canine narcolepsy were identified in Collies, Dachshunds, Beagles, Fox Terriers and in several mixed breeds (109).
Genetic transmission in Dobermans and Labradors has been well established as autosomal recessive with full penetrance (109, 114). Puppies born from narcoleptic Doberman Pinscher-Labrador Retriever crosses are all affected. Thus, both breeds are likely to have mutations at the same locus, coined canarc-1. (109, 114). As in human cases (89), the disease onset in familial cases is earlier than in sporadic cases (113, 115). In sporadic cases, the disease (cataplexy onset) begins as early as seven weeks and as late as seven years old (113), suggesting the acquired nature of the disease in these cases. In Dobermans, affected dogs display spontaneous complete cataplexy as early as four weeks, but almost always by six months (109, 115). Symptom severity increases until 5-6 months of age (with females being more affected during development), and it appears to decrease slowly and then remain stable through old age (115, 116).
Genetic canine narcolepsy was thought to be an invaluable model in searching for narcolepsy genes since it is possible that the canine narcolepsy gene (its equivalent or genes with a functional relationship with the canine narcolepsy gene) may also be involved in some human cases.
8. Pathophysiology of narcolepsy
8.1. Pathophysiological consideration of the symptoms of narcolepsy
The similarity between cataplexy and REM sleep atonia (the presence of frequent episodes of hypnagogic hallucinations and of sleep paralysis, and the propensity for narcoleptics to go directly from wakefulness into REM sleep [i.e., SOREMPs]), suggests that narcolepsy is primarily a “disease of REM sleep” (117). This hypothesis may, however, be too simplistic and does not explain the presence of sleepiness during the day and the short latency to both NREM and REM sleep during nocturnal and nap recordings. Another complementary hypothesis is that narcolepsy results from the disruption of the control mechanisms of both sleep and wakefulness or, in other words, of the vigilance-state boundary problems (118). According to this hypothesis, a cataplectic attack represents an intrusion of REM sleep atonia during wakefulness, while the hypnagogic hallucinations appear as dream-like imagery taking place in the waking state, especially at sleep onset in patients who frequently have SOREMPs.
Cataplexy is associated with an inhibition of the monosynaptic H-reflex and the polysynaptic deep tendon reflexes (50). In control subjects, it is only during REM sleep that the H-reflex is totally suppressed. This finding highlights the relationship between the inhibition of motor processes during REM sleep and the sudden atonia and areflexia seen during cataplexy. Studies in canine narcolepsy, however, suggest that the mechanisms for the induction of cataplexy are different from those for REM sleep (110). Furthermore, an extended human study confirmed that cataplexy correlates much more highly to hypocretin-deficient narcolepsy (see below), in contrast to other REM sleep-related phenomena (16). Cataplexy may, thus, be viewed as somewhat distinct from other REM-related symptoms and as a hypocretin-deficiency pathological phenomenon. The fact that patients with other sleep disorders, such as sleep apnea, and even healthy controls, can manifest SOREMPs, hypnagogic hallucinations, and sleep paralysis when their sleep/wake patterns are sufficiently disturbed, yet these subjects never develop cataplexy, provides further support to the proposal that cataplexy may be unrelated to other REM-associated symptoms (19, 63, 119, 120). Although cataplexy and REM sleep atonia have great similarity and possibly share a common executive system, it is not necessary for the regulatory mechanism of both states to be identical. The mechanism of emotional triggering of cataplexy remains undetermined.
8.2. HLA typing in narcolepsy and other susceptible genes
In Japan, 1984, Juji et al. (121) reported that 100% of narcoleptics were positive for HLA DR2, and a close association subsequently was confirmed in Europe (122, 123) and also in North America (124). However, several DR2-negative cases have also been reported, suggesting the presence of other predisposing genes (125-127). The HLA gene complex maps on short arm of the chromosome six. It is divided into three subregions: HLA class I, II, and III. HLA plays a key role in the recognition and processing of foreign antigens by the immune system. HLA DR2 is implicated in several autoimmune diseases such as insulin-dependent diabetes mellitus, and multiple sclerosis (see the section of narcolepsy and immune system). More specific antisera were used to better characterize HLA DR2, and it was shown that the serological haplotype, DR15-DQ6 (DR2-DQw1 subtype) was associated with narcolepsy (128). Furthermore, the amplification of the polymorphic exon (second exon) of the HLA DQA and DQB genes by polymerase chain reaction has shown that all Caucasian narcoleptic subjects have the same alleles DRB1*1501, DQA1*0102 and DQB1*0602 (129). However, in African American narcoleptics the susceptibility to narcolepsy is more closely associated with the DQ6 subtype than with the DR15. In fact, only 70% of African American narcoleptics are DR15 positive, whereas they are almost all positive for the DQ6 (130-133). The most specific marker of narcolepsy in a number of different ethnic groups studied to date is DQB1*0602 (38).
Studies of HLA association were also conducted in narcoleptic subjects without cataplexy. An association with DQB1*0602 was found in only 40.9% of cases. This result clearly shows the close association between DQB1*0602 and the presence of cataplexy (132). In normal subjects, a significant reduction in REM sleep latency was noted when they were DQB1*0602 positive (38). Furthermore, HLA-DQB1*0602 homozygosity doubles to quadruples the risk for narcolepsy (134), and the relative risk for narcolepsy varies in DQB1*0602 heterozygote subjects according to the second allele located in trans to DQB1*0602 (135).
However, it was reported that approximately 25% of familial narcoleptic cases (including narcoleptic subjects and subjects presenting only recurrent isolated sleep episodes) are negative for DQB1*0602 (136), supporting the existence of one or more genes with high penetrance not associated with HLA. These genes could be located using systematic genome screening methods in extended families. The single published study showed a significant link in 4q13-q21 (lod score of 3.09) in eight small multigenerational families of narcoleptics, which favors the implication of other genes such as the CLOCK gene and the GABA β-1 receptor gene in these families (137). The candidate gene strategy has also been used. Along this line, it has been reported that the tumor necrosis factor-alpha gene with thymine residue at position −857 in its promoter region (TNF-alpha[−857T]) are associated with human HLA-DRB1*1501-positive (138) and -negative narcolepsy (139). In addition, a different distribution of the catechol-O-methyl transferase genotype, a key enzyme in dopaminergic and noradrenergic degradation, was found as well as a correlation between this phenotype and the severity of narcolepsy in female narcoleptic subjects (140).
8.3. Dog leukocyte antigen (DLA) and canine narcolepsy
Since human narcolepsy-cataplexy is specifically associated with the HLA gene HLA DQ*0602 (and DR15), three populations of dogs were tested to determine whether a specific DLA allele was present in affected animals as in narcoleptic humans. These included genetically narcoleptic Doberman Pinschers and Labrador Retrievers, and small breed dogs with sporadic narcolepsy. Unlike humans, narcoleptic dogs tested do not share any single DLA locus reactivity, suggesting that a specific MHC class II haplotype is not a requirement for the disease (141) (Table 2). In further experiments, a human HLA-DRb hybridization probe was used on DNA from narcoleptic dogs to determine whether there was an association between the DLA allele and susceptibility to narcolepsy (142). This probe detected polymorphisms in both Doberman Pinschers and Labrador Retrievers. Results of this study also excluded the possibility of a close linkage between DLA and the canarc-1 locus in these narcoleptic dogs (142). However, it now appears that more extensive studies including other DLA regions, specifically in “sporadic dogs,” examine the involvement of histocompatibility mechanisms in the development of hypocretin ligand-deficient narcolepsy in this species.
8.4. Narcolepsy and Immune System
An abundant amount of research into the HLA association in narcolepsy has already been conducted and has yielded important discoveries. However, the research has yet to provide clinically meaningful information. This work, therefore, is only very briefly addressed in this review. A remarkably high HLA association with narcolepsy was discovered in the early 1980s (121). Since the time of this initial finding, a variety of research across multiple ethnic groups has corroborated the existence of this strong HLA association. The most specific marker of narcolepsy in a number of different ethnic groups studied to date is DQB1*0602 (38). This association is seen in an average of approximately 90% of those with unequivocal cataplexy (132). Importantly, this association is substantially lower (approximately 40%) in individuals who have narcolepsy without cataplexy (Table 1).
The strong association between HLA type and narcolepsy with cataplexy raises the possibility that narcolepsy is an autoimmune disease (143). Some earlier studies testing a variety of serological tests in narcoleptics yielded higher levels of antistreptolysine 0 and anti-DNase antibodies in narcoleptics than in controls (144, 145). There is, however, no strong evidence of the inflammatory processes or immune abnormalities associated with narcolepsy (143), and studies have found no classical auto-antibodies and no increase in oligoclonal CSF bands in narcoleptics (146). Typical autoimmune pathologies (e.g., erythrocyte sedimentation rates, serum immunoglobulin levels, C-reactive protein levels, complement levels and lymphocyte subset ratios) are apparently normal in narcoleptic patients (147). Recent studies by Black et al. examined the presence of many neuron-specific and organ-specific auto-antibodies, but any auto-antibody was not associated with narcolepsy patients (HLA DQB1*0602-positive and -negative) (148). Furthermore, the same authors tested for immunoglobulin (Ig)G reactive to preprohypocretin and its major cleavage products (including hypocretin 1 and 2) in serum or CSF in DQB1*0602-positive narcoleptic subjects with cataplexy, but no evidence for IgG reactive to preprohypocretin or its cleavage products was found in narcoleptic subjects (149).
8.5. Discovery of canine narcolepsy gene (canarc-1)
Screening of genetic markers, including mini-satellite probes and functional candidate gene probes, revealed that canarc-1 cosegregates with a homolog of the switch region of the human immunoglobulin μ heavy-chain gene (Sμ) (114). The genuine Sμ segments are involved in a complex somatic recombination process, allowing individual B cells to switch immunoglobulin classes upon activation (see (114)). Fluorescence in situ hybridization (FISH) indicates that canarc-1 is located on a different canine chromosome from the canine immunoglobulin switch loci (150). Sequence analysis of the Sμ-like marker indicates that the Sμ-like marker has high homology to the true gene but is not a functional part of the immunoglobulin switch machinery (151). Thus, positional cloning of the region where the Sμ-like marker is located was initiated.
After 10 years of research, canarc-1 was finally identified, and narcolepsy in Dobermans and Labradors was found to be caused by a mutation in the hypocretin receptor 2 gene (Hcrtr 2) (Fig. 3) (Table 2). The mutations in Dobermans and Labradors were found in the same gene, but different locus, and both mutations cause exon skipping deletions in the Hcrtr 2 transcripts and the loss of function of Hcrtr 2, and, thus, impairs postsynaptic hypocretin neurotransmission. Therefore, it appears that these mutations occur independently in both breeds. Another mutation in Hcrtr 2 (152) was also found in a new narcoleptic family of Dachshunds, but the reason that Hcrtr 2 mutation often occurs in canines (no human case was yet identified) is not yet known.
Fig. 3. Genomic organization of the canine Hcrtr 2 locus.

The Hcrtr 2 gene is encoded by 7 exons. Sequence of exon-intron boundary at the site for the deletion of the transcript revealed that the canine short interspersed nucleotide element (SINE) was inserted 35 bp upstream of the 5′ splice donor site of the fourth encoded exon in narcoleptic Doberman Pinschers. This insertion falls within the 5′ flanking intronic region needed for pre-mRNA Lariat formation and proper splicing, causing exon 3 to be spliced directly to exon 5, and exon 4 to be omitted. This mRNA potentially encodes a non-functional protein with 38 amino acids deleted within the 5th transmembrane domain, followed by a frameshift and a premature stop codon at position 932 in the encoded RNA. In narcoleptic Labradors, the insertion was found 5 bp downstream of the 3′ splice site of the fifth exon, and exon 5 is spliced directly to exon 7, omitting exon 6.
Almost simultaneously (along with the discovery of the canine narcolepsy gene), a report that preprohypocretin (preproorexin) knockout mice also exhibited a narcolepsy-like phenotype, including sleep fragmentation and episodes of behavioral arrest similar to cataplexy in canine narcolepsy (9), was made by a group led by Dr. Yanagisawa (Table 2). Considering how similar human and canine narcolepsy are at the phenotypic level, it was thought that abnormalities in the hypocretin system are likely to be involved in some human cases, either at the functional or the genetic level.
9. Hypocretin (orexin) transmission and human narcolepsy
9.1. Mutation screening of hypocretin-related genes in human narcolepsy
After extensive screening (especially in familial and early-onset human narcolepsy), it was demonstrated that mutations in hypocretin-related genes are rare: only a single case with early-onset (6 months of age) was found to be associated with a single-point mutation in the preprohypocretin gene (15) (Table 2). This result was, however, not surprising, considering the fact that a large majority of human narcolepsy cases are sporadic. Even in rare familial cases of narcolepsy, it is unlikely that a high-penetrant single gene (like hypocretin-related-genes) is involved.
9.2. Hypocretin deficiency in human narcolepsy
Despite the lack of genetic abnormalities in the hypocretin system, it was found that the large majority (85-90%) of patients with narcolepsy-cataplexy have low or undetectable hypocretin-1 ligand in their CSF (13, 14) (Fig. 4). Postmortem human studies (utilizing a few brains) have confirmed hypocretin-ligand deficiency (both hypocretin 1 and 2) in the narcoleptic brain (15, 153) (Fig. 4) (Table 2).
Fig. 4. (a) Structures of mature hypocretin-1 (orexin-A) and hypocretin-2 (orexin-B) peptides. (b) Schematic representation of the hypocretin (orexin) system. (c) Projections of hypocretin neurons in the rat brain and relative abundances of Hcrtr 1 and 2.
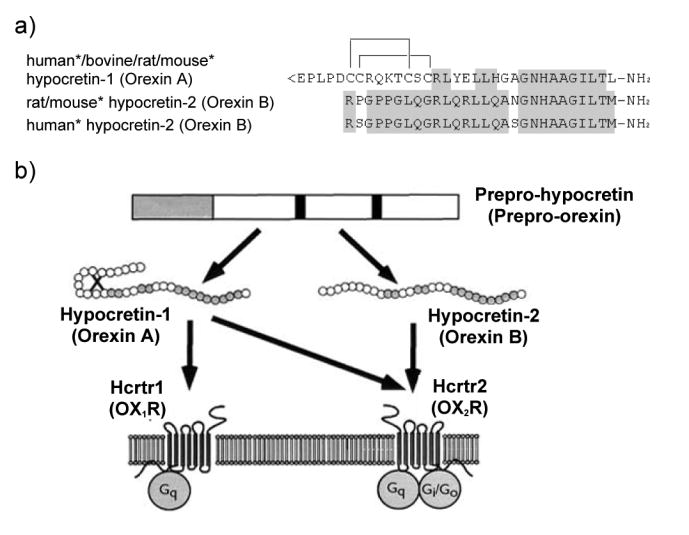
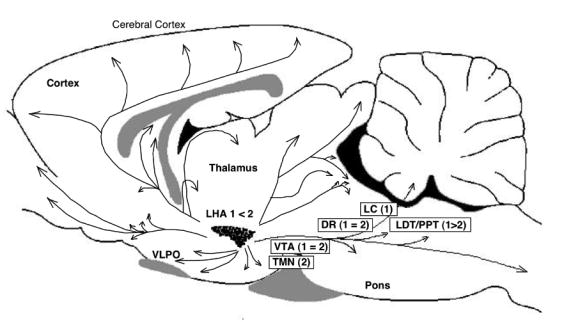
(a) The topology of the two intrachain disulfide bonds in orexin-A is indicated in the above sequence. The shaded areas indicate the amino acid identities. Asterisks indicate that human and mouse sequences were deduced from the respective cDNA sequences and not from purified peptides. Hypocretin-1 (orexin-A) and hypocretin-2 (orexin-B) are derived from a common precursor peptide, prepro-hypocretin (prepro-orexin). (b) The actions of hypocretins are mediated via two G protein-coupled receptors named hypocretin receptor 1 (Hcrtr 1) and hypocretin receptor 2 (Hcrtr 2), also known as orexin-1 (OX1R) and orexin-2 (OX2R) receptors, respectively. Hcrtr 1 is selective for hypocretin-1, whereas Hcrtr 2 is nonselective for both hypocretin 1 and hypocretin 2. Hcrtr 1 is coupled exclusively to the Gq subclass of heterotrimeric G proteins, whereas in vitro experiments suggest that Hcrtr 2 couples with Gi/o, and/or Gq. (adapted from Sakurai 2003) (c) Hypocretin-containing neurons project to these previously identified monoaminergic and cholinergic and cholinoceptive regions where hypocretin receptors are enriched. Impairments of hypocretin input may, thus, result in cholinergic and monoaminergic imbalance and generation of narcoleptic symptoms. VTA, ventral tegmental area; SN, substantia nigra; LC, locus coeruleus; LDT, laterodorsal tegmental nucleus; PPT, pedunculopontine tegmental nucleus; PRF, pontine reticular formation; BF, basal forebrain; VLPO, ventrolateral preoptic nucleus; LHA, lateral hypothalamic area; TMN; tuberomamillary nucleus; DR, dorsal raphe.
Hypocretin deficiency has also been observed in sporadic cases of canine narcolepsy (7 out of 7 currently studied; the results of 4 cases are reported), suggesting that the pathophysiology in these animals mirrors that of most human cases (154) (Table 2).
Such low levels of CSF hypocretin-1 are relatively specific for narcolepsy-cataplexy but are also seen in a few other neurological conditions, such as a subset of patients with Guillain-Barré syndrome and Ma2 positive paraneoplastic syndrome (155, 156). Since these conditions are clinically distinct from narcolepsy, low CSF hypocretin levels in these conditions do not confound their diagnostic values.
Among the patients with narcolepsy (and its spectrum) and other hypersomnias, hypocretin deficiency is closely associated with the occurrence of cataplexy and HLA-DQ1*0602 among narcoleptic subjects (16, 157, 158) (Fig. 6).
Fig. 6. Lumbar CSF hypocretin-1 concentrations in controls, narcoleptics, and other pathologies.

Each point is the concentration of hypocretin-1 in the crude (unfiltered) lumbar CSF of a single individual. Represented are controls (samples taken both during night and day) and narcoleptics, including those with typical cataplexy, with atypical cataplexy, with cataplexy but who are HLA-negative, and without cataplexy, as well as narcolepsy family probands. Individuals with hypersomnia due to idiopathic hypersomnia, periodic hypersomnia, or hypersomnia due to secondary etiology are also shown, as are those with other diagnostically described sleep disorders (obstructive sleep apnea (n=17), restless legs syndrome (n=12), insomnia (n=12)) and those with a variety of neurological disorders. Specific pathologies are described for individuals with low (<110 pg/mL) or intermediate (110 – 200 pg/mL) concentrations of hypocretin-1. Data is derived from (16).
It should be also noted that even when a very strict criteria for cataplexy is applied, about 10% of narcolepsy-cataplexy patients have normal CSF hypocretin-1 (14, 16, 158). Whether or not hypocretin neurotransmission is abnormal in these rare cases is presently unknown. Considering the fact that hypocretin production and hypocretin neurons appeared to be normal in Hcrtr 2-mutated narcoleptic Dobermans (154), it is possible that deficiencies in hypocretin receptors and a downstream pathway may exist in some of these patients. However, this cannot be currently tested.
9.3. Diagnostic value of CSF hypocretin-1 measures and general considerations of the assay
When used to assess patients for narcolepsy, low CSF hypocretin-1 appears to be a more specific test than the MSLT and CSF hypocretin-1 levels (Table 1). Therefore, low CSF hypocretin-1 levels (less than 110 pg/ml) were also included for the second revision of ICSD (17). Previously, no specific and sensitive diagnostic test for narcolepsy based on the pathophysiology of the disease was available, and the final diagnosis was often delayed for several years after the disease onset, typically adolescence (83). Many patients with narcolepsy and related EDS disorders are, therefore, likely to obtain immediate benefit from this new specific diagnostic test. Furthemore, hypocretin agonists may be promising in the treatment of narcolepsy (see the section for the future directions).
CSF hypocretin-1 levels are currently measured by the direct radioimmunoassay (RIA) (in 100 μl) without any extraction. Hypocretin-1 signals in the CSF were very stable, and the levels measured by RIA were not altered in the CSF samples that were frozen and thawed up to six times nor in the CSF samples kept at room temperature for 72 hours (159). Although it was not fully determined whether the levels measured by RIA correspond to their biological activities of hypocretin-1 in the CSF samples (especially if CSF samples were kept under varied conditions), the “net” immunoreactive-hypocretin signals (without consideration of their biological activities) may be sufficient for the diagnostic purpose of hypocretin-deficient narcolepsy. There was also no concentration gradient in hypocretin-1 levels up to 12ml in human lumbar CSF (159), making no strict selection of the CSF fraction for this diagnostic test. Thus, most of the authors are currently using direct assay for measuring CSF hypocretin-1 levels for diagnostic purposes.
Nevertheless, a large inter-assay variation of RIA is still the one of the major concerns. In our experience, the variation was as high as 20% even in short-term assessments and up to 100% for long-term assessments. Therefore, the standardization of the assay condition is critical (see detail in Nishino (160)). It is recommended that all comparative samples (most typically control human CSF samples) be run in a single RIA. If this is not possible, reference samples (such as stock CSF samples with known hypocretin-1 values), in addition to the peptide standard, should be included in each assay and the values adjusted accordingly. In this regard, we suggest using 30% of the normal CSF hypocretin value obtained in the same lab (rather than the published values) as the cut-off point for the diagnosis of the hypocretin-deficient condition.
At the 5th International Symposium on Narcolepsy (Ascona, 2004), the standardization of CSF hypocretin measures was discussed during a workshop for the diagnostic guideline for narcolepsy. The panel discussed the possibility of providing (1) monoclonal antibody and RIA development kits, (2) hypocretin standards, and (3) reference CSF samples (ideally 2 different concentrations, such as 150 and 300 pg/ml) to the worldwide laboratories that carry out the assay.
9.4. Symptomatic cases of narcolepsy and EDS, and the hypocretin system
CSF hypocretin-1 measurements were also carried out in a limited number of symptomatic cases of narcolepsy/EDS, including narcolepsy/EDS associated with tumors (n=5), head trauma (n=3), vascular disorders (n=5), encephalopathies (n=3), degeneration (n=30), demyelinating disorder (n=7), genetic/congenital disorders (n=11) and others (n=2) (85).
Reduced CSF hypocretin-1 levels were seen in most symptomatic narcolepsy cases of EDS with various etiologies and EDS in these cases is sometimes reversible with an improvement of the causative neurological disorder and an improvement of the hypocretin status, such as acute disseminated encephalomyelitis (ADEM) and multiple sclerosis cases (161-164). It is also noted that some symptomatic EDS cases (with Parkinson's disease and thalamic infarction) appeared, but they are not linked with hypocretin ligand deficiency (104, 165, 166). In contrast to idiopathic narcolepsy cases, an occurrence of cataplexy is not closely associated with hypocretin ligand deficiency in symptomatic cases (85).
Since CSF hypocretin measures are still experimental, cases with sleep abnormalities/cataplexy are habitually selected for CSF hypocretin measures and should be further studied if all or a large majority of cases with low CSF hypocretin-1 levels with CNS interventions exhibit EDS/cataplexy.
10. Hypocretin /orexin system and sleep regulation
Hypocretins/orexins were only recently discovered (in 1998, one year prior to the cloning of the canine narcolepsy gene) by two independent research groups. One group called the peptides “hypocretin” because of their primary hypothalamic localization and similarities with the hormone “secretin” (12). The other group called the molecules “orexin” after observing that central administration of these peptides increased appetite in rats (11).
Hypocretins/orexins (hypocretin-1 and hypocretin-2/Orexin A and Orexin B) are cleaved from a precursor preprohypocretin (prepro-orexin) peptide (11, 12). Hypocretin-1 with 33 residues contains four cysteine residues forming two disulfide bonds. Hypocretin-2 consists of 28 amino acids and shares similar sequence homology especially at the C-terminal side but has no disulfide bonds (a linear peptide) (11) (Fig. 5). There are two G-protein-coupled hypocretin receptors, Hcrtr 1 and Hcrtr 2, also called orexin receptor 1 and 2 (OX1R and OX2R), and distinct distribution of these receptors in the brain is known: Hcrtr 1 is abundant in the locus coeruleus (LC). Hypocretin Hcrtr 2 is found in the tuberomamillary nucleus (TMN) and basal forebrain. Both receptor types are found in the midbrain raphe nuclei and mesopontine reticular formation (167) (Fig. 5).
Fig. 5. (a) CSF hypocretin-1 levels in narcoleptic and control subjects. (b) Preprohypocretin mRNA in situ hybridization in the hypothalamus of control and narcoleptic subjects.
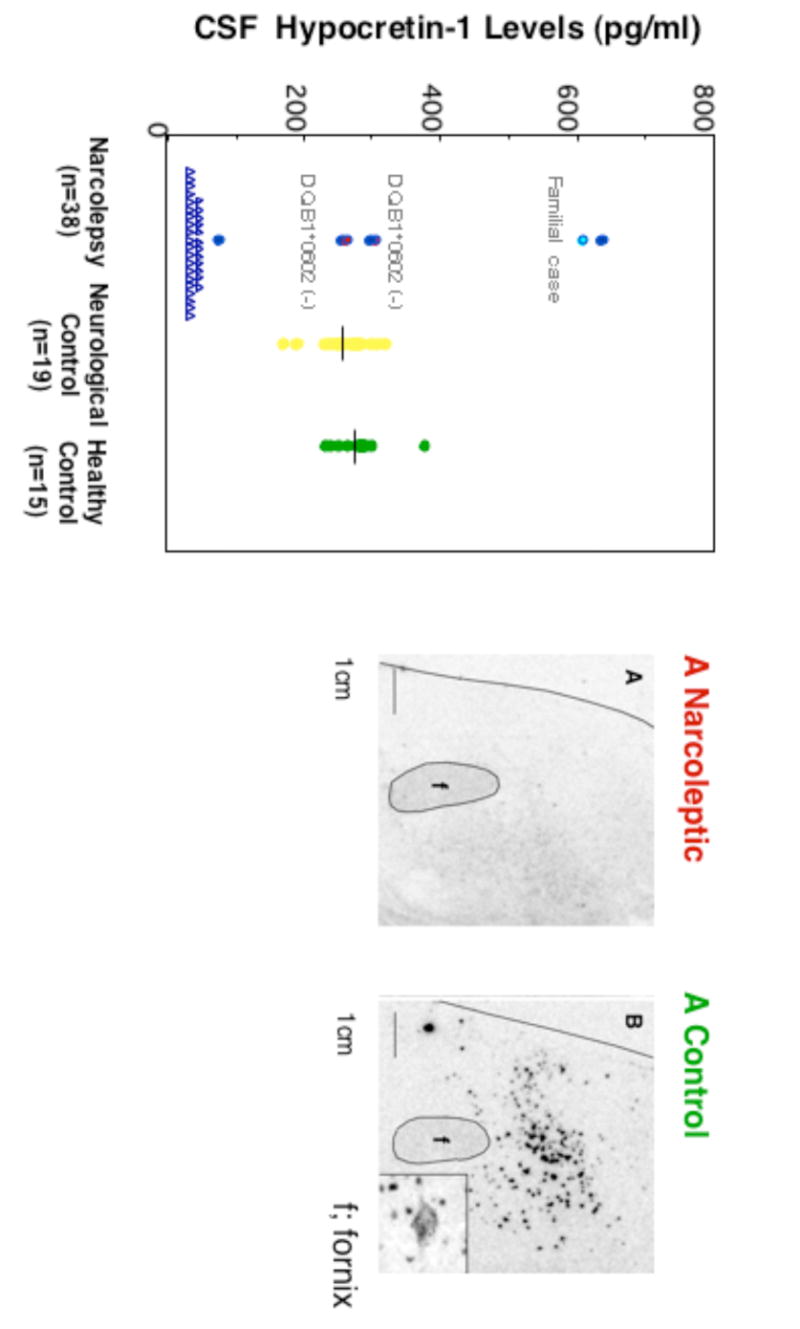
(a) CSF hypocretin-1 levels are undetectably low in most narcoleptic subjects (84.2%). Note that two HLA DqB1*0602-negative and one familial case have normal or high CSF hypocretin levels. (b) Preprohypocretin transcripts are detected in the hypothalamus of control (B) but not narcoleptic subjects (A). Melanin-concentrating hormone (MCH) transcripts are detected in the same region in control and narcoleptic sections (data not shown).
Hypocretins-1 and -2 are produced exclusively by a well-defined group of neurons localized in the lateral hypothalamus. The neurons project to the olfactory bulb, cerebral cortex, thalamus, hypothalamus and brainstem, particularly the LC, raphe nucleus and to the cholinergic nuclei (the laterodorsal tegmental and pedunculopontine tegmental nuclei) and cholinoceptive sites (such as pontine reticular formation) thought to be important for sleep regulation (168) (Fig. 5).
A series of recent studies have now shown that the hypocretin system is a major excitatory system that affects the activity of monoaminergic (dopamine [DA], norepinephrine [NE], serotonin [5-HT] and histamine) and cholinergic systems with major effects on vigilance states (169, 170) (Fig. 5). It is, thus, likely that a deficiency in hypocretin neurotransmission induces an imbalance between these classical neurotransmitter systems, with primary effects on sleep-state organization and vigilance. Indeed, DA and/or NE contents have been reported to be high in several brain structures in narcoleptic Dobermans and in human narcolepsy postmortem brains (8, 171). These changes are possibly due to compensatory mechanisms, since the drugs that enhance dopaminergic neurotransmission (such as amphetamine-like stimulants and modafinil [for EDS]) and NE neurotransmission (such as noradrenaline uptake blockers [for cataplexy]) are needed to treat the symptoms of narcolepsy (8). Histamine is another monoamine implicated in the control of vigilance, and the histaminergic system is also likely to indirectly mediate the wake-promoting effects of hypocretin (172-174). Interestingly, brain histamine contents both in Hcrtr 2-mutated and ligand-deficient narcoleptic dogs are dramatically reduced (175). Furthermore, two impendent human studies also demonstrated that CSF histamine contents are significantly lower in hypocretin-deficient narcolepsy than those in controls (175, 176). Interestingly, low CSF hypocretin levels were also observed in other primary hypersominias, such as narcolepsy without cataplexy and idiopathic hypersomnia (hypocretin non-deficient hypersomnias), but histamine levels in patients with sleep apnea are in the normal range (176). Although the functional role of CSF histamine is not presently well understood, this finding is very interesting since altered central histamine transmission may be involved in broader categories of hypersomnia and is not limited to hypocretin-deficient narcolepsy. It is, thus, interesting to further evaluate the involvement of the histaminergic system in the pathophysiology of narcolepsy/EDS and therapeutic applications of histaminergic compounds (177).
Many measurable activities (brain and body) and compounds manifest rhythmic fluctuations over a 24-hour period. Whether or not hypocretin tone changes with zeitgeber time was assessed by measuring extracellular hypocretin-1 levels in the rat brain CSF across 24-hour periods, using in vivo dialysis (178-180). The results demonstrate the involvement of a slow diurnal pattern of hypocretin neurotransmission regulation (as in the homeostatic and/or circadian regulation of sleep). Hypocretin levels increase during the active periods and are highest at the end of the active period, and the levels decline with the onset of sleep. Furthermore, sleep deprivation increases hypocretin levels (178-180).
Recent electrophysiological studies have shown that hypocretin neurons are active during wakefulness and reduce their activity during slow wave sleep (181, 182). Neuronal activity during REM sleep is the lowest, but intermittent increases in the activity associated with body movements or phasic REM activity are observed (181, 182). In addition to this short-term change, the results of microdialysis experiments also suggest that basic hypocretin neurotransmission fluctuates across the 24-hour period and slowly builds up toward the end of the active period. Adrenergic LC neurons are typical wake-active neurons involved in vigilance control, and it has been recently demonstrated that basic firing activity of wake-active LC neurons also significantly fluctuates across various circadian times (183).
Several acute manipulations such as exercise, low glucose utilization in the brain, as well as forced wakefulness, increase hypocretin levels (169, 179, 184). It is, therefore, hypothesized that a build-up/acute increase of hypocretin levels may counteract homeostatic sleep propensity that typically increases during the daytime and during forced wakefulness (179). Due to the lack of increase in hypocretin tone, narcoleptic subjects may not be able to stay awake for prolonged periods and do not respond to various alerting stimuli. Conversely, reduction of the hypocretin tone at sleep onset may contribute to the profound deep sleep that normally inhibits REM sleep at sleep onset, and the lack of this system in narcolepsy may allow the occurrence of REM sleep at sleep onset.
11. Treatments and pharmacological aspects of narcolepsy
Non-pharmacological treatments (i.e., by behavioral modification) are often reported to be useful additions to the clinical management of narcoleptic patients (185-188). Regular napping usually relieves sleepiness for one to two hours (185) and is the treatment of choice for some patients, but this often has negative social and professional consequences. Exercising to avoid obesity, keeping a regular sleep-wake schedule, or having a supportive social environment (e.g., patient group organizations and support groups) is also helpful. In almost all cases, however, pharmacological treatment is needed, and 94% of patients reported using medications in a recent survey by a patient group organization (189).
11.1. Current pharmacological treatment for human narcolepsy
For EDS, amphetamine-like CNS stimulants or modafinil (nonamphetamine stimulant, mechanisms of action is debated) are used most often (Table 4). These compounds possess wake-promoting effects in narcoleptic subjects as well as in control populations, but very high doses are required to normalize abnormal sleep tendency during daytime for narcolepsy (190). For consolidating nighttime sleep, benzodiazepine hypnotics or gamma hydroxybutyrate (GHB) are occasionally used, and nighttime administration of GBH reduces EDS and cataplexy during the daytime (191, 192). GHB was once classified as a schedule I control substance in 2000 but has been recently approved for the treatment of narcolepsy. Since amphetamine-like stimulants and modafinil have little effect on cataplexy, tricyclic antidepressants, such as imipramine or clomipramine, are used in addition to control cataplexy (8). However, these compounds can cause a number of side effects, such as dry mouth, constipation or impotence. GHB is also used for the treatment of cataplexy, but the mechanisms of GHB are currently unknown. The antidepressants and GHB used are also effective for the other REM sleep phenomena.
11.2. Pharmacological control of cataplexy and EDS: Findings from the canine experiments
Along with the discovery of canarc-1, biomedical research in narcolepsy has been also greatly facilitated using narcoleptic canines (107, 113). Based on the data obtained from the canine model, current understanding of the neuropharmacological control of cataplexy and excessive sleepiness (as well as some prospects for the new treatment of narcolepsy) is discussed. For more details on the neuropharmacological results, please refer to a review article by Nishino and Mignot (8).
12.2.1. REM Sleep/Cataplexy and Narcolepsy
After the discovery of SOREMPs in narcoleptic patients (193), narcolepsy has often been regarded as a disorder of REM sleep generation. REM sleep usually appears 90 minutes after sleep onset and re-appears every 90 minutes in humans (30 minutes in dogs). Therefore, it was thought that in narcolepsy REM sleep can intrude in active wake or at sleep onset, resulting in cataplexy, sleep paralysis and hypnagogic hallucinations, and these three symptoms are often categorized as “dissociated manifestations of REM sleep” (see (8)). Abnormal generation of REM sleep might, therefore, be central to narcolepsy, but this has not been previously demonstrated experimentally. We have, therefore, analyzed the REM sleep and cataplexy cyclicity in narcoleptic and control canines to observe whether the cyclicity at which REM sleep occurs is disturbed in narcoleptic canines (110). Interval histograms for REM sleep episodes revealed that a clear 30-minute cyclicity exists in both narcoleptic and control animals, suggesting that the system controlling REM sleep generation is intact in narcoleptic dogs. In contrast to REM sleep, cataplexy can be elicited any time upon emotional stimulation (i.e., no 30-minute cyclicity is observed) (110).
These results, together with the results of extensive human study, show that cataplexy is closely associated with hypocretin-deficient status (cataplexy appears now to be a unique pathological condition caused by a loss of hypocretin neurotransmission)(16), suggesting that the mechanisms for the triggering of cataplexy and REM sleep are distinct. However, previous electrophysiological data has also demonstrated various similarities between REM sleep atonia and cataplexy (194). Since H-reflex activity (one of the monosynaptic spinal electrically induced reflexes) profoundly diminishes or disappears during both REM sleep and cataplexy, it is likely that the motor inhibitory components of REM sleep are also responsible for the atonia during cataplexy (194). Thus, the executive systems for the induction of muscle atonia during cataplexy and REM sleep are likely to be the same. This interpretation is also supported by the pharmacological findings that most compounds that significantly reduce or enhance REM sleep, reduce and enhance cataplexy, respectively. However, some exceptions, such as discrepant effects of D2/D3 antagonists on REM sleep and cataplexy, also exist (see below for more detail) (195).
12.2.2. Monoaminergic and cholinergic Interactions and cataplexy
The importance of increased cholinergic activity in triggering REM sleep or REM sleep atonia is well established (see (196)). Similarly, activation of the cholinergic systems using the acetylcholinesterase inhibitor physostigmine also greatly exacerbates cataplexy (197). This cholinergic effect is mediated by muscarinic receptors since muscarinic stimulation aggravates cataplexy, while its blockade suppresses it, and nicotinic stimulation or blockade has no effect (197).
Monoaminergic transmission is also critical for the control of cataplexy. All therapeutic agents currently used to treat cataplexy (i.e., antidepressants or monoamine oxidase inhibitors [MAOIs]) are known to act on these systems. Furthermore, whereas a subset of cholinergic neurons are activated during REM sleep, the firing rate of monoaminergic neurons in the brainstem (such as in the LC and the raphe magnus) are well known to be dramatically depressed during this sleep stage (198, 199). Using canine narcolepsy, it was recently demonstrated that adrenergic LC activity is also reduced during cataplexy (200). In contrast, dopaminergic neurons in the ventral tegmental area (VTA) and substantia nigra do not significantly change their activity during natural sleep cycles (201, 202).
Since cataplexy in dogs can be easily elicited and quantified, the canine narcolepsy model has been intensively used to dissect the mode of action of currently used anticataplectic medications. The most commonly prescribed anticataplectic mediations in humans are tricyclic antidepressants. These compounds, however, have a complex pharmacological profile that includes monoamine uptake inhibition (dopamine [DA], epinephrine, norepinephrine [NE] and serotonin [5-HT]), anticholinergic, alpha-1 adrenergic antagonistic and antihistaminergic effects. It is, thus, difficult to conclude which one of these pharmacological properties is actually involved in their therapeutic effects.
We, therefore, first studied the effects of a large number (a total of 17 compounds) of uptake blockers/release enhancers specific for the adrenergic, serotonergic or dopaminergic system, and adrenergic uptake inhibition was found to be the key property involved in the anticataplectic effect (203). Serotonergic uptake blockers were only marginally effective at high doses and the dopaminergic uptake blockers were completely ineffective. Interestingly, it was later found that these dopamine uptake inhibitors had potent alerting effects in canine narcolepsy (8).
We also compared the effects of several antidepressants with those of their demethylated metabolites. Many antidepressant compounds (most typically tricyclics) are known to metabolize significantly by a hepatic first pass into their demethylated metabolites that have longer half-lives and higher affinities for adrenergic uptake sites (204). During chronic drug administration, these demethylated metabolites accumulate (204) and can, thus, be involved in the drug's therapeutic action. The effects of five available antidepressants (i.e., amitriptyline, imipramine, clomipramine, zimelidine, and fluoxetine) were compared with those of their respective demethylated metabolites (i.e., nortriptyline, desipramine, desmethylclomipramine, norzimelidine and norfluoxetine) (205). In all cases, the demethylated metabolites were found to be more active on cataplexy than were the parent compounds. We also found that the active dose of all anticataplectic compounds tested positively and correlated with the in vitro potency of each compound to the adrenergic transporter but not with that of the serotonergic transporter (205). In fact, the anticataplectic effects were negatively correlated with the in vitro potency for serotonergic uptake inhibition, but this may be biased since potent adrenergic uptake inhibitors included in the study have a relatively low affinity to serotonergic uptake sites.
Although the results presented were obtained from inbred Hcrtr-2 mutated narcoleptic Dobermans, similar findings have been obtained in more diverse cases of sporadic canine narcolepsy in various breeds donated to our colony (see (197)). Protryptiline and desipramine (two compounds with no significant serotonergic uptake-blocking properties) have also been shown to be very effective for the treatment of human cataplexy (186, 206-208). Thus, the preferential involvement of the adrenergic system for the modes of action of anticataplectic effects of antidepressants, are suggested, regardless of the form of deficit in hypocretin neurontransmission (receptor mutation vs. hypocretin-ligand deficiency).
In order to dissect receptor subtypes that significantly modify cataplexy, more than 200 compounds with various pharmacological properties (cholinergic, adrenergic, dopaminergic, serotonergic, prostaglandins, opioids, benzodiazepines, GABA-ergics and adenosinergics) have also been studied in the narcoleptic canine model (see (8) for details). Although many compounds such as M2 antagonists, alpha-1 agonists, alpha-2 antagonists, dopaminergic D2/D3antagonists, 5HT1a agonists, TRH analogs, prostaglandin E2, and L type Ca2+ channel blockers reduce cataplexy, very few compounds significantly aggravate cataplexy (8). Since unpleasant side effects of drugs generally reduce cataplexy in these animals, we assume the cataplexy-aggravating effects are more specific. Stimulation of muscarinic M2 (non-M1) receptors significantly aggravates cataplexy, and among the monoaminergic receptors, stimulation of the postsynaptic adrenergic alpha-1b receptors (209) and presynaptic alpha-2 receptors (210) was also found to aggravate cataplexy, a result consistent with a primary adrenergic control of cataplexy. We also found that small doses of DA D2/D3 agonists significantly aggravated cataplexy and induced significant behavioral sedation/drowsy states in these animals (211, 212). These pharmacological findings parallel neurochemical abnormalities previously reported in canine narcolepsy, namely significant increases in alpha-2 receptors in the LC (213), D2 receptors in the amygdala, nucleus accumbens (214) and M2 receptors in the pons (215). To date, no other receptor ligands (i.e., adenosinergic, histaminergic or GABA-ergic) have been found to aggravate cataplexy, but thalidomide (an old hypnotic with an immunomodulatory property) significantly aggravates cataplexy, but the mechanisms involved in this effect are not presently known (216).
The sites of action of D2/D3 agonists were also investigated by local drug perfusion experiments, and a series of experiments identified the acting sites for these compounds. These include dopaminergic nuclei or cell groups, such as the VTA (212), SN (217) and A11 (218) (a diencephalic DA cell group that directly project to the spinal ventral horn), suggesting a direct involvement of DA cell groups and DA cell body autoreceptors for the regulation of cataplexy. The cataplexy-inducing effects of D2/D3 agonists are, however, difficult to reconcile considering the fact that dopaminergic uptake blockers have absolutely no effect on cataplexy (203). We believe that D2/D3 receptor mechanisms are more specifically involved in the control of sleep-related motor tonus (i.e., cataplexy or muscle atonia without phasic REM events) than those for active REM sleep. A recent finding in canine narcolepsy that sulpiride (a D2/D3 antagonist) significantly suppresses cataplexy but has no effect on REM sleep also supports this notion (195). It should be also noted that D2/3 agonists are clinically used for the treatment of human periodic leg movements during sleep (PLMS) (219). Involuntary leg movements during sleep are often associated with restless leg syndrome (RLS) and disturbed nighttime sleep. As reported in humans (220), narcoleptic Dobermans often exhibit PLMS-like movements (221), and it appears that there may be an overlap in the pathophysiological mechanisms between cataplexy and PLMS, and the dopaminergic system (i.e., D2/D3 receptor mechanisms) may be specifically involved both symptoms.
The effects on cataplexy by cholinergic stimulation in various brain regions were also examined in narcoleptic and in control canines. Local injection or perfusion of carbachol (a predominantly muscarinic agonist) into the pontine reticular formation (PRF) was found to aggravate canine cataplexy in a dose-dependent fashion (222). The results obtained in the PRF with cholinergic agonists were somewhat expected considering the well established role of the pontine cholinergic systems in the regulation of REM sleep and REM sleep atonia. Surprisingly, however, we also found that the local injection/perfusion of carbachol unilaterally or bilaterally into the basal forebrain (rostral to the preoptic area, in the vertical or horizontal limbs of the diagonal band of Broca and medial septum) dose-dependently aggravated cataplexy and induced long-lasting episodes of muscle atonia accompanied by desynchronized EEG in narcoleptic canines (223). Physostigmine was also found to aggravate cataplexy when injected into the same site, thus suggesting that fluctuations in endogenous levels of acetylcholine in this structure may be sufficient to induce cataplexy (223). The carbachol injections did not induce cataplexy in normal animals, but rather induced wakefulness.
The basal forebrain is anatomically connected with the limbic system, which is regarded as a critical circuit for integrating emotions. Furthermore, basal forebrain neurons are known to respond to the arousing property of appetitive stimuli (224), which we use to induce cataplexy in narcoleptic dogs. Considering the fact that emotional excitation is an alerting stimulus in normal animals but induces cataplexy in narcoleptic animals, the basal forebrain may be involved in triggering a paradoxical reaction to emotions-atonia rather than wakefulness in narcoleptic animals. These results also suggest that more global brain structures (than those for REM sleep generation) are involved in the induction of cataplexy. Cataplexy is now demonstrated to be closely associated with the loss of hypocretin neurotransmission. Global and persistent cholinergic/monoaminergic imbalance due to the loss of hypocretin neurotransmission may be required for the occurrence of cataplexy, and cataplexy could not be induced simply by an increase in REM sleep propensity and/or vigilance state instability that occurs in various disease conditions (such as depression) or in some physiological conditions (such as REM sleep deprivation). The findings that REM sleep abnormalities and sleep fragmentation are often seen in other sleep disorders such as narcolepsy without cataplexy, sleep apnea, and even in healthy subjects when their sleep patterns are disturbed but who never develop cataplexy, further supports this hypothesis. The mechanism for emotional triggering of cataplexy remains to be studied, but multiple brain sites and multiple functional and anatomical systems are likely to be involved.
12.2.3. Dopaminergic transmission and EEG arousal
Narcoleptic Dobermans were also used for a series of pharmacological experiments to dissect modes of action of wake-promoting compounds. Amphetamine-like CNS stimulants currently used clinically for the management of sleepiness in narcolepsy enhance monoaminergic transmission presynaptically. However, these compounds also lack pharmacological specificity. In order to study the mode of action of these wake-promoting compounds on daytime sleepiness, the stimulant properties of several dopaminergic and adrenergic uptake inhibitors were quantified and compared to the effects of amphetamine and modafinil using four-hour daytime polygraphic recordings (225). In spite of their lack of effect on cataplexy, all dopaminergic uptake inhibitors induced significant EEG arousal (Fig. 4). In contrast, nisoxetine and desipramine, two potent adrenergic uptake inhibitors, had little effect on EEG arousal but significantly suppressed REM sleep. Furthermore, the in vivo potency of DA uptake inhibitors on EEG arousal correlates well with their in vitro affinity to the DA transporter (DAT), but not to the norepinephrine transporter (NET). These results are consistent with the hypothesis that the presynaptic modulation of dopaminergic transmission is a key property mediating the EEG arousal effects of these compounds. Interestingly, we also found that modafinil binds to the DAT site with low affinity (225, 226), similar to the affinity range for amineptine (a DA uptake-inhibitor which also enhances EEG arousal in our model). Thus, the DAT binding property may also contribute to the stimulant properties of modafinil. We recently demonstrated that the wake-promoting effect of modafinil and amphetamine were completely absent in mice lacking DAT (i.e., DAT knockout mice) (227). These results clearly indicate that DAT is required for the mediation of the wake-promoting effect of modafinil (and amphetamine), but other pharmacological properties may also be involved in the wake-promoting effects of modafinil (228, 229).
Amphetamine (which showed a potent EEG arousal property) has, however, a relatively low DAT binding affinity (230), suggesting that other mechanisms, such as monoamine release (exchange diffusion through the DAT) by amphetamine, are also involved in the mechanism of EEG arousal. Amphetamine is also reported to block the vascular monoamine transporter 2 (VMAT 2) and to induce reverse-transport of monoamines into the synaptic area (231). To further assess the net effects of amphetamine on monoaminergic neurotransmission, we measured DA and NE efflux together with the wake-promoting effects of amphetamine-analogs and isomers in the canine narcolepsy model (232). Polygraphic recordings demonstrated that d-amphetamine was about twice as potent as l-amphetamine, and was six times more potent than l-methamphetamine in increasing wakefulness, while d-amphetamine and l-amphetamine were equipotent in reducing REM sleep and cataplexy, and l-methamphetamine was about half as potent as l- and d-amphetamine (232). By measurements of extracellular levels of DA and NE, we found that d-amphetamine was more potent in increasing DA efflux than l-amphetamine, and l-methamphetamine had little effect. In contrast, there was no significant difference in the potencies of theses three derivatives on NE efflux. Thus, the potencies of amphetamine isomers/analogs on wakefulness correlated well with DA, but not NE efflux in the brain of narcoleptic dogs, and this further exemplifies the importance of the DA system for the pharmacological control of EEG arousal (232).
However, the involvement of the dopaminergic systems in the regulation of the sleep/wake process has not been given much attention, mostly due to early electrophysiological findings demonstrating that dopaminergic neurons in the ventral tegmental area (VTA) and substantia nigra do not change their activity significantly during the sleep cycle (202), in contrast to noradrenergic cells of the LC or serotonergic cells of the raphe which increase firing in wake versus sleep. Although the firing patterns of DA neurons during slow wave sleep are different from those during wake or REM sleep, DA release in the LC and amygdala measured with microdialysis experiments failed to demonstrate the state-dependent change (233). These experimental results led most investigators to believe that adrenergic tone was more important than dopaminergic transmission for the control of EEG arousal.
Therefore, the dopaminergic system may not be so important for the normal sleep/wake cycle regulation but may play a key role for forced wakefulness by motivation and/or by stimulants, and the hypocretin-dopaminergic interaction may also be involved in these alerting mechanisms. In clinical conditions, a disturbance of this mechanism may be more troublesome since it may induce intolerable sleepiness in situations requiring motivated wakefulness.
An involvement of dopaminergic system in intolerable sleepiness is also noted in some pathological conditions, such as Parkinson's disease. Frequent sleep attacks, and associations with accidents are reported by patients with Parkinson's disease treated with DA D2/D3 agonists (234, 235); this class of compounds induces drowsy states and cataplexy in the canine model of narcolepsy (211, 212).
12.2.4. Hypocretin replacement therapy
Finally, a canine narcolepsy model (both familial and sporadic narcoleptic dogs) was used for the evaluation of replacement/supplement therapies of hypocretin ligands (236). Using a ligand-deficit sporadic narcoleptic dog, we have assessed the effects on cataplexy from intravenous (IV) administration of hypocretin-1 (6 – 384 μg/kg). In a separate experiment, we found that a small portion of hypocretin-1 penetrates to the CNS (by assessing the increase in CSF hypocretin levels) after high doses (96- 386 μg/kg) of IV administration of hypocretin-1 (236). However, hypocretin-1 at these high-dose ranges only produced a short-lasting anticataplectic effect (236). Thus, development of centrally penetrable hypocretin agonists (i.e., small molecular synthetic agonists) is likely to be necessary for the human application.
13. Hypocretin deficiency and narcoleptic phenotype
Human studies have demonstrated that the occurrence of cataplexy is closely associated with hypocretin deficiency (16) (Fig. 6). Furthermore, the hypocretin deficiency was already observed at very early stages of the disease (just after the onset of EDS), even before the occurrences of clear cataplexy (237). Occurrences of cataplexy are rare in acute symptomatic cases of EDS associated with a significant hypocretin deficiency (see (85)); therefore, it appears that a chronic and selective deficit of hypocretin neurotransmission may be required for the occurrence of cataplexy. A possibility of an involvement of a secondary neurochemical change for the occurrence of cataplexy cannot be ruled out. If some of these changes are irreversible, hypocretin supplement therapy may only have limited effects on cataplexy.
Sleepiness in narcolepsy is most likely due to the difficulty in maintaining wakefulness as normal subjects do. The sleep pattern of narcoleptic subjects is also fragmented; they exhibit insomnia (frequent wakening) at night. This fragmentation occurs across 24 hours, and, thus, the loss of hypocretin signaling is likely to play a role of this vigilance stage stability (see (238)), but other mechanism may also be involved in EDS in narcoleptic subjects. One of the most important characteristics of EDS in narcolepsy is that sleepiness is reduced, and patients feel refreshed after a short nap, but this does not last long as they become sleepy within a short period of time. Hypocretin-1 levels in the extracellular space and in the CSF of rats significantly fluctuate across 24 hours, and build up toward the end of the active periods (179). Several manipulations (such as sleep deprivation, exercise and long-term food deprivation) are also known to increase hypocretin tonus (178, 179, 239). Thus, the lack of this hypocretin build-up (or increase) caused by circadian time and by various alerting stimulations may also play a role for EDS associated with hypocretin-deficient narcolepsy.
14. Future directions
(1) The observation of low CSF hypocretin-1 levels is very specific to narcolepsy when compared to other sleep or neurological disorders. Measuring CSF hypocretin-1 is, thus, rapidly becoming a new diagnostic tool for the condition. The availability of this test is also challenging our view of the nosology of narcolepsy. As emphasized by Honda et al. (89), narcolepsy with cataplexy may be more a homogeneous etiological entity, as reflected by low CSF hypocretin, to be differentiated from narcolepsy without cataplexy or the related syndrome of idiopathic hypersomnia, with generally normal hypocretin levels. In the second revised ICSD, narcolepsy with cataplexy and narcolepsy without cataplexy will be coded separately. Low CSF hypocretin-1 will also be considered as a positive diagnostic result for narcolepsy-cataplexy
(2) Since most narcolepsy-cataplexy subjects (about 90% of idiopathic cases) are hypocretin-ligand deficient, hypocretin agonists may be promising in the treatment of narcolepsy. In this respect, the development of small-molecular and centrally penetrant (i.e., non-peptide) hypocretin agonists is likely to be necessary (236). A consideration is the possible absence of functional hypocretin receptors many years after the disease onset. Cell transplantation, using embryonic hypothalamic cells or neural stem cells, and gene therapy (preprohypocretin /orexin gene transfer using various vectors) might also be used to cure the disease in the future.
(3) Although cataplexy is now known to be closely associated with hypocretin deficiency in narcolepsy, the pathophysiological mechanisms underlying the occurrence of cataplexy are largely unknown. The observation that prepubertal narcolepsy-cataplexy cases are almost always hypocretin-deficient suggests that hypocretin deficiency occurs at cataplexy onset (240). Considering the fact that acute ablation of hypocretin ligands by focal hypothalamic lesions associated with immune-related inflammatory encephalophaties, such as in multiple sclerosis and acute disseminated encephalomyelitis (ADEM), rarely induce cataplexy (see (85)), chronic and selective loss of hypocretin ligand may be required to exhibit cataplexy. The mechanisms of emotional induction of cataplexy remain to be studied.
(4) The causes/mechanisms of the ligand deficiency in human narcolepsy remain unknown, but it is believed to be a result of the acquired cell death of hypocretin neurons. This is likely because (1) the onset of most sporadic cases of human narcolepsy is around puberty, later than those for the genetic animal models, (2) the only known human hypocretin gene mutation had a very early onset at six months of age, and (3) postnatal ablation of hypocretin neurons in mice (241) induces a phenotype that most resembles human narcolepsy. The mechanisms of the hypocretin cell death, especially in relation to HLA positivity, should, therefore, be determined to prevent and/or rescue the disease. The current hypothesis, that narcolepsy is an autoimmune disease, is still unsubstantiated.
(5) Hypocretins are involved in various other hypothalamic functions such as feeding, energy homeostasis and neuroendocrine regulation (169, 170). Narcolepsy now appears to be a more complex condition than simply a sleep disorder (18), and the disease is likely to be associated with various hypothalamic dysfunctions due to hypocretin deficiency. Narcolepsy is, thus, an important model to study the fundamental hypothalamic mechanisms linking sleep regulation, energy homeostasis and/or feeding (18).
Fig. 7. Effects of various DA and NE uptake inhibitors and amphetamine-like stimulants on the EEG arousal of narcoleptic dogs and correlation between in vivo EEG arousal effects and in vitro DAT binding affinities.

The effects of compounds on daytime sleepiness was studied using 4-hr daytime polygraphic recordings (10:00-14:00) in 4-5 narcoleptic animals. Two doses were studied for each compound. All DA uptake inhibitors and CNS stimulants dose-dependently increased EEG arousal and reduced slow wave sleep (SWS) when compared to vehicle treatment. In contrast, nisoxetine and desipramine (two potent NE uptake inhibitors) had no significant effect on EEG arousal when doses that completely suppressed REM sleep were injected. Compounds with both adrenergic and dopaminergic effects (nomifensine, mazindol, D-amphetamine) were active on both EEG arousal (left panel) and REM sleep. The effects of the two doses studied for each stimulant was used to construct a rough dose-response curve (left panel). The drug dose that increased the time spent in wakefulness by 40% more than the baseline (vehicle session) was then estimated for each compound. The order of potency of the compounds obtained was: mazindol > (amphetamine) > nomifensine > GBR 12,909 > amineptine> (modafinil) > bupropion. In vitro DA transporter (DAT) binding was performed using [3H]-WIN 35,428 onto canine caudate membranes and demonstrated that the affinity of these DA uptake inhibitors varied widely between 6.5 nM and 3.3 mM. In addition, it was also found that both amphetamine and modafinil have a low but significant affinity (same range as amineptine) for the DAT. A significant correlation between in vivo and in vitro effects was observed for all 5 DA uptake inhibitors and modafinil (right panel).
Table.
Characterization of narcolepsy in human, dog and mice.
| Onset | Symptoms/Phenotype | Transmission | Association with HLA / DLA | Abnormality in Hypocretin system | ||
|---|---|---|---|---|---|---|
| Human | Sporadic | Adolescent | EDS, Cataplexy, SP, HH SOREMPs, Sleep Fragmentation Obesity (+) | ? | DQB1*0602 (+) (90-95%) | Hypocretin Ligand Deficiency (∼90%) |
| Familial | Earlier onset than sporadic cases | EDS, Cataplexy, SP, HH SOREMPs, Sleep Fragmentation Obesity (+) | ? | DQB1*0602 (+) (75-80%) | Hypocretin Ligand Deficiency (∼75%) | |
| Hypocretin Mutant (the only one case is identified) | Extremely early onset (6 mo) | EDS Severe Cataplexy | DeNovo Mutant (?) Dominant | DQB1*0602 (−) | Mutation in Preprohypocretin Gene Hypocretin ligand deficiency | |
|
| ||||||
| Dog | Sporadic (17 breeds) | 7wk-7yr | Cataplexy Short Sleep Latency SOREMPs | ? | (−) | Hypocretin Ligand Deficiency |
| Familial Dobermans, Laboradors, Dachshund | earlier than 6 mo | Cataplexy Short Sleep Latency SOREMPs | Autosomal Recessive 100% of penetrance | (−) | Mutation in Hcrtr2 gene | |
|
| ||||||
| Mice | Hypocretin KO | ∼4wk | Cataplexy, SOREMPs Short Sleep Latency Obesity (?) | Autosomal Recessive 100% of penetrance | Hypocretin Ligand Deficiency | |
| Hypocretin Cell Death Hypocretin/Ataxin-3 transgenic | ∼6wk | Cataplexy, SOREMPs Short Sleep Latency Obesity (++) | Autosomal Dominant 100% of penetrance | Hypocretin/Dynorphin Deficiency | ||
| Hcrtr1 KO | Fragmented Sleep Obesity (?) | Absence of Hcrtr1 | ||||
| Hcrtr2 KO | Cataplexy, SOREMPs Short Sleep Latency Obesity (?) | Autosomal Recessive 100% of penetrance | Absence of Hcrtr2 | |||
| Double Receptor KO | Same Sleep Phenotype as that of Preprohypocretin KOMice, Obesity (?) | Recessive for each receptor gene | Absence of Hcrtr1 and 2 | |||
EDS, Excessive Daytime Sleepiness, SP, Sleep Paralysis; HH, Hypnagogic Hallucination; SOREMPs, Sleep onset REM periods; KO,knockout; HLA, Human leukocyto antigen' DLA, Dog Leukocyto Antigen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gélineau JBE. De la narcolepsie. Gazette des hôpitaux. 1880;53:626–628. [Google Scholar]
- 2.Billiard M, Besset A, Cadilhac J. The clinical and polygraphic development of narcolepsy. In: Guilleminault C, Lugaresi E, editors. Sleep/wake disorders: natural history, epidemiology and longterm evolution. New York: Raven Press; 1983. pp. 171–185. [Google Scholar]
- 3.Sonka K, Tafti M, Billiard M. Narcolepsy and aging. In: Smiru S, Frauceschi M, Ferini-Strambi L, editors. Sleep and aging. Milano: Masson; 1991. pp. 181–186. [Google Scholar]
- 4.Guilleminault C. Narcolepsy Syndrome. In: Kryger MH, Roth T, Dement WC, editors. Principles and Practice of Sleep Medicine. 2. Philadelphia: W. B. Saunders; 1994. pp. 549–561. [Google Scholar]
- 5.Hublin C, Kaprio J, Partinene M, Koskenvuo M, Heikkila K, Koskimies S, et al. The prevalence of narcolepsy: an epidemiological study of the Finnish twin cohort. Ann Neurol. 1994;35:709–716. doi: 10.1002/ana.410350612. [DOI] [PubMed] [Google Scholar]
- 6.Tashiro T, Kanbayashi T, Hishikawa Y. An epidemiological study of narcolepsy in Japanese. The 4th International Symposium on Narcolepsy; 1994; Tokyo, Japan. 1994. p. 13. [Google Scholar]
- 7.Guilleminault C, Mignot E, Grumet FC. Familial patterns of narcolepsy. Lancet. 1989;2(8676):1376–9. doi: 10.1016/s0140-6736(89)91977-6. [DOI] [PubMed] [Google Scholar]
- 8.Nishino S, Mignot E. Pharmacological aspects of human and canine narcolepsy. Prog Neurobiol. 1997;52:27–78. doi: 10.1016/s0301-0082(96)00070-6. [DOI] [PubMed] [Google Scholar]
- 9.Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 10.Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98(3):365–76. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 11.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 12.De Lecea L, Kilduff TS, Peyron C, Gao XB, Foye PE, Danielson PE, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355(9197):39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- 14.Nishino S, Ripley B, Overeem S, Nevsimalova S, Lammers GJ, Vankova J, et al. Low CSF hypocretin (orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol. 2001;50:381–388. doi: 10.1002/ana.1130. [DOI] [PubMed] [Google Scholar]
- 15.Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6(9):991–7. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- 16.Mignot E, Lammers GJ, Ripley B, Okun M, Nevsimalova S, Overeem S, et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002;59(10):1553–62. doi: 10.1001/archneur.59.10.1553. [DOI] [PubMed] [Google Scholar]
- 17.ICSD-2, editor. Diagnostic and coding manual. 2. Westchester, Illinois: American Academy of Sleep Medicine; 2005. ICSD-2-International classification of sleep disorders. [Google Scholar]
- 18.Nishino S. The hypocretin/orexin system in health and disease. Biol Psychiatry. 2003;54(2):87–95. doi: 10.1016/s0006-3223(03)00349-4. [DOI] [PubMed] [Google Scholar]
- 19.Ohayon MM, Priest RG, Caulet M, Guilleminault C. Hypnagogic and hypnopompic hallucinations: pathological phenomena? Br J Psychiatry. 1996;169(4):459–67. doi: 10.1192/bjp.169.4.459. [DOI] [PubMed] [Google Scholar]
- 20.Ondzé B, Lubin S, Lavandier B, Kohler F, Mayeux D, Billiard M. Frequency of narcolepsy in the population of a French departement. Sleep. 1999;22(Supplement):S121. [Google Scholar]
- 21.Roth B. Narcolepsy and Hypersomnia. Basel: Karger; 1980. [Google Scholar]
- 22.Ohayon MM, Priest RG, Zulley J, Smirne S, Paiva T. Prevalence of narcolepsy symptomatology and diagnosis in the European general population. Neurology. 2002;58(12):1826–33. doi: 10.1212/wnl.58.12.1826. [DOI] [PubMed] [Google Scholar]
- 23.Dement WC, Carskadon M, Ley R. The prevalence of narcolepsy II. Sleep Res. 1973;2:147. [Google Scholar]
- 24.Silber MH, Krahn LE, Olson EJ, Pankratz VS. The epidemiology of narcolepsy in Olmsted County, Minnesota: a population-based study. Sleep. 2002;25(2):197–202. doi: 10.1093/sleep/25.2.197. [DOI] [PubMed] [Google Scholar]
- 25.Solomon P. Narcolepsy in Negroes. Dis Nerv Sys. 1945;6:179–183. [Google Scholar]
- 26.Honda Y. Census of narcolepsy, cataplexy and sleep life among teen-agers in Fujisawa city. Sleep Res. 1979;8:191. [Google Scholar]
- 27.Lavie P, Peled R. Narcolepsy is a rare disease in Isreal. Sleep. 1987;10(6):608–609. [PubMed] [Google Scholar]
- 28.Dauvilliers Y, Montplaisir J, Molinari N, Carlander B, Ondze B, Besset A, et al. Age at onset of narcolepsy in two large populations of patients in France and Quebec. Neurology. 2001;57(11):2029–33. doi: 10.1212/wnl.57.11.2029. [DOI] [PubMed] [Google Scholar]
- 29.Westphal C. Eigenthümliche mit Einschläfen verbundene Anfälle. Arch Psychiat. 1877;7:631–635. [Google Scholar]
- 30.Krabbe E, Magnussen G. Familial narcolepsy. Acta Psychiatr Scand. 1942;17:149–173. [Google Scholar]
- 31.Yoss RE, Daly DD. Narcolepsy. Med Clin North Am. 1960;44(4):953–967. doi: 10.1016/s0025-7125(16)33982-7. [DOI] [PubMed] [Google Scholar]
- 32.Nevsimalova-Bruhova S, Roth B. Heredofamilial aspects of narcolepsy and hypersomnia. Schweiz Arch Neurol Neurochir Psychiat. 1972;110:45–54. [PubMed] [Google Scholar]
- 33.Kessler S, Guilleminault C, Dement WC. A family study of 50 REM narcoleptics. Acta Neurol Scandinav. 1979;50:503–512. doi: 10.1111/j.1600-0404.1974.tb02796.x. [DOI] [PubMed] [Google Scholar]
- 34.Baraitser M, Parkes JD. Genetic study of narcoleptic syndrome. J Med Genet. 1978;15:254–259. doi: 10.1136/jmg.15.4.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honda Y, Asaka A, Tanimura M, Furusho T. A genetic study of narcolepsy and excessive daytime sleepiness in 308 families with a narcolepsy or hypersomnia proband. In: Guilleminault C, Lugaresi E, editors. Sleep/wake Disorders: Natural History, Epidemiology and Long Term Evolution. New York: Raven Press; 1983. [Google Scholar]
- 36.Honda Y. Clinical features of narcolepsy. In: Honda Y, Juji T, editors. HLA in narcolepsy. Berlin: Springer-Verlag; 1988. pp. 24–57. [Google Scholar]
- 37.Billiard M, Pasquie-Magnetto V, Heckman M, Carlander B, Besset A, Billiard M. Family studies in narcolepsy. Sleep. 1994;17:S-54–S-59. doi: 10.1093/sleep/17.suppl_8.s54. [DOI] [PubMed] [Google Scholar]
- 38.Mignot E. Genetic and familial aspects of narcolepsy. Neurology. 1998;50 1:S16–S22. doi: 10.1212/wnl.50.2_suppl_1.s16. [DOI] [PubMed] [Google Scholar]
- 39.Hublin C, Partinen M, Kaprio J, Koskenvuo M, Guilleminault C. Epidemiology of narcolepsy. Sleep. 1994;17:S7–S12. doi: 10.1093/sleep/17.suppl_8.s7. [DOI] [PubMed] [Google Scholar]
- 40.Lankford DA, Wellman JJ, O'Hara C. Posttraumatic narcolepsy in mild to moderate closed head injury. Sleep. 1994;17:S25–S28. doi: 10.1093/sleep/17.suppl_8.s25. [DOI] [PubMed] [Google Scholar]
- 41.Orellana C, Villemin E, Tafti M, Carlander B, Beset A, Billiard M. Life events in the year preceding the onset of narcolepsy. Sleep. 1994;17:S50–53. doi: 10.1093/sleep/17.suppl_8.s50. [DOI] [PubMed] [Google Scholar]
- 42.Guilleminault C. Narcolepsy and its differential diagnosis. In: Guilleminault C, editor. Sleep and it disorders in children. New York: Raven Press; 1987. pp. 181–194. [Google Scholar]
- 43.Broughton R, Ghanem Q. The impact of compound narcolepsy on the life of the patient. In: Guilleminault C, D WC, Passouant P, editors. Narcolepsy. New York: Spectrum; 1976. pp. 201–220. [Google Scholar]
- 44.Dement WC. Daytime sleepiness and sleep “attacks”. In: Guilleminault C, Dement WC, Passouant P, editors. Narcolepsy. New York: Spectrum Publications Inc; 1976. pp. 17–42. [Google Scholar]
- 45.Cohen FL, Smith KM. Learning and memory in narcoleptic patients and controls. Sleep Res. 1989;18:117. [Google Scholar]
- 46.Rogers AE, Rosenberg RS. Test of memory in narcoleptics. Sleep. 1990;13:42–52. [PubMed] [Google Scholar]
- 47.Parkes JD, Baraitser M, Marsden CD, Asselman P. Natural history, symptoms and treatment of the narcoleptic syndrome. Acta Neurol Scand. 1975;52:337–353. doi: 10.1111/j.1600-0404.1975.tb05830.x. [DOI] [PubMed] [Google Scholar]
- 48.Yoss RE, Daly DD. Criteria for the diagnosis of the narcoleptic syndrome. Proc Staff Meet Mayo. 1957;32:320–328. [PubMed] [Google Scholar]
- 49.Kales A, Soldates CR, Bixler EO. Narcolepsy-cataplexy II Psychosocial consequences and associated psychopathology. Arch Neurol. 1982;39:169–171. doi: 10.1001/archneur.1982.00510150039009. [DOI] [PubMed] [Google Scholar]
- 50.Guilleminault C, Wilson RA, Dement WC. A study on cataplexy. Arch Neurol. 1974;31:255–261. doi: 10.1001/archneur.1974.00490400069008. [DOI] [PubMed] [Google Scholar]
- 51.Henneberg R. Uber genuine narkolepsie. Neurol Zbl. 1916;30:282–290. [Google Scholar]
- 52.Löwenfeld L. Uber Narkolepsie. Munch Med Wochenschr. 1902;49:1041–1045. [Google Scholar]
- 53.Daniels LE. Narcolepsy. Medicine. 1934;13(1):1–122. [Google Scholar]
- 54.Wilson SAK. The narcolepsies. Annual Congress Assoc Phys. 1927 June 3;:63–109. [Google Scholar]
- 55.Gelb M, Guilleminault C, Kraemer H, Lin S, Moon S, Dement WC, et al. Stability of cataplexy over several months-information for the design of therapeutic trials. Sleep. 1994;17:265–273. doi: 10.1093/sleep/17.3.265. [DOI] [PubMed] [Google Scholar]
- 56.Passouant P, Baldy-Moulinier M, Assilloux C. Eata de mal cataplectique au cours d'ume maladie de Gelineau, influence de la clomipramine. Rev Neurol. 1970;123:56–50. [PubMed] [Google Scholar]
- 57.Hishikawa Y, Shimizu T. Physiology of REM sleep, cataplexy, and sleep paralysis. In: Fahn S, Hallet M, Lüders HO, Marsden CDü, editors. Negative Motor Phenomena. Philadelphia: Lippincot-Raven Publishers; 1995. pp. 245–271. [PubMed] [Google Scholar]
- 58.Rosenthal L, Merlotti L, Young D, Zorick F, Wittig R, Roehrs T, et al. Subjective and polysomnographic characteristics of patients diagnosed with narcolepsy. Gen Hosp Psychiatry. 1990;12:191–197. doi: 10.1016/0163-8343(90)90078-q. [DOI] [PubMed] [Google Scholar]
- 59.Hishikawa Y. Sleep paralysis. In: Guilleminault C, D WC, Passpouant P, editors. Narcolepsy. New York: Spectrum; 1976. pp. 97–124. [Google Scholar]
- 60.Parkes JD, Fenton G, Struthers G, Curzon G, Kantamaneni BD, Buxton BH, et al. Narcolepsy and Cataplexy. Clinical features, treatment and cerebrospinal fluid findings. Q J Med. 1974;172:525–536. [PubMed] [Google Scholar]
- 61.Rosenthal C. Uber das aufreten von halluzinatorisch-kataplektischem angstsyndrom, wachanfallen und ahnlichen storungen bei schizophrenen. Mschr Psychiat. 1939;102:11. [Google Scholar]
- 62.Dahlitz M, Parkes JD. Sleep paralysis. Lancet. 1993;341:406–407. doi: 10.1016/0140-6736(93)92992-3. [DOI] [PubMed] [Google Scholar]
- 63.Fukuda K, Miyasita A, Inugami M, Ishihara K. High prevalence of isolated sleep paralysis: Kanashibari phenomenon in Japan. Sleep. 1987;10(3):279–286. doi: 10.1093/sleep/10.3.279. [DOI] [PubMed] [Google Scholar]
- 64.Goode B. Sleep paralysis. Arch Neurol. 1962;6(3):228–234. doi: 10.1001/archneur.1962.00450210056006. [DOI] [PubMed] [Google Scholar]
- 65.Ribstein M. Hypnagogic hallucinations. In: Guilleminault C, Dement WC, Passouant P, editors. Narcolepsy. New York: Spectrum; 1976. pp. 145–160. [Google Scholar]
- 66.Chetrit M, Besset A, Damci D, Lelarge C, Billiard M. Hypnogogic hallucinations associated with sleep onset REM period in narcolepsy-cataplexy. J Sleep Res. 1994;3 1:43. [Google Scholar]
- 67.Hishikawa Y, Wakamatsu H, Furuya E, Sugita Y, Masaoka S, Kaneda H, et al. Sleep satiation in narcoleptic patients. Electroencephalogr Clin Neurophysiol. 1976;41:1–18. doi: 10.1016/0013-4694(76)90210-8. [DOI] [PubMed] [Google Scholar]
- 68.Broughton R, Dunham W, Newman J, Lutley K, Dushesne P, Rivers M. Ambulatory 24 hour sleep-wake monitoring in narcolepsy-cataplexy compared to matched control. Electroenceph Clin Neurophysiol. 1988;70:473–481. doi: 10.1016/0013-4694(88)90145-9. [DOI] [PubMed] [Google Scholar]
- 69.Montplaisir J, Billard M, Takahashi S, Bell IR, Guilleminault C, Dement WC. Twenty-four-hour recording in REM-narcoleptics with special reference to nocturnal sleep disruption. Biol Psych. 1978;13(1):78–89. [PubMed] [Google Scholar]
- 70.Godbout R, Montplaisir J. Comparison of sleep parameters in narcoleptics with and without periodic movements of sleep. In: Koella WP, Ruther E, Schulz H, editors. Sleep '84. Gustav: Fischer Verlag; 1985. pp. 380–382. [Google Scholar]
- 71.Mosko SS, Shampain DS, Sassin JF. Nocturnal REM latency and sleep disturbance in narcolepsy. Sleep. 1984;7:115–125. doi: 10.1093/sleep/7.2.115. [DOI] [PubMed] [Google Scholar]
- 72.Mayer G, Pollmächer T, Meier-Ewert K, Schulz H. Zur Einschätzung des Behinderungsgrades bei Narkolepsie. Gesundh-Wes. 1993;55:337–342. [PubMed] [Google Scholar]
- 73.Schenck CH, Mahowald MW. Motor Dyscontrol in narcolepsy: Rapid-Eye-Movement (REM) sleep without atonia and REM Sleep Behavior Disorder. Annals of Neurology. 1992;32(1):3–10. doi: 10.1002/ana.410320103. [DOI] [PubMed] [Google Scholar]
- 74.Chokroverty S. Sleep apnea in narcolepsy. Sleep. 1986;9(1):250–253. doi: 10.1093/sleep/9.1.250. [DOI] [PubMed] [Google Scholar]
- 75.Guilleminault C, Dement WC, Passouant P, editors. Narcolepsy. New York: Spectrum Publications; 1976. [Google Scholar]
- 76.Honda Y, Doi Y, Ninomiya R, Ninomiya C. Increased frequency of non-insulin-dependent diabetes mellitus among narcoleptic patients. Sleep. 1986;9(1):254–259. doi: 10.1093/sleep/9.1.254. [DOI] [PubMed] [Google Scholar]
- 77.Schuld A, Hebebrand J, Geller F, Pollmacher T. Increased body-mass index in patients with narcolepsy. Lancet. 2000;355(9211):1274–5. doi: 10.1016/S0140-6736(05)74704-8. [DOI] [PubMed] [Google Scholar]
- 78.Lammers GJ, Pijl H, Iestra J, Langius JAE, Buunk G, Meinders AE. Spontaneous food choice in narcolepsy. Sleep. 1996;19(1):75–76. doi: 10.1093/sleep/19.1.75. [DOI] [PubMed] [Google Scholar]
- 79.Mayer G, Hellmann F, Leonhard E, Meier-Ewert K. Circadian temperature and activity rhythms in unmedicated narcoleptic patients. Pharmacol Biochem Behav. 1997;58(2):395–402. doi: 10.1016/s0091-3057(97)00241-4. [DOI] [PubMed] [Google Scholar]
- 80.Sachs C, Kaisjer L. Autonomic control of cardiovascular reflexes in Narcolepsy. Journal of Neurology, Neurosurgery, and Psychiatry. 1980;43:535–539. doi: 10.1136/jnnp.43.6.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Broughton R, Ghanem Q, Hishikawa Y, Sugita Y, Nevsimalova S, Roth B. Life effects of narcolepsy in 180 patients from North America, Asia and Europe compared to matched controls. Le J Can Sci Neurol. 1981;8(4):299–303. doi: 10.1017/s0317167100043419. [DOI] [PubMed] [Google Scholar]
- 82.Aldrich MS. Automobile accidents in patients with sleep disorders. Sleep. 1989;12:487–494. doi: 10.1093/sleep/12.6.487. [DOI] [PubMed] [Google Scholar]
- 83.Alaila SL. Life effects of narcolepsy: measures of negative impact, social support and psychological well-being. In: Goswanmi M, Pollak CP, Cohen FL, Thorpy MJ, Kavey NB, editors. Loss, Grief and Care: Psychosocial Aspects of Narcolepsy. New York: Haworth Press; 1992. pp. 1–22. [Google Scholar]
- 84.Roth B, Nevsimalova S. Depression in narcolepsy and hypersomnia. Schweitz Arch Neurol Neurochir Psychiat. 1975;116:291–300. [PubMed] [Google Scholar]
- 85.Nishino S, K T. Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications In the hypothalamic hypocretin/orexin system. Sleep Med Rev. 2005;9(4):269–310. doi: 10.1016/j.smrv.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 86.Black JE, Brooks SN, Nishino S. Narcolepsy and syndromes of primary excessive daytime somnolence. Semin Neurol. 2004;24(3):271–82. doi: 10.1055/s-2004-835069. [DOI] [PubMed] [Google Scholar]
- 87.Smolik P, Roth B. Kleine-Levin syndrome ethiopathogenesis and treatment. Acta Univ Carol Med Monogr. 1988;128:5–94. [PubMed] [Google Scholar]
- 88.Billiard M. Kleine Levin syndrome. In: Krieger MH, Roth T, Dement WC, editors. Principles and Practice of Sleep Medicine. Philadelphia: WB Saunders Company; 1989. pp. 377–378. [Google Scholar]
- 89.Honda Y. Clinical features of Narcolepsy: Japanese Experience. In: Honda Y, Juji T, editors. HLA in Narcolepsy. 1988. pp. 24–57. [Google Scholar]
- 90.Richardson GS, Carskadon MA, Flagg W, van den Hoed J, Dement WC, Mitler MM. Excessive daytime sleepiness in man: multiple sleep latency measurement in narcoleptic and control subjects. Electroencephalogr Clin Neurophysiol. 1978;45:621–627. doi: 10.1016/0013-4694(78)90162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chervin RD, Kraemer HC, Guilleminalt C. Correlates of sleep latency on the multiple sleep latency test in a clinical population. Electroencephalogr Clin Neurophysiol. 1995;95:147–153. doi: 10.1016/0013-4694(95)00075-a. [DOI] [PubMed] [Google Scholar]
- 92.Carskadon MA, Dement WC, Mitler MM, Roth T, Westbrook PR, Keenan S. Guidelines for the multiple sleep latency test (MSLT): a standard measure of sleepiness. Sleep. 1986;9:519–524. doi: 10.1093/sleep/9.4.519. [DOI] [PubMed] [Google Scholar]
- 93.American Sleep Disorders Association. The clinical use of the multiple sleep latency test. Sleep. 1992;15(3):268–276. doi: 10.1093/sleep/15.3.268. [DOI] [PubMed] [Google Scholar]
- 94.Lammers GJ, Van Dijk JG. The Mutiple Sleep Latency Test: A paradoxical test? Clinical Neurology and Neurosurgery. 1992;94(suppl):S108–S110. doi: 10.1016/0303-8467(92)90040-a. [DOI] [PubMed] [Google Scholar]
- 95.Moscovitch A, Partinen M, Guilleminault C. The positive diagnosis of narcolepsy and narcolepsy's borderland. Neurology. 1993;43:55–60. doi: 10.1212/wnl.43.1_part_1.55. [DOI] [PubMed] [Google Scholar]
- 96.Aldrich MS. The neurobiology of narcolepsy-cataplexy. Prog Neurobiol. 1993;41:533–541. doi: 10.1016/0301-0082(93)90042-q. [DOI] [PubMed] [Google Scholar]
- 97.Broughton R, Duschene P, Aguirre M, Newman J, Lutley K. A single nap is as acurate as the MSLT in diagnosing EDS in narcolepsy-cataplexy. Sleep Res. 1988;17:150. [Google Scholar]
- 98.Raynal D. Polygraphic aspects of narcolepsy. In: Guilemminault C, D WC, Passouant P, editors. Narcolepsy. New York: Spectrum publications, Inc; 1976. pp. 669–684. [Google Scholar]
- 99.Roth B, Nevsimalova S, Sonka K, Docekal P. An alternative to the multiple sleep latency test to determine sleepiness in narcolepsy and hypersomnia: Polygraphic scores of sleepiness. Sleep. 1986;9(1):243–245. [PubMed] [Google Scholar]
- 100.Billiard M, Salva MQ, DeKoninck J, Besset A, Touchon J, Cadihac J. Daytime sleep characterisitcs and their relationships with night sleep in the narcoleptic patient. Sleep. 1986;9(1):167–174. doi: 10.1093/sleep/9.1.167. [DOI] [PubMed] [Google Scholar]
- 101.Genton P, Benlakhel K, Disdier P, Leprince Y, Lavernhe G, Viallet F, et al. Diagnosis of narcolepsy-cataplexy: importance of continuous recording in ambulatory EEG. Report of 20 cases. Neurophysiologie Clinique. 1995;25(4):187–195. doi: 10.1016/0987-7053(96)80174-2. [DOI] [PubMed] [Google Scholar]
- 102.Mitler MM, Gujavarty KS, Browman CP. Maintenance of wakefulness test: a polysomnographic technique for evaluating treatment in patients with excessive somnolence. Electroenchephalogr Clin Neurophysiol. 1982;53:658–661. doi: 10.1016/0013-4694(82)90142-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mitler MM, Walsleben J, Sangal RB, Hirshkowitz M. Sleep latency on the maintenance of wakefulness test (MWT) for 530 patients with narcolepsy while free of psychoactive drugs. Electroencephalogr Clin Neurophysiol. 1998;107(1):33–8. doi: 10.1016/s0013-4694(98)00044-3. [DOI] [PubMed] [Google Scholar]
- 104.Ripley B, Overeem S, Fujiki N, Nevsimalova S, Uchino M, Yesavage J, et al. CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology. 2001;57(12):2253–8. doi: 10.1212/wnl.57.12.2253. [DOI] [PubMed] [Google Scholar]
- 105.Bassetti C, Aldrich MS. Idiopathic hypersomnia. A series of 42 patients. Brain. 1997;120(Pt 8):1423–35. doi: 10.1093/brain/120.8.1423. [DOI] [PubMed] [Google Scholar]
- 106.Aldrich M. The clinical spectrum of narcolepsy and idiopathic hypersomnia. Neurology. 1996;46:393–401. doi: 10.1212/wnl.46.2.393. [DOI] [PubMed] [Google Scholar]
- 107.Mitler MM, Boysen BG, Campbell L, Dement WC. Narcolepsy-cataplexy in a female dog. Exp Neurol. 1974;45(2):332–40. doi: 10.1016/0014-4886(74)90122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Knecht CD, Oliver JE, Redding R, Selcer R, Johnson G. Narcolepsy in a dog and a cat. J Am Vet Med Assoc. 1973;162(12):1052–3. [PubMed] [Google Scholar]
- 109.Foutz A, Mitler M, Cavalli-Sforza L, Dement WC. Genetic factors in canine narcolepsy. Sleep. 1979;1(4):413–421. doi: 10.1093/sleep/1.4.413. [DOI] [PubMed] [Google Scholar]
- 110.Nishino S, Riehl J, Hong J, Kwan M, Reid M, Mignot E. Is narcolepsy REM sleep disorder? Analysis of sleep abnormalities in narcoleptic Dobermans. Neuroscience Research. 2000;38(4):437–446. doi: 10.1016/s0168-0102(00)00195-4. [DOI] [PubMed] [Google Scholar]
- 111.Kaitin KI, Kilduff TS, Dement WC. Sleep fragmentation in genetically narcoleptic dogs. Sleep. 1986;9:116–119. doi: 10.1093/sleep/9.1.116. [DOI] [PubMed] [Google Scholar]
- 112.Kaitin KI, Kilduff TS, Dement WC. Evidence for excessive sleepiness in canine narcoleptics. Electroencephalogr Clin Neurophysiol. 1986;64(5):447–54. doi: 10.1016/0013-4694(86)90078-7. [DOI] [PubMed] [Google Scholar]
- 113.Baker TL, Foutz AS, McNerney V, Mitler MM, Dement WC. Canine model of narcolepsy: genetic and developmental determinants. Exp Neurol. 1982;75(3):729–42. doi: 10.1016/0014-4886(82)90038-3. [DOI] [PubMed] [Google Scholar]
- 114.Mignot E, Wang C, Rattazzi C, Gaiser C, Lovett M, Guilleminault C, et al. Genetic linkage of autosomal recessive canine narcolepsy with a mu immunoglobulin heavy-chain switch-like segment. Proc Natl Acad Sci U S A. 1991;88(8):3475–3478. doi: 10.1073/pnas.88.8.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Riehl J, Nishino S, Cederberg R, Dement WC, Mignot E. Development of cataplexy in genetically narcoleptic Dobermans. Exp Neurol. 1998;152(2):292–302. doi: 10.1006/exnr.1998.6847. [DOI] [PubMed] [Google Scholar]
- 116.Riehl J, Choi S, Mignot E, Nishino S. Changes with age in severity of cataplexy and in sleep/wake fragmentation in narcoleptic Doberman pinschers. Sleep. 1999;22S:S3. APSS abstract. [Google Scholar]
- 117.Dement W, Rechtschaffen A, Gulevich G. The nature of the narcoleptic sleep attack. Neurology. 1966;16(1):18–33. doi: 10.1212/wnl.16.1.18. [DOI] [PubMed] [Google Scholar]
- 118.Broughton R, Valley V, Aguirre M, Roberts J, Suwalski W, Dunham W. Excessive daytime sleepiness and pathophysiology of narcolepsy-cataplexy: a laboratory perspective. Sleep. 1986;9(1):105–215. doi: 10.1093/sleep/9.1.205. [DOI] [PubMed] [Google Scholar]
- 119.Bishop C, Rosenthal L, Helmus T, Roehrs T, Roth T. The frequency of multiple sleep onset REM periods among subjects with no excessive daytime sleepiness. Sleep. 1996;19:727–730. doi: 10.1093/sleep/19.9.727. [DOI] [PubMed] [Google Scholar]
- 120.Aldrich MS, Chervin RD, Malow BA. Value of the multiple sleep latency test (MSLT) for the diagnosis of narcolepsy. Sleep. 1997;20(8):620–629. [PubMed] [Google Scholar]
- 121.Juji T, Satake M, Honda Y, Doi Y. HLA antigens in Japanese patients with narcolepsy. All the patients were DR2 positive. Tissue Antigens. 1984;24:316–319. doi: 10.1111/j.1399-0039.1984.tb02144.x. [DOI] [PubMed] [Google Scholar]
- 122.Langdon N, Welsh KI, Van Dam M, Vaughan RW, Parkes D. Genetic markers in Narcolepsy. Lancet. 1984;2:1178–1180. doi: 10.1016/s0140-6736(84)92742-9. [DOI] [PubMed] [Google Scholar]
- 123.Billiard M, Seignalet J. Extraordinary assoication between HLA-DR2 and narcolepsy. Lancet. 1985;1:226–227. [PubMed] [Google Scholar]
- 124.Poirier G, Monplaisir J, Décary F, Momège D, Lebrun A. HLA antigens in narcolepsy and idiopathic central nervous. Sleep. 1986;9(1):153–158. doi: 10.1093/sleep/9.1.153. [DOI] [PubMed] [Google Scholar]
- 125.Andreas-Zietz A, Keller E, Scholz S, Albert E, Roth B, Nevsimalova S, et al. DR2 negative narcolepsy. Lancet. 1986;2:684–685. doi: 10.1016/s0140-6736(86)90188-1. September 20, 1986. [DOI] [PubMed] [Google Scholar]
- 126.Guilleminault C, Grumet C. HLA-DR2 and Narcolepsy: not all narcoleptic-cataplectic patients are DR2. Human Immunology. 1986;17:1–2. doi: 10.1016/0198-8859(86)90068-6. [DOI] [PubMed] [Google Scholar]
- 127.Mueller-Eckhardt G, Meier-Ewert K, Schendel DJ, Reinecker FB, Multhoff G, Mueller-Eckhardt C. HLA and narcolepsy in a German population. Tissue Antigens. 1986;28(3):163–9. doi: 10.1111/j.1399-0039.1986.tb00476.x. [DOI] [PubMed] [Google Scholar]
- 128.Honda Y, Matsuki K. Genetic aspects of narcolepsy. In: Thorpy M, editor. Handbook of Sleep Disorder. New York: Marcel Dekker, Inc; 1990. pp. 217–234. [Google Scholar]
- 129.Kuwata S, Tokunaga K, Jin F, Juji T, Sasaki T, Honda Y. To the editor: “NARCOLEPSY”. The new england journal of medicine. 1991:270–272. [Google Scholar]
- 130.Matsuki K, Grumet FC, Lin X, Guilleminault C, Dement WC. DQ rather than DR gene marks susceptibility to narcolepsy. Lancet. 1992;339:1052. doi: 10.1016/0140-6736(92)90571-j. [DOI] [PubMed] [Google Scholar]
- 131.Mignot E, Lin X, Arrigoni J, Macaubas C, Olive F, Hallmayer J, et al. DQB1*0602 and DQA1*0102 (DQ1) are better markers than DR2 for narcolepsy in caucasian and black americans. Sleep. 1994;17:S60–S67. doi: 10.1093/sleep/17.suppl_8.s60. [DOI] [PubMed] [Google Scholar]
- 132.Mignot E, Hayduk R, Black J, Grumet FC, Guilleminault C. HLA Class II studies in 509 narcoleptic patients. Sleep Res. 1997;26:433. [Google Scholar]
- 133.Mignot E, Kimura A, Lattermann A, Lin X, Yasunaga S, Mueller-Eckhardt G, et al. Extensive HLA Class II studies in 58 non DRB1*15 (DR2) narcoleptic patients with cataplexy. Tissue Antigens. 1997;49:329–341. doi: 10.1111/j.1399-0039.1997.tb02761.x. [DOI] [PubMed] [Google Scholar]
- 134.Pelin Z, Guilleminault C, Rish N, Grumet FC, Mignot E. HLA-DQB1*0602 homozygosity increases relative risk for narcolepsy but not disease severity in two ethnic groups. Tissue Antigens. 1998;1998(51):96–100. doi: 10.1111/j.1399-0039.1998.tb02952.x. [DOI] [PubMed] [Google Scholar]
- 135.Mignot E, Lin L, Rogers W, Honda Y, Qiu X, Lin X, et al. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am J Hum Genet. 2001;68(3):686–99. doi: 10.1086/318799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mignot E, Meehan J, Grumet FC, Hallmayer J, Guilleminault C, Hesla PE, et al. HLA studies in sporadic and familial narcolepsy. 13th Congress of the European Sleep Rseach Society; 1996; Brussel, Belgium. 1996. In press. [Google Scholar]
- 137.Nakayama J, Miura M, Honda M, Miki T, Honda Y, Arinami T. Linkage of human narcolepsy with HLA association to chromosome 4p13-q21. Genomics. 2000;65(1):84–6. doi: 10.1006/geno.2000.6143. [DOI] [PubMed] [Google Scholar]
- 138.Hohjoh H, Terada N, Nakayama T, Kawashima M, Miyagawa T, Honda Y, et al. Case-control study with narcoleptic patients and healthy controls who, like the patients, possess both HLA-DRB1*1501 and -DQB1*0602. Tissue Antigens. 2001;57(3):230–5. doi: 10.1034/j.1399-0039.2001.057003230.x. [DOI] [PubMed] [Google Scholar]
- 139.Wieczorek S, Gencik M, Rujescu D, Tonn P, Giegling I, Epplen JT, et al. TNFA promoter polymorphisms and narcolepsy. Tissue Antigens. 2003;61(6):437–42. doi: 10.1034/j.1399-0039.2003.00068.x. [DOI] [PubMed] [Google Scholar]
- 140.Dauvilliers Y, Neidhart E, Lecendreux M, Billiard M, Tafti M. MAO-A and COMT polymorphisms and gene effects in narcolepsy. Mol Psychiatry. 2001;6(4):367–72. doi: 10.1038/sj.mp.4000911. [DOI] [PubMed] [Google Scholar]
- 141.Dean RR, Kilduff TS, Dement WC, Grumet FC. Narcolepsy without unique MHC class II antigen association: Studies in the canine model. Hum Immunol. 1989;25:27–35. doi: 10.1016/0198-8859(89)90067-0. [DOI] [PubMed] [Google Scholar]
- 142.Mignot E, Nishino S, Hunt Sharp LH, Arrigoni J, Siegel JM, Reid MS, et al. Heterozygosity at the canarc-1 locus can confer susceptibility for narcolepsy: induction of cataplexy in heterozygous asymptomatic dogs after administration of a combination of drugs acting on monoaminergic and cholinergic systems. J Neurosci. 1993;13:1057–1064. doi: 10.1523/JNEUROSCI.13-03-01057.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mignot E, Guilleminault C, Grumet FC, Dement WC. Is narcolepsy an autoimmune disease?. In: Smirne S, Francesci M, Ferini-Strambi L, Zucconi M, editors. Sleep, Hormones, and the Immune System; Proceedings of the Third Milano International Symposium; September 18-19; Milan: Masson; 1992. pp. 29–38. [Google Scholar]
- 144.Billiard M, Laaberki MF, Reygrobellet C, Seignalet J, Brissaud L, Besset A. Elevated antibodies to streptococcal antigens in narcoleptic subjects. Sleep Res. 1989;18:201. [Google Scholar]
- 145.Montplaisir J, Poirier G, Lapierre O, Montplaisir S. Streptococcal antibodies in Narcolepsy and idiopathic hypersomnia. Sleep Res. 1989;18:271. [Google Scholar]
- 146.Frederickson S, Carlander B, Billiard M, Link H. CSF immune variable in patients with narcolepsy. Acta Neurol Scand. 1990;81:253–254. doi: 10.1111/j.1600-0404.1990.tb00978.x. [DOI] [PubMed] [Google Scholar]
- 147.Matsuki K, Juji T, Honda Y. Immunological features of Narcolepsy in Japan. In: Honda Y, Juji T, editors. HLA in Narcolepsy. 1988. pp. 150–157. [Google Scholar]
- 148.Black JL, 3rd, Krahn LE, Pankratz VS, Silber M. Search for neuron-specific and nonneuron-specific antibodies in narcoleptic patients with and without HLA DQB1*0602. Sleep. 2002;25(7):719–23. doi: 10.1093/sleep/25.7.719. [DOI] [PubMed] [Google Scholar]
- 149.Black JL, 3rd, Silber MH, Krahn LE, Avula RK, Walker DL, Pankratz VS, et al. Studies of Humoral Immunity to Preprohypocretin in Human Leukocyte Antigen DQB1*0602-Positive Narcoleptic Subjects with Cataplexy. Biol Psychiatry. 2005 doi: 10.1016/j.biopsych.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 150.Dutra AS, Mignot E, Puck JM. Establishing FISH methodology to study synthenic regions between canine and human chromosomes. Am J Hu Genet. 1995;57:A112. [Google Scholar]
- 151.Faraco J, Lin X, Li R, Hinton L, Lin L, Mignot E. Genetic studies in narcolepsy, a disorder affecting REM sleep. J Hered. 1999;90(1):129–32. doi: 10.1093/jhered/90.1.129. [DOI] [PubMed] [Google Scholar]
- 152.Hungs M, Fan J, Lin L, Lin X, Maki RA, Mignot E. Identification and functional analysis of mutations in the hypocretin (orexin) genes of narcoleptic canines. Genome Res. 2001;11(4):531–9. doi: 10.1101/gr.gr-1610r. [DOI] [PubMed] [Google Scholar]
- 153.Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27(3):469–74. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ripley B, Fujiki N, Okura M, Mignot E, Nishino S. Hypocretin levels in sporadic and familial cases of canine narcolepsy. Neurobiol of Dis. 2001;8(3):525–534. doi: 10.1006/nbdi.2001.0389. [DOI] [PubMed] [Google Scholar]
- 155.Nishino S, Kanbayashi T, Fujiki N, Uchino M, Ripley B, Watanabe M, et al. CSF hypocretin levels in Guillain-Barre syndrome and other inflammatory neuropathies. Neurology. 2003;61(6):823–5. doi: 10.1212/01.wnl.0000081049.14098.50. [DOI] [PubMed] [Google Scholar]
- 156.Overeem S, Dalmau J, Bataller L, Nishino S, Mignot E, Verschuuren J, et al. Hypocretin-1 CSF levels in anti-Ma2 associated encephalitis. Neurology. 2004;62(1):138–40. doi: 10.1212/01.wnl.0000101718.92619.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kanbayashi T, Inoue Y, Chiba S, Aizawa R, Saito Y, Tsukamoto H, et al. CSF hypocretin-1 (orexin-A) concentrations in narcolepsy with and without cataplexy and idiopathic hypersomnia. J Sleep Res. 2002;11(1):91–3. doi: 10.1046/j.1365-2869.2002.00284.x. [DOI] [PubMed] [Google Scholar]
- 158.Krahn LE, Pankratz VS, Oliver L, Boeve BF, Silber MH. Hypocretin (orexin) levels in cerebrospinal fluid of patients with narcolepsy: relationship to cataplexy and HLA DQB1*0602 status. Sleep. 2002;25(7):733–6. doi: 10.1093/sleep/25.7.733. [DOI] [PubMed] [Google Scholar]
- 159.Ripley B, Overeem S, Fujiki N, Nevsimalova S, Uchino M, Yesavage J, et al. CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology. 2001;57:2253–2258. doi: 10.1212/wnl.57.12.2253. [DOI] [PubMed] [Google Scholar]
- 160.Nishino S. Hypocretin measurements in the CSF, blood and brain tissues: basic to clinical applications. In: Nishino S, Sakurai T, editors. The Orexin/Hypocretin System: Its Physiology And Pathophysiology. Totowa, New Jersey: Humana; 2005. [Google Scholar]
- 161.Yoshikawa S, Suzuki S, Yamamoto K, Shichiri M, Miyamoto R, Kuroda K, et al. A case of acute disseminated encephalomyelitis associated with hypersomnia and low CSF hypocretin levels. Pediatric Neurology. 2004 doi: 10.1016/j.pediatrneurol.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 162.Nozaki H, Katada S, Sato M, Tanaka K, Nishizawa M. A case with hypersomnia and paresthesia due to diffuse MS lesions from hypothalamus to spine. Rinsho Shinkeigaku. 2004;44(1):59. [Google Scholar]
- 163.Kato T, Kanbayashi T, Yamamoto K, Nakano T, Shimizu T, Hashimoto T, et al. Hypersomnia and low CSF hypocretin-1 (orexin-A) concentration in a patient with multiple sclerosis showing bilateral hypothalamic lesions. Intern Med. 2003;42(8):743–5. doi: 10.2169/internalmedicine.42.743. [DOI] [PubMed] [Google Scholar]
- 164.Oka Y, Kanbayashi T, Mezaki T, Iseki K, Matsubayashi J, Murakami G, et al. Low CSF hypocretin-1/orexin-A associated with hypersomnia secondary to hypothalamic lesion in a case of multiple sclerosis. J Neurol. 2004;251(7):885–6. doi: 10.1007/s00415-004-0442-z. [DOI] [PubMed] [Google Scholar]
- 165.Tohyama J, Kanazawa O, Akasaka N, Kamimura T. A case of bilateral paramedian thalamic infarction in childhood with the sensory disturbance and the sensory loss of taste. No To Hattatsu. 2004;36(1):65–9. [PubMed] [Google Scholar]
- 166.Overeem S, van Hilten JJ, Ripley B, Mignot E, Nishino S, Lammers GJ. Normal hypocretin-1 levels in Parkinson's disease patients with excessive daytime sleepiness. Neurology. 2002;58(3):498–9. doi: 10.1212/wnl.58.3.498. [DOI] [PubMed] [Google Scholar]
- 167.Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, et al. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435(1):6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- 168.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18(23):9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Willie JT, Chemelli RM, Sinton CM, Yanagisawa M. To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci. 2001;24:429–58. doi: 10.1146/annurev.neuro.24.1.429. [DOI] [PubMed] [Google Scholar]
- 170.Taheri S, Zeitzer JM, Mignot E. The role of hypocretins (orexins) in sleep regulation and narcolepsy. Annu Rev Neurosci. 2002;25:283–313. doi: 10.1146/annurev.neuro.25.112701.142826. [DOI] [PubMed] [Google Scholar]
- 171.Nishino S, Fujiki N, Ripley B, Sakurai E, Kato M, Watanabe T, et al. Decreased brain histamine contents in hypocretin/orexin receptor-2 mutated narcoleptic dogs. Neurosci Lett. 2001;313(3):125–8. doi: 10.1016/s0304-3940(01)02270-4. [DOI] [PubMed] [Google Scholar]
- 172.Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, Watanabe T, et al. Arousal effect of orexin A depends on activation of the histaminergic system. Proc Natl Acad Sci U S A. 2001;98(17):9965–70. doi: 10.1073/pnas.181330998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yamanaka A, Tsujino N, Funahashi H, Honda K, Guan JL, Wang QP, et al. Orexins activate histaminergic neurons via the orexin 2 receptor. Biochem Biophys Res Commun. 2002;290(4):1237–45. doi: 10.1006/bbrc.2001.6318. [DOI] [PubMed] [Google Scholar]
- 174.Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001;21(23):9273–9. doi: 10.1523/JNEUROSCI.21-23-09273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nishino S, Sakurai E, Nevisimalova S, Vankova J, Yoshida Y, Watanabe T, et al. CSF histamine content is decreased in hypocretin-deficient human narcolepsy. Sleep. 2002;25(suppl):A476. [Google Scholar]
- 176.Kanbayashi T, Kodama T, Hondo H, Sato S, Miyazaki N, Kuroda K, et al. CSF histamine and noradrenaline contents in narcolepsy and other sleep disorders. Sleep. 2004;27(Abstarct Suppliment):A236. [Google Scholar]
- 177.Shiba T, Fujiki N, Wisor J, Edgar D, Sakurai T, Nishino S. Wake promoting effects of thioperamide, a histamine H3 antagonist in orexin/ataxin-3 narcoleptic mice. Sleep. 2004;(suppl):A241–A242. [Google Scholar]
- 178.Fujiki N, Yoshida Y, Ripley B, Honda K, Mignot E, Nishino S. Changes in CSF hypocretin-1 (orexin A) levels in rats across 24 hours and in response to food deprivation. NeuroReport. 2001;12(5):993–7. doi: 10.1097/00001756-200104170-00026. [DOI] [PubMed] [Google Scholar]
- 179.Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, et al. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14(7):1075–81. doi: 10.1046/j.0953-816x.2001.01725.x. [DOI] [PubMed] [Google Scholar]
- 180.Zeitzer JM, Buckmaster CL, Parker KJ, Hauck CM, Lyons DM, Mignot E. Circadian and homeostatic regulation of hypocretin in a primate model: implications for the consolidation of wakefulness. J Neurosci. 2003;23(8):3555–60. doi: 10.1523/JNEUROSCI.23-08-03555.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46(5):787–98. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25(28):6716–20. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Aston-Jones G, Chen S, Zhu Y, Oshinsky ML. A neural circuit for circadian regulation of arousal. Nature Neurosci. 2001;4(7):732–8. doi: 10.1038/89522. [DOI] [PubMed] [Google Scholar]
- 184.Wu MF, John J, Maidment N, Lam HA, Siegel JM. Hypocretin release in normal and narcoleptic dogs after food and sleep deprivation, eating, and movement. Am J Physiol Regul Integr Comp Physiol. 2002;283(5):R1079–86. doi: 10.1152/ajpregu.00207.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Roehrs T, Zorick F, Wittig R, Paxton C, Sicklesteel J, Roth T. Alerting effects of naps in patients with narcolepsy. Sleep. 1986;9(1):194–199. doi: 10.1093/sleep/9.1.194. [DOI] [PubMed] [Google Scholar]
- 186.Thorpy MJ, Goswami M. Treatment of narcolepsy. In: Thorpy MJ, editor. Handbook of Sleep Disorders. New York: Marcel Dekker, Inc; 1990. pp. 235–258. [Google Scholar]
- 187.Mullington J, Broughton R. Scheduled naps in the management of daytime sleepiness in narcolepsy-cataplexy. Sleep. 1993;16(5):444–456. doi: 10.1093/sleep/16.5.444. [DOI] [PubMed] [Google Scholar]
- 188.Rogers AE. Problems and coping strategies identified by narcoleptic patients. J Neurosurg Nursing. 1984;16(6):326–334. doi: 10.1097/01376517-198412000-00009. [DOI] [PubMed] [Google Scholar]
- 189.Association AN. The Eye Opener. 1992. Stimulant medication survey; pp. 1–3. [Google Scholar]
- 190.Mitler MM, Aldrich MS, Koob GF, Zarcone VP. Narcolepsy and its treatment with stimulants. Sleep. 1994;17(4):352–371. [PubMed] [Google Scholar]
- 191.Lammers GJ, Arends J, Declerck AC, Ferrari MD, Schouwink G, Troost J. Gamma-hydroxybutyrate and narcolepsy: a double-blind placebo-controlled study. Sleep. 1993;16(3):216–220. doi: 10.1093/sleep/16.3.216. [DOI] [PubMed] [Google Scholar]
- 192.Scrima L, Hartman PG, Johnson FH, Hiller FC. Efficacy of gamma-hydroxybutyrate versus placebo in treating narcolepsy-cataplexy: double-blind subjective measures. Biol Psychiatry. 1989;26:331–343. doi: 10.1016/0006-3223(89)90048-6. [DOI] [PubMed] [Google Scholar]
- 193.Vogel G. Studies in psychophysiology of dreams III. The dream of narcolepsy. Arch Gen Psychiatry. 1960;3:421–428. doi: 10.1001/archpsyc.1960.01710040091011. [DOI] [PubMed] [Google Scholar]
- 194.Guilleminault C. Cataplexy. Narcolepsy (Advances in Sleep Research Vol. 3) 1976:125–143. [Google Scholar]
- 195.Okura M, Riehl J, Mignot E, Nishino S. Sulpiride, a D2/D3 Blocker, Reduces Cataplexy but not REM Sleep in Canine Narcolepsy. Neuropsychopharmacology. 2000;23(5):528–538. doi: 10.1016/S0893-133X(00)00140-8. [DOI] [PubMed] [Google Scholar]
- 196.Siegel JM. Brainstem mechanisms generating REM sleep. In: Kryger MH, Roth T, Dement WC, editors. Principles and Practice of Sleep Medicine. Philadelphia: W. B. Saunders Company; 1994. pp. 125–144. [Google Scholar]
- 197.Baker TL, Dement WC. Canine narcolepsy-cataplexy syndrome: evidence for an inherited monoaminergic-cholinergic imbalance. In: McGinty DJ, Drucker-Colin R, Morrison A, Parmeggiani PL, editors. Brain Mechanisms of Sleep. New York: Raven Press; 1985. pp. 199–233. [Google Scholar]
- 198.Trulson ME, Jacobs BL. Raphe unit activity in freely moving cats: Correlation with level of behavioral arousal. Brain Res. 1979;163:135–150. doi: 10.1016/0006-8993(79)90157-4. [DOI] [PubMed] [Google Scholar]
- 199.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wu MF, Gulyani SA, Yau E, Mignot E, Phan B, Siegel JM. Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience. 1999;91(4):1389–99. doi: 10.1016/s0306-4522(98)00600-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Miller JD, Farber J, Gatz P, Roffwarg H, German DC. Activity of mesencephalic dopamine and non-dopamine neurons across stages of sleep and waking in the rat. Brain Res. 1983;273:133–141. doi: 10.1016/0006-8993(83)91101-0. [DOI] [PubMed] [Google Scholar]
- 202.Trulson ME, Preussler DW, Howell GA. Activity of substantia nigra units across the sleep-waking cycle in freely moving cats. Neurosci Lett. 1981;26:183–188. doi: 10.1016/0304-3940(81)90346-3. [DOI] [PubMed] [Google Scholar]
- 203.Mignot E, Renaud A, Nishino S, Arrigoni J, Guilleminault C, Dement WC. Canine cataplexy is preferentially controlled by adrenergic mechanisms: evidence using monoamine selective uptake inhibitors and release enhancers. Psychopharmacology. 1993;113(1):76–82. doi: 10.1007/BF02244337. [DOI] [PubMed] [Google Scholar]
- 204.Peet M, Coppen A. The pharmacokinetics of antidepressant drugs: relevance to their therapeutic effect. In: Paykel ES, Coppen A, editors. Psychopharmacology of Affective Disorders. Oxford: Oxford University Press; 1979. pp. 91–107. [Google Scholar]
- 205.Nishino S, Arrigoni J, Shelton J, Dement WC, Mignot E. Desmethyl metabolites of serotonergic uptake inhibitors are more potent for suppressing canine cataplexy than their parent compounds. Sleep. 1993;16(8):706–12. doi: 10.1093/sleep/16.8.706. [DOI] [PubMed] [Google Scholar]
- 206.Schmidt H, Clark R, Hyman R. Protriptyline: an effective agent in the treatment of Narcolepsy-cataplexy syndrome and hypersomnia. Am J Psychiatry. 1977;134:183–185. doi: 10.1176/ajp.134.2.183. [DOI] [PubMed] [Google Scholar]
- 207.Hishikawa Y, Ida H, Nakai K, Kaneko Z. Treatment of narcolepsy with imipramine (tofranil) and desmethylimipramine (pertofran) J Neuro Sci. 1965;3:453–461. doi: 10.1016/0022-510x(66)90001-3. [DOI] [PubMed] [Google Scholar]
- 208.Mitler MM, Hajdukovic R, Erman M, Koziol JA. Narcolepsy. J Clin Neurophysiol. 1990;7(1):93–118. doi: 10.1097/00004691-199001000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nishino S, Fruhstorfer B, Arrigoni J, Guilleminault C, Dement WC, Mignot E. Further characterization of the alpha-1 receptor subtype involved in the control of cataplexy in canine narcolepsy. J Pharmacol Exp Ther. 1993;264:1079–1084. [PubMed] [Google Scholar]
- 210.Nishino S, Haak L, Shepherd H, Guilleminault C, Sakai T, Dement WC, et al. Effects of central alpha-2 adrenergic compounds on canine narcolepsy, a disorder of rapid eye movement sleep. J Pharmacol Exp Ther. 1990;253:1145–1152. [PubMed] [Google Scholar]
- 211.Nishino S, Arrigoni J, Valtier D, Miller JD, Guilleminault C, Dement WC, et al. Dopamine D2 mechanisms in canine narcolepsy. J Neurosci. 1991;11:2666–2671. doi: 10.1523/JNEUROSCI.11-09-02666.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Reid MS, Tafti M, Nishino S, Sampathkumaran R, Siegel JM, Dement WC, et al. Local administration of dopaminergic drugs into the ventral tegmental area modulate cataplexy in the narcoleptic canine. Brain Res. 1996;733:83–100. doi: 10.1016/0006-8993(96)00541-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fruhstorfer B, Mignot E, Bowersox S, Nishino S, Dement WC, Guilleminault C. Canine narcolepsy is associated with an elevated number of α2 receptors in the locus coeruleus. Brain Res. 1989;500:209–214. doi: 10.1016/0006-8993(89)90315-6. [DOI] [PubMed] [Google Scholar]
- 214.Bowersox S, Kilduff T, Faul K, Dement WC, Ciaranello RD. Brain dopamine receptor levels elevated in canine narcolepsy. Brain Res. 1987;402:44–48. doi: 10.1016/0006-8993(87)91045-6. [DOI] [PubMed] [Google Scholar]
- 215.Boehme R, Baker T, Mefford I, Barchas J, Dement WC, Ciaranello R. Narcolepsy: cholinergic receptor changes in an animal model. Life Sci. 1984;34:1825–1828. doi: 10.1016/0024-3205(84)90675-1. [DOI] [PubMed] [Google Scholar]
- 216.Kanbayashi T, Nishino S, Tafti M, Hishikawa Y, Dement WC, Mignot E. Thalidomide, a hypnotic with immune modulating properties, increases cataplexy in canine narcolepsy. NeuroReport. 1996;12:1881–1886. doi: 10.1097/00001756-199608120-00002. [DOI] [PubMed] [Google Scholar]
- 217.Honda K, Riehl J, Mignot E, Nishino S. Dopamine D3 agonists into the substantia nigra aggravate cataplexy but do not modify sleep. NeuroReport. 1999;10(17):3717–24. doi: 10.1097/00001756-199911260-00046. [DOI] [PubMed] [Google Scholar]
- 218.Okura M, Fujiki N, Kita I, Honda K, Yoshida Y, Mignot E, et al. The roles of midbrain and diencephalic dopamine cell groups in the regulation of cataplexy in narcoleptic Dobermans. Neurobiol Dis. 2004;16(1):274–82. doi: 10.1016/j.nbd.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 219.Happe S, Trenkwalder C. Role of dopamine receptor agonists in the treatment of restless legs syndrome. CNS Drugs. 2004;18(1):27–36. doi: 10.2165/00023210-200418010-00003. [DOI] [PubMed] [Google Scholar]
- 220.Wittig R, Zorick F, Piccione P, Sicklesteel J, Roth T. Narcolepsy and disturbed nocturnal sleep. Clin Electroencephalogr. 1983;14(3):130–4. doi: 10.1177/155005948301400306. [DOI] [PubMed] [Google Scholar]
- 221.Okura M, Fujiki N, Ripley B, Takahashi S, Amitai N, Mignot E, et al. Narcoleptic canines display periodic leg movements during sleep. Psychiatry Clin Neurosci. 2001;55(3):243–4. doi: 10.1046/j.1440-1819.2001.00842.x. [DOI] [PubMed] [Google Scholar]
- 222.Reid MS, Tafti M, Geary J, Nishino S, Siegel JM, Dement WC, et al. Cholinergic mechanisms in canine narcolepsy: I. Modulation of cataplexy via local drug administration into pontine reticular formation. Neuroscience. 1994;59:511–522. doi: 10.1016/0306-4522(94)90173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nishino S, Tafti M, Reid MS, Shelton J, Siegel JM, Dement WC, et al. Muscle atonia is triggered by cholinergic stimulation of the basal forebrain: implication for the pathophysiology of canine narcolepsy. J Neurosci. 1995;15(7 Pt 1):4806–4814. doi: 10.1523/JNEUROSCI.15-07-04806.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rolls ET, Sanghera MK, Roper-Hall A. The latency of activation of neurons in the lateral hypothalamus and substantia innominata during feeding in the monkey. Brain Res. 1979;164:121–135. doi: 10.1016/0006-8993(79)90010-6. [DOI] [PubMed] [Google Scholar]
- 225.Nishino S, Mao J, Sampathkumaran R, Honda K, Dement WC, Mignot E. Differential effects of dopaminergic and noradrenergic uptake inhibitors on EEG arousal and cataplexy of narcoleptic canines. Sleep Res. 1996;25:317. [Google Scholar]
- 226.Mignot E, Nishino S, Guilleminault C, Dement WC. Modafinil binds to the dopamine uptake carrier site with low affinity. Sleep. 1994;17:436–437. doi: 10.1093/sleep/17.5.436. [DOI] [PubMed] [Google Scholar]
- 227.Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. Dopaminergic role in stimulant-induced wakefulness. J Neurosci. 2001;21(5):1787–94. doi: 10.1523/JNEUROSCI.21-05-01787.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Saper CB, Scammell TE. Modafinil: A drug in search of a mechanism. Sleep. 2004;27(1):11–12. [PubMed] [Google Scholar]
- 229.Gallopin T, Luppi PH, Rambert FA, Frydman A, Fort P. Effect of the wake-promoting agent modafinil on sleep-promoting neurons from the ventrolateral preoptic nuclueus: an in vitro pharmacologic study. Sleep. 2004;27(1):19–25. [PubMed] [Google Scholar]
- 230.Nishino S, Mao J, Sampathkumaran R, Shelton J, Mignot E. Increased dopaminergic transmission mediates the wake-promoting effects of CNS stimulants. Sleep Research Online. 1998;1:49–61. http://www.sro.org/1998/Nishino/49/ [PubMed]
- 231.Segal DS, Kuczenski R. Behavioral pharmacology of amphetamine. In: Cho AK, Segal DS, editors. Amphetamine and its Analogs: Psychopharmacology, Toxicology and Abuse. San Diego: Academic Press; 1994. pp. 115–150. [Google Scholar]
- 232.Kanbayashi T, Honda K, Kodama T, Mignot E, Nishino S. Implication of dopaminergic mechanisms in the wake-promoting effects of amphetamine: a study of D- and L-derivatives in canine narcolepsy. Neuroscience. 2000;99(4):651–659. doi: 10.1016/s0306-4522(00)00239-6. [DOI] [PubMed] [Google Scholar]
- 233.Shouse MN, Staba RJ, Saquib SF, Farber PR. Monoamines and sleep: microdialysis findings in pons and amygdala. Brain Res. 2000;860(12):181–9. doi: 10.1016/s0006-8993(00)02013-8. [DOI] [PubMed] [Google Scholar]
- 234.Rye D, Bliwize DL, Dihenia B, Grecki P. Daytime sleepiness in Parkinson's disease. J Sleep Res. 2000;9:63–69. doi: 10.1046/j.1365-2869.2000.00201.x. [DOI] [PubMed] [Google Scholar]
- 235.Frucht S, Rogers JD, Greene PE, Gordon MF, Fahn S. Falling asleep at the wheel: motor vehicle mishaps in persons taking pramipexole and ropinirole. Neurology. 1999;52(9):1908–10. doi: 10.1212/wnl.52.9.1908. [DOI] [PubMed] [Google Scholar]
- 236.Fujiki N, Ripley B, Yoshida Y, Mignot E, Nishino S. Effects of IV and ICV hypocretin-1 (orexin A) in hypocretin receptor-2 gene mutated narcoleptic dogs and IV hypocretin-1 replacement therapy in a hypocretin ligand deficient narcoleptic dog. Sleep. 2003;6(8):953–959. doi: 10.1093/sleep/26.8.953. [DOI] [PubMed] [Google Scholar]
- 237.Tsukamoto H, Ishikawa T, Fujii Y, Fukumizu M, Sugai K, Kanbayashi T. Undetectable levels of CSF hypocretin-1 (orexin-A) in two prepubertal boys with narcolepsy. Neuropediatrics. 2002;33(1):51–2. doi: 10.1055/s-2002-23601. [DOI] [PubMed] [Google Scholar]
- 238.Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726–31. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- 239.Martins PJ, D'Almeida V, Pedrazzoli M, Lin L, Mignot E, Tufik S. Increased hypocretin-1 (orexin-a) levels in cerebrospinal fluid of rats after short-term forced activity. Regul Pept. 2004;117(3):155–8. doi: 10.1016/j.regpep.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 240.Kanbayashi T, Yano T, Ishiguro H, Kawanishi K, Chiba S, Aizawa R, et al. Hypocretin-1 (orexin-A) levels in human lumbar CSF in different age groups: infants to elderly persons. Sleep. 2002;25(3):337–9. doi: 10.1093/sleep/25.3.337. [DOI] [PubMed] [Google Scholar]
- 241.Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30(2):345–54. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- 242.Fujiki N, Morris L, Mignot E, Nishino S. Analysis of onset location, laterality and propagation of cataplexy in canine narcolepsy. Psychiatry Clin Neurosci. 2002;56(3):275–276. doi: 10.1046/j.1440-1819.2002.00978.x. [DOI] [PubMed] [Google Scholar]