

Abstract
Background and purpose:
We explored the stereoselective activation of the P2Y11 receptor, stably expressed and tagged with GFP, in 1321N1 cells, in comparison to its closest homologue, the P2Y1 receptor.
Experimental approach:
The potency of several chiral ATP analogues was determined by measuring increases in intracellular calcium concentration ([Ca2+]i). In a series of ATP-α-B and ATP-α-S analogues, a non-bridging oxygen atom of P α was substituted by BH3 or sulphur, respectively, introducing a chiral center at P α. The pairs of diastereoisomers (A and B isomers) were each applied as purified compounds.
Key results:
The (B) isomers (ATP-α-B Sp isomers and ATP-α-S Rp isomers) of all derivatives tested were more potent at the P2Y11 receptor than the corresponding (A) isomers (ATP-α-B Rp isomers and ATP-α-S Sp isomers) and the parent compounds. This characteristic of the P2Y11 receptor is opposite to the behaviour of the same diastereoisomers at the P2Y1 receptor, at which the (A) isomers are more active.
Conclusions and implications:
The distinctly opposite diastereoselective activity of ATP derivatives at the P2Y11 and the P2Y1 receptor will allow the deciphering of structural differences of the ligand recognition sites between these receptor subtypes and may aid in the development of subtype-selective agonists. Moreover, ATP-α-B diastereoisomers are not active at the P2Y2 receptor. Thus, they are compounds suitable for distinguishing the functional contribution of the two ATP-activated P2Y receptors, the P2Y2 and P2Y11 receptor, in physiological or pathophysiological responses of cells.
Keywords: α-phosphate-modified ATP analogues, calcium release, chiral ATP analogues, diastereoselectivity, purinergic receptor, purinoceptor
Introduction
The family of P2Y receptors comprises eight functionally cloned members (Burnstock and Knight, 2004) that represent two phylogenetically different subgroups. One group contains the receptors that couple to Gi proteins (P2Y12,13,14) and the other group consists of receptors that mainly couple to Gq proteins (P2Y1,2,4,6,11) (Costanzi et al., 2004). Among these proteins, the P2Y1 and P2Y11 receptors are found to be the closest homologues, sharing 33% identical amino acids (Communi et al., 1997). However, both receptors display differences in their pharmacological properties despite being exclusively activated by adenine nucleotides. The most striking difference is the preference of the human P2Y1 receptor for adenosine diphosphates over triphosphates, which is opposite to that seen at the human P2Y11 receptor. Moreover, the P2Y1 receptor is characterized by the high potency of 2-methlythio adenosine 5′-diphosphate (2-MeS-ADP) or 2-methylthio adenosine 5′-triphosphate (2-MeS-ATP) (Palmer et al., 1998), whereas at the P2Y11 receptor these agonists are only weakly potent (Communi et al., 1997). Further changes of the phosphate chain and the ribose moiety increase the potency of the 2-alkylthio-ATP derivatives for the P2Y11 receptor as was observed for ARC-67085MX (2-propylthio-β,γ-dichloromethylene-d-ATP) (Communi et al., 1999; Wilkin et al., 2001; White et al., 2003). Furthermore, modified ribose as in d-ATP and 2′(3′)-O-(4-benzoylbenzoyl)adenosine 5′-triphosphate (BzATP) results in potent ligands at the P2Y11 receptor (Burnstock and Knight, 2004), whereas at the P2Y1 receptor BzATP shows antagonistic activity (Vigne et al., 1999). A selective agonist for the P2Y1 receptor is MRS2365 ((N)-methanocarba-2MeSADP), in which a pseudo-ribose, consisting of a bicyclic structure fused into the (N)-methanocarba modification, replaces the ribose moiety (Chhatriwala et al., 2004). The constrained northern conformation of the pseudo-ribose leads to increased potency at the P2Y1 receptor in general and to a preserved potency at the P2Y11 receptor, whereas the corresponding (S) isomers display greatly reduced potency at both receptors (Kim et al., 2002).
Adenosine phosphorothioates (ATP-β-S (adenosine 5′-[β-thio]triphosphate), ATP-γ-S (adenosine 5′-[γ-thio]triphosphate)) are able to activate both receptors, with the P2Y11 receptor preferring the γ- and the P2Y1 receptor the β-phosphorothioates as ligands. The action of adenosine 5′-O-(1-thiotriphosphate) (Adenosine 5′-[α-thio]triphosphate (ATP-α-S)) was more closely investigated. Through substitution of one of the non-bridging oxygen atoms of Pα by sulfur, a new chiral center in the ATP molecule is introduced. The resulting diastereoisomers were separated, and we have shown before that these ATP-α-S diastereoisomers display a diastereoselective activity at the P2Y1 receptor (Major et al., 2004; Major and Fischer, 2004). Therefore, we postulated a stereoselective recognition site for the nucleotide at the P2Y1 receptor. We recently synthesized chiral adenosine 5′-[α-borano]triphosphate (ATP-α-B) analogues, where a borano group (BH3) substitutes a non-bridging oxygen at Pα, Likewise, these analogues proved agonists at the P2Y1 receptor, and one chiral isomer was clearly preferred at this receptor (Nahum et al., 2002).
In the present study, we aimed at characterizing the preference of the P2Y11 receptor for different ATP-α-S and ATP-α-B diastereoisomers (Figure 1). Here, we report the clear preference of the P2Y11-GFP (green fluorescent protein) receptor for the (B) isomers (ATP-α-B Sp isomers and ATP-α-S Rp isomers) of the chiral derivatives tested. This stereoselective discrimination is in contrast to the chiral preferences of the P2Y1 receptor. In addition, at the P2Y11 receptor, the (B) isomers showed higher potency as compared to their parent compounds.
Figure 1.

(a) Structure of the ATP analogues 1 to 8 (studied in Tables 1 and 2): 1. R=H, X=O−; 2. R=SMe, X=O−; 3. R=Cl, X=O−; 4. R=H, X=BH3−; 5. R=SMe, X=BH3−; 6. R=Cl, X=BH3−; 7. R=H, X=S; 8. R=SMe; X=S. The substitution at Pα results in two diastereoisomers for each analogue, called (A) and (B) isomers. (b) Absolute configuration around the Pα of the diastereoisomers: the Rp/Sp configuration is assigned to the (A/B) isomers of the borano-modified and the (B/A) isomers of the thio-substituted analogues, as the group priorities around Pα are opposite for the ATP-α-B and ATP-α-S derivatives. The charge distributions around Pα are indicated in (a).
These findings add substantially to the already existing knowledge about the structural determinants of ATP that are required for ligand preference by different P2Y receptor subtypes. These new data may ultimately lead us to the development of P2Y receptor subtype-selective agonists.
Materials and methods
Cell culture and transfection
The DNA of the human P2Y11 receptor (GenBankTM/EBI accession number AF030335) was kindly provided by Dr D Communi (Brussels). The complete DNA of the receptor was subcloned between the EcoRI and BamHI sites of pEGFPN1 (Clontech, Takara Bio Europe, St. Germain-en Laye, France) expression vector. The DNA of the human P2Y1 receptor (GenBankTM/EBI accession number NM 002563) was placed between the EcoRI and SmaI sites of the pEGFPN1 expression vector. 1321N1 human astrocytoma cells were transfected with the recombinant plasmids using FuGENE 6 transfection reagent as per the manufacturer's protocol (Roche, Mannheim, Germany). Transfected cells were selected with 0.5 mg/ml G418 (geneticine) and grown at 37°C in 10% CO2 in high-glucose Dulbecco's modified Eagles' medium (DMEM) supplemented with 5% fetal calf serum (FCS), 100 U/ml penicillin and 100 IU/ml streptomycin. G418 was kept throughout in the medium to achieve a stable expression of the P2Y1-GFP or P2Y11-GFP receptor fusion protein. The GFP tag was used to monitor successful expression and localization of the receptor at the plasma membrane of the cells.
[Ca2+]i measurements
The cells were plated on coverslips (diameter=22 mm), and single cell measurement was done after 3 days, when the cells were 30–50% confluent. The changes of free intracellular Ca2+ concentration ([Ca2+]i) were measured, as described before (Ubl et al., 1998) by preincubation of the cells with 2 μM fura-2AM at 37°C for 30 min in NaHBS (HEPES-buffered saline solution: 145 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 25 mM glucose and 20 mM HEPES/Tris pH 7.4) and then stimulating the cells under continuous superfusion of pre-warmed NaHBS at 37°C with different concentrations of various agonists. Fluorescence intensity was recorded alternatively at 340 and 380 nm excitation and 520 nm emission. Changes were monitored in single cells bathed in a perfusion chamber which was placed on the microscope stage of a fluorescence imaging system from TILL Photonics with an × 40/1.30 oil immersion objective and a flow rate of 1 ml/min (Vöhringer et al., 2000).
Data analysis
We analyzed the fluorescence ratio R at the two wavelengths, 340 and 380 nm, R=ΔF340/F380. The resulting data were further analyzed with the Excel program applying basal value subtraction at the calcium traces and calculating the peak height for each cell. Concentration–response data obtained from average values from 40 to 70 single cells were further analyzed to derive median effective concentration (EC50) values (half-maximal response) using the SigmaPlot program (Systat, Erkrath, Germany). The EC50 values were calculated using the following equation with a standard slope:
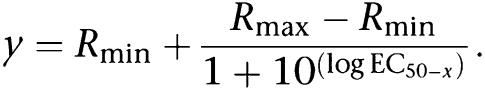 |
Curve fitting was performed using the same equation. If the free curve fit extended beyond the experimental data, the maximal response (Rmax) was adjusted to the plateau value, which was experimentally obtained for the other experiments. Average results are presented as means±s.e.m. from the number of assays shown in the text.
Materials
Geneticine (G418 sulphate) from Calbiochem, Darmstadt, Germany; ATP, ATPγS, BzATP (Sigma, Deisenhofen); Rp/Sp-ATP-α-S (Biolog, Bremen), 2-MeS-ATP (Biotrend, Köln); DMEM, penicillin/streptomycin (10 000/10 000 U/ml), trypsin/ethylenediaminetetraacetic acid (0.05%/0.02%), FCS (Seromed, Biochrom, Berlin); cell culture dishes (Nunc, Wiesbaden); cover slips (22 mm) (OmniLab); Fura 2-AM (Biomol, Hamburg/Molecular Probes, Eugene, OR, USA). The novel agonists that were used in this study were synthesized as described by Nahum et al. (2002).
Results
Heterologous expression of the P2Y11-GFP receptor in 1321N1 cells
The 1321N1 astrocytoma cell line lacks endogenously expressed P2Y receptors (Lazarowski et al., 1995; Gendron et al., 2003), which we also confirmed for the cell batch used in these experiments. We found that wild-type cells as well as mock-transfected 1321N1 cells did not display any Ca2+ responses when challenged with 100 μM ADP or ATP (data not shown).
In this study, we have used the GFP-tagged human P2Y11 receptor expressed in 1321N1 cells. To demonstrate functional expression of the P2Y11-GFP receptor in stably transfected cells, various agonists were tested for their ability to induce an increase in [Ca2+]i. The cells were monitored for the change in fluorescence intensity of the calcium indicator fura-2 after agonist stimulation, as described in the Materials and methods section. We obtained EC50 values±s.e.m (n=3–4, as indicated) for ATPγS, BzATP, ATP and 2-MeS-ATP that were 1.3±0.3 μM (4), 1.5±0.3 μM (3), 3.0±0.87 μM (4) and 13.8±5.58 μM (3), respectively. The order of potency was found to be ATPγS=BzATP>ATP>2-MeS-ATP, which is similar to previously reported findings (Communi et al., 1999).
Diastereoselective activation of the P2Y11-GFP receptor
We tested several borano-modified diastereoisomers of ATP (Figure 1a) for their stereoselective action at the P2Y11 receptor by analyzing the responses of [Ca2+]i. The concentration–response curves obtained for the different ATP-α-B derivatives and the parent compounds are displayed in Figure 2a–c. The corresponding (A) and (B) isomers of the ATP-α-B derivatives exhibited a clear difference in potency for elevation of [Ca2+]i. All the different ATP-α-B diastereoisomeric pairs (ATP-α-B (A/B), 2-MeS-ATP-α-B (A/B), 2-chloroadenosine 5′-triphosphate (2-Cl-ATP)-α-B (A/B)) displayed the same stereoselective preference in activating the P2Y11 receptor. All (B) isomers were found to be more potent than the (A) isomers ranging from 3- to 10-fold, as seen by the EC50 values in Table 1.
Figure 2.

Concentration–response curves for ATP-α-B analogues and parent compounds in inducing intracellular [Ca2+]i rise in 1321N1 cells stably expressing the P2Y11 receptor GFP fusion protein. Cells preincubated with 2 μM fura-2-AM were stimulated with varying concentrations of agonists and the change in fluorescence (ΔF340 nm/F380 nm) was detected. Data represent the mean±s.e.m. from 40 to 70 single cells and were obtained in at least three separate experiments. (a) Curves for 2-unsubstituted ATP-α-B diastereoisomers. (b) Data obtained with the 2-MeS-ATP-α-B isomers. The maxima of the curve for the (A) isomer and the parent compound have been fixed, as described in Materials and methods. (c) Curves for 2-Cl-ATP-α-B diastereoisomers. The ATP curve is shown for comparison as well as the incomplete curve for 2-Cl-ATP. The maxima of the curve for both isomers have been fixed as described in Materials and methods.
Table 1.
Potencies of ATP-α-B analogues and ATP-α-S analogues, respectively, and the parent compounds at P2Y11 and P2Y1 receptor for [Ca2+]i release
| Nucleotide | Analogue no.a | Isomer | Absolute configuration |
Receptor |
|
|---|---|---|---|---|---|
|
P2Y11 |
P2Y1b |
||||
| Potency (EC50 value; μM) | |||||
| ATP | 1 | 2.83±0.82 (4) | 0.20±0.04 (4) | ||
| 2-MeS-ATP | 2 | 13.8±5.58 (3) | 0.001±0.001 (3) | ||
| 2-Cl-ATP | 3 | >30 μM | ND | ||
| Pα-borano analogues | |||||
| ATP-α-B | 4 | A | Rp | 2.38±0.56 (6) | 0.12±0.02 (3) |
| B | Sp | 0.34±0.05 (6) | 1.20±0.18 (3) | ||
| 2-MeS-ATP-α-B | 5 | A | Rp | 1.35±0.52 (3) | 0.002±0.001 (3) |
| B | Sp | 0.26±0.03 (4) | 0.06±0.01 (3) | ||
| 2-Cl-ATP-α-B | 6 | A | Rp | 4.19±3.41 (3) | 0.004±0.002 (3) |
| B | Sp | 0.47±0.25 (3) | 0.04±0.01 (3) | ||
| Pα-thio analogues | |||||
| ATP-α-S | 7 | A | Sp | 1.71±0.55 (4) | 0.009±0.002 (3) |
| B | Rp | 0.27±0.06 (5) | 0.07±0.02 (3) | ||
| 2-MeS-ATP-α-S | 8 | A | Sp | 2.64±1.10 (6) | 0.001±0.001 (3) |
| B | Rp | 0.64±0.12 (5) | 0.02±0.005 (3) | ||
Abbreviations: ATP-α-B, adenosine 5′-[α-borano]triphosphate; ATP-α-S, adenosine 5′-[α-thio]triphosphate; [Ca2+]i, intracellular-free calcium concentration; 2-Cl-ATP, 2-chloroadenosine 5′-triphosphate; EC50, median effective concentration; 2-MeS-ATP, 2-methylthio adenosine 5′-triphosphate
Data were obtained from concentration–response curves, such as shown in Figure 2 or 4 and are expressed as EC50 values±s.e.m. (μM). They represent the mean and standard error and were obtained from at least three separate experiments (n=3–6, as indicated) with 40 to 70 single cells per concentration of agonist investigated.
For the GFP-tagged P2Y11 receptor stably expressed in 1321N1 cells, data are derived from the analysis of the concentration–response curves displayed in Figures 2 and 4.
See formula scheme in Figure 1.
For the GFP-tagged P2Y1 receptor, data are from our previous analysis (see reference Major et al., 2004; Nahum et al., 2002), where the receptor was analyzed similarly stably expressed in HEK293 cells.
The (B) isomer of ATP-α-B (EC50=340±53.0 nM) displayed a seven-fold higher potency than ATP (EC50=2.83±0.82 μM) and its corresponding (A) isomer (EC50=2.38±0.56 μM) was equipotent with ATP (Figure 2a). The tendency that the (B) isomers are preferred by the P2Y11 receptor was the same for all the other ATP-α-B derivatives tested. The (B) isomer of 2-MeS-ATP-α-B was the most potent ligand of all compounds tested in this study with an EC50 value of 260±33.0 nM. The corresponding (A) isomer (EC50=1.35±0.52 μM) was five-fold less potent, but displayed a much higher potency than 2-MeS-ATP (EC50=13.8±5.58 μM). The difference was one order of magnitude (Figure 2b).
The 2-Cl-substitution had a negative influence on the potency of ATP-α-B at the P2Y11 receptor (Figure 2c). The (A) isomer of 2-Cl-ATP-α-B (EC50=4.19±3.41 μM) displayed the lowest potency of all the diastereoisomers used in this study. Only the (B) isomer (EC50=467±246 nM) showed a potency that was comparable to that of the other α-borano (B) isomers. The parent compound 2-Cl-ATP, where the phosphate moiety was left unchanged, was too weak an agonist at the P2Y11 receptor to determine a complete concentration–response curve using single cell calcium measurements. At a concentration of 100 μM, we only obtained 60% of the maximal ATP response. Therefore, in Figure 2c, the concentration–effect curve of ATP is displayed for comparison as well as the incomplete curve for 2-Cl-ATP (EC50>30 μM).
The action of the ATP-α-B diastereoisomers at the P2Y11 receptor presented here stands in contrast to the diastereoselectivity at the P2Y1 receptor, as seen previously by us. At the P2Y1 receptor, the (A) isomers were more potent than the (B) isomers in inducing a calcium response (Nahum et al., 2002) or in causing receptor endocytosis (Tulapurkar et al., 2004, 2005). For comparison, the EC50 values for the ATP-α-B derivatives at the P2Y1 receptor are also included in Table 1. However, it should be noted that in that study the P2Y1 receptor was heterologously expressed in HEK293 cells. For a better and more valid comparison, we additionally investigated the action of 2-Cl-ATP-α-B isomers at the P2Y1-GFP receptor protein stably expressed in 1321N1 cells (Figure 3a). It was not possible to carry out a complete investigation of the potencies of all ligands used in this study, as the amount of compounds was not sufficient. The stereoselectivity of the 2-Cl-ATP-α-B diastereoisomers at the P2Y1 receptor was identical for this receptor, whether the receptor was expressed in 1321N1 cells or in HEK293 cells. The (A) isomer displayed a higher potency (EC50=20±5.0 nM, n=3) than the (B) isomer (EC50=649±51.7 nM, n=3). Moreover, the difference in potency of the (A) isomer at the P2Y1 receptor compared to the potency at the P2Y11 receptor is striking. This characteristic and the stereoselectivity are consistent with the findings from our previous study and therefore independent of the receptor expression system used.
Figure 3.

Concentration–response curves obtained with 1321N1 cells stably expressing the P2Y1 receptor GFP fusion protein for ATP-α-B/S analogues in inducing [Ca2+]i rise. Cells preincubated with 2 μM fura-2-AM were stimulated with varying concentrations of agonists and the change in fluorescence (ΔF340 nm/F380 nm) was detected. Data represent the mean±s.e.m. from 40 to 70 single cells and were obtained in at least three separate experiments. (a) Curves for 2-Cl-ATP-α-B diastereoisomers. The maximum of the curve for the (B) isomer has been fixed as described in Materials and methods. (b) Curves for 2-unsubstituted ATP-α-S diastereoisomers.
Furthermore, we investigated the action of the adenosine 5′-O-(1-thiotriphosphate) derivatives (ATP-α-S) (Figure 1a) at the P2Y11 receptor. The concentration–response curves for each ATP-α-S diastereoisomeric pair are presented in Figure 4a and b, in comparison to the respective parent compound, which has an unchanged phosphate moiety. The two different pairs (ATP-α-S (A/B) and 2-MeS-ATP-α-S (A/B)) displayed the same diastereoselective activity at the P2Y11 receptor as the ATP-α-B derivatives. The (B) isomers were found to be more potent than the (A) isomers, as shown by the summary of the resulting EC50 values in Table 1. However, the difference between these diastereoisomers was not as pronounced as with the α-borano-substituted ATP analogues.
Figure 4.

Concentration–response curves for ATP-α-S analogues and parent compounds in inducing [Ca2+]i rise in 1321N1 cells stably expressing the P2Y11 receptor GFP fusion protein. Cells preincubated with 2 μM fura-2-AM were stimulated with varying concentrations of agonists and the change in fluorescence (ΔF340 nm/F380 nm) was detected. Data represent the mean±s.e.m. from 40 to 70 single cells and were obtained in at least three separate experiments. (a) Curves for 2-unsubstituted ATP-α-S diastereoisomers. The maximum of the curve for the (A) isomer has been fixed as described in Materials and methods. (b) Curves for 2-MeS-ATP-α-S diastereoisomers. The maximum of the 2-MeS-ATP curve has been fixed as described in Materials and methods.
The (B) isomer of ATP-α-S (EC50=270±64.0 nM) was found to be the most potent of the α-sulfur-substituted compounds tested, whereas the corresponding (A) isomer with an EC50 value of 1.71±0.55 μM displayed a six-fold lower potency (Figure 4a). However, both substances had a higher potency at the P2Y11 receptor than ATP itself. The (B) isomer of 2-MeS-ATP-α-S (EC50=640±128 nM) showed only a four-fold increase in potency, compared to its corresponding (A) isomer (EC50=2.64±1.10 μM) but was found to be 20-fold more potent than the parent compound, 2-MeS-ATP (EC50=13.8±5.58 μM) (Figure 4b). Nevertheless, the maxima of the concentration–effect curves for both isomers were lower than the other agonist-evoked maxima, which makes a direct comparison of the these EC50 values difficult.
Importantly, the stereoselective activity at the P2Y11 receptor is again in contrast to the activity of the α-thio-modified compounds at the P2Y1 receptor. We have found that the P2Y1 receptor prefers the (A) over the (B) isomers of these substances (Major et al., 2004). The EC50 values for the ATP-α-S derivatives at the P2Y1 receptor are also included in Table 1. Again, in that study the P2Y1 receptor was stably expressed in HEK293 cells. Therefore, we also investigated the action of ATP-α-S diastereoisomers at the P2Y1-GFP receptor stably expressed in 1321N1 cells (Figure 3b). Like for the 2-Cl-ATP-α-B isomers, we obtained similar results with 1321N1 cells as with HEK293 cells concerning the stereoselective preference. The (A) isomer (EC50=21.9±10.1 nM, n=6) of ATP-α-S was more potent than the (B) isomer (EC50=108±14.7 nM, n=5).
Discussion
In this study, we investigated the potency of different novel diastereomeric analogues of ATP (Figure 1) at the P2Y11 receptor. Through substitution of one of the non-bridging oxygen atoms of Pα by borane or sulfur, a new chiral center in the ATP molecule was introduced. The resulting diastereoisomers were separated and the absolute configuration around the Pα was assigned (Major et al., 2004). The Rp configuration was attributed to the (A) isomers of the borano-modified and the (B) isomers of the thio-substituted analogues. This difference in assignment is due to the group priorities around Pα, which are opposite for the ATP-α-B and ATP-α-S derivatives. Sulfur is of higher priority in the chemical nomenclature compared to oxygen, and borane is of lower priority than oxygen (Figure 1b). Here, we determined the increase in [Ca2+]i in 1321N1 astrocytoma cells stably expressing a P2Y11 receptor GFP fusion protein upon stimulation with these analogues. The compounds tested activated the P2Y11 receptor with an EC50 value in the low micromolar range.
The diastereoisomers showed significant changes in the potencies at the P2Y11 receptor as compared to their parent compounds ATP, 2-MeS-ATP and 2-Cl-ATP. Specifically, the introduction of a borane/sulfur group resulted in more potent ligands at the P2Y11 receptor. The (B) isomer of ATP-α-B was eight-fold more potent than ATP. For the (B) isomer of 2-MeS-ATP-α-B, the change in potency was 50-fold compared to 2-MeS-ATP. The same was probably true for the (B) isomer of the 2-chloro-substituted ATP-α-B isomers. The parent compound 2-Cl-ATP was too weak an agonist to determine a complete concentration–response curve with the amount of drug available. Nevertheless, the difference in potency between the borano and unsubstituted 2-Cl-ATP derivative is at least 30-fold.
The significant effect of the borane substitution on the potency of the ATP derivatives for the P2Y11 receptor is in contrast to the findings at the P2Y1 receptor. At the P2Y1 receptor, the introduction of borane into ATP or 2-MeS-ATP did not create more potent ligands (Nahum et al., 2002), whereas the introduction of sulfur at Pα of ATP lead to an increase in potency at the P2Y1 receptor (Major et al., 2004). However, the 2-MeS-ATP-α-S analogues were as potent as 2-MeS-ATP itself. This demonstrates that the introduction of sulfur at Pα leads to an increased potency of ATP at the P2Y1 receptor but is not able to further increase the potency of 2-methylthioether derivatives, which are already very potent agonists at this receptor. This underlines that the 2-methylthio substitution plays a pivotal role in the potency of the ATP derivatives at the P2Y1 receptor (Schachter et al., 1996; Palmer et al., 1998). In contrast, at the P2Y11 receptor both sulfur- and borano-substitution increased the potency of ATP as well as 2-MeS-ATP and 2-Cl-ATP itself. Moreover, the shift in potency was more distinct for the (B) isomers of 2-MeS-ATP-α-B/S derivatives, compared to the parent compound than for the (B) isomers of the 2-unsubstituted ATP-α-B/S analogues compared to ATP. This shows that the introduction of a borane/sulfur group at Pα determines the potency of the derivative at the P2Y11 receptor overriding the negative influence of a substituent at position 2 of the ATP molecule.
Interestingly, at the P2Y11 receptor, for all Pα-substituted derivatives the corresponding (B) diastereoisomers were found to be more potent than the (A) isomers. This stereoselective action of the ligands is opposite to that found at the P2Y1 receptor which prefers the (A) isomers (Nahum et al., 2002; Major et al., 2004). In contrast, the P2Y2 and P2Y4 receptors display the same stereoselectivity as P2Y11 receptor regarding the activity of α-thio diastereoisomers. At both P2Y2 and P2Y4 receptors, the Rp-UTP-α-S isomer was more potent than the Sp isomer but their potency was weak compared to UTP (Jacobson et al., 2006). All the receptors mentioned above belong to one phylogenetic subgroup (Costanzi et al., 2004). Surprisingly, the amino-acid sequence of the P2Y11 receptor protein is more closely related to the P2Y1 receptor, although besides the P2Y2 receptor the human P2Y11 receptor is an ‘ATP-receptor'. Still, the pharmacological profiles of both ATP-preferring receptors differ much more than the profiles of the P2Y11 and P2Y1 receptor (Burnstock and Knight, 2004). Both receptors are activated by adenine nucleotides exclusively and are blocked by Reactive Blue, whereas the P2Y2 receptor can also be activated by uridine nucleotides and shows an affinity for PPADS not shared by the P2Y1/11 receptors (Burnstock and Knight, 2004). Another P2Y receptor at which the action of α-thio diastereoisomers had been investigated is the P2Y12 receptor. The ATP-α-S isomers were found to be antagonists at the P2Y12 receptor with only a slightly higher affinity for the (A) isomer (Cusack and Hourani, 1982). As the antagonistic action of the ATP-α-S isomers at the P2Y12 receptor was determined by measuring adenylyl cyclase activity in platelets, it is rather difficult to compare these results with our investigations.
Furthermore, the P2Y11 receptor displays the same diastereoselectivity for borano-/thiophosphate nucleotide analogues as the catalytic site of ecto-nucleotidase (NTPDase) 1 (CD39, EC 3.6.1.5) (Cusack et al., 1983; Nahum et al., 2002). The ATP-α-B (B) isomers were found to be about 10 times less stable in an assay to test the enzymatic stability regarding NTPDase 1 than the corresponding (A) isomers, but they showed a hydrolysis rate that was still half that of ATP itself. The 1321N1 cells possess an ectonucleotidase activity characteristic for NTPDase 1 with Km value of 66 μM±13 for ATP hydrolysis (Lazarowski et al., 1997). If we take this into consideration, the potencies found for all (B) isomers tested in this study might be influenced by competition for binding to the receptor and to the NTPDase 1. However, if we consider the experimental design of the measurements in this study, where we have a continuous flow application of the agonists, and the much higher affinity of the P2Y11 receptor for ATP as compared to the NTPDase 1, competition in binding does certainly not affect our analysis.
An interesting observation made here is that the ATP-α-B/-S analogues appear to be more potent at the P2Y1 receptor as compared to the P2Y11 receptor (Table 1). As both receptors, the P2Y1 and P2Y11 receptor, were expressed in different cell systems, it is difficult to make an absolute comparison of the EC50 values. Therefore, we also stably expressed the P2Y1-GFP receptor in the 1321N1 cells. In these cells, we found a rank order of potency for the 2-Cl-ATP-α-B and ATP-α-S analogues comparable to that in HEK293 cells. The tendency that the ligands are overall more potent at the P2Y1 receptor than at the P2Y11 receptor was also observed. This underlines that our data show the stereoselectivity and receptor subtype selectivity of the compounds tested.
The (A) isomer of the 2-MeS-ATP-α-B derivatives is the most selective of all the analogues tested here, being more active at the P2Y1 receptor, comparable to the selectivity of the (A) isomer of 2-Cl-ATP-α-B. This large receptor-subtype selectivity is due to the combination of modifications on both C2 and the phosphate chain of the ATP scaffold. This finding, in addition to our previous report (Tulapurkar et al., 2004) showing the lack of any activity of the 2-MeS-ATP-α-B diastereoisomers at the P2Y2 receptor, suggests the possibility of a selective activation of the P2Y1 receptor by the (A) isomer of these compounds in cells/organs, which also express P2Y11 and P2Y2 receptors. Moreover, the ATP-α-B derivatives that had no activity at the P2Y2 receptor are compounds suitable for distinguishing the functional contribution of the two ATP-activated P2Y receptors, the P2Y2 and P2Y11 receptor, in physiological or pathophysiological responses of cells to ATP.
In summary, we have found a diastereoselective activity of ATP-α-B and ATP-α-S diastereoisomers at the P2Y11 receptor demonstrated by the preference of the (B) isomers. The diastereoselectivity is opposite at the P2Y1 receptor that prefers the (A) isomers of these compounds. This shows that both receptors prefer different diastereoisomers of the chiral ATP analogues, although the receptors are close homologues within the P2Y receptor family, sharing >50% of the amino-acid residues presumably involved in ligand recognition. These findings add to the understanding of the structural and conformational determinants of nucleotides which activate different P2Y receptors. The different diastereoselectivity of these receptors may enable us to provide a more detailed insight into the structure–activity relationships of these P2Y receptors. This knowledge will undoubtedly be of particular importance for the development of subtype-specific agonists or antagonists, which may be considered as potentially attractive drug candidates.
Acknowledgments
This work is supported by Deutsche Forschungsgemeinschaft (DFG) and BMBF/MOS (grant no 1812). We are thankful to Dr Rainer Schäfer (FAN GmbH, Magdeburg, Germany) for critical and helpful comments concerning the manuscript. We also thank Mrs K Christoph, Mrs I Laubinger for excellent technical assistance. We thank Dr D Communi for the P2Y11 receptor cDNA clone.
Abbreviations
- 1321N1
human astrocytoma cell line
- ATP-α-B
adenosine 5′-[α-borano]triphosphate
- ATP-α-S
adenosine 5′-[α-thio]triphosphate
- ATP-β-S
adenosine 5′-[β-thio]triphosphate
- ATP-γ-S
adenosine 5′-[γ-thio]triphosphate
- BzATP
2′(3′)-O-(4-benzoylbenzoyl)adenosine 5′-triphosphate
- 2-Cl-ATP
2-chloroadenosine 5′-triphosphate
- [Ca2+]i
intracellular-free calcium concentration
- DMEM
Dulbecco's modified Eagles' medium
- FCS
fetal calf serum
- G418
geneticine
- GFP
green fluorescent protein
- NaHBS
HEPES-buffered saline solution
- 2-MeS-ATP
2-methylthio adenosine 5′-triphosphate
- 2-MeS-ADP
2-methlythio adenosine 5′-diphosphate
- NTPDase
ecto-nucleotidase
Conflict of interest
The authors state no conflict of interest.
References
- Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- Chhatriwala M, Ravi RG, Patel RI, Boyer JL, Jacobson KA, Harden TK. Induction of novel agonist selectivity for the ADP-activated P2Y1 receptor versus the ADP-activated P2Y12 and P2Y13 receptors by conformational constraint of an ADP analog. J Pharmacol Exp Ther. 2004;311:1038–1043. doi: 10.1124/jpet.104.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communi D, Govaerts C, Parmentier M, Boeynaems JM. Cloning of a human purinergic P2Y receptor coupled to phospholipase C and adenylyl cyclase. J Biol Chem. 1997;272:31969–31973. doi: 10.1074/jbc.272.51.31969. [DOI] [PubMed] [Google Scholar]
- Communi D, Robaye B, Boeynaems JM. Pharmacological characterization of the human P2Y11 receptor. Br J Pharmacol. 1999;128:1199–1206. doi: 10.1038/sj.bjp.0702909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack NJ, Hourani SM. Adenosine 5-diphosphate antagonists and human platelets: no evidence that aggregation and inhibition of stimulated adenylate cyclase are mediated by different receptors. Br J Pharmacol. 1982;76:221–227. doi: 10.1111/j.1476-5381.1982.tb09210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack NJ, Pearson JD, Gordon JL. Stereoselectivity of ectonucleotidases on vascular endothelial cells. Biochem J. 1983;214:975–981. doi: 10.1042/bj2140975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron FP, Neary JT, Theiss PM, Sun GY, Gonzalez FA, Weisman GA. Mechanisms of P2X7 receptor-mediated ERK1/2 phosphorylation in human astrocytoma cells. Am J Physiol Cell Physiol. 2003;284:C571–C581. doi: 10.1152/ajpcell.00286.2002. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Costanzi S, Ivanov AA, Tchilibon S, Besada P, Gao ZG, et al. Structure activity and molecular modeling analyses of ribose- and base-modified uridine 5′-triphosphate analogues at the human P2Y2 and P2Y4 receptors. Biochem Pharmacol. 2006;71:540–549. doi: 10.1016/j.bcp.2005.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg AK, Erlinge D, et al. Methanocarba modification of uracil and adenine nucleotides: high potency of Northern ring conformation at P2Y1, P2Y2, P2Y4, and P2Y11 but not P2Y6 receptors. J Med Chem. 2002;45:208–218. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarowski ER, Homolya L, Boucher RC, Harden TK. Identification of an ecto-nucleoside diphosphokinase and its contribution to interconversion of P2 receptor agonists. J Biol Chem. 1997;272:20402–20407. doi: 10.1074/jbc.272.33.20402. [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Watt WC, Stutts MJ, Boucher RC, Harden TK. Pharmacological selectivity of the cloned human P2U-purinoceptor: potent activation by diadenosine tetraphosphate. Br J Pharmacol. 1995;116:1619–1627. doi: 10.1111/j.1476-5381.1995.tb16382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major DT, Fischer B. Molecular recognition in purinergic receptors. 1. A comprehensive computational study of the h-P2Y1-receptor. J Med Chem. 2004;47:4391–4404. doi: 10.1021/jm049772m. [DOI] [PubMed] [Google Scholar]
- Major DT, Nahum V, Wang Y, Reiser G, Fischer B. Molecular recognition in purinergic receptors. 2. Diastereoselectivity of the h-P2Y1-receptor. J Med Chem. 2004;47:4405–4416. doi: 10.1021/jm049771u. [DOI] [PubMed] [Google Scholar]
- Nahum V, Zündorf G, Levesque SA, Beaudoin AR, Reiser G, Fischer B. Adenosine 5′-O-(1-boranotriphosphate) derivatives as novel P2Y1 receptor agonists. J Med Chem. 2002;45:5384–5396. doi: 10.1021/jm020251d. [DOI] [PubMed] [Google Scholar]
- Palmer RK, Boyer JL, Schachter JB, Nicholas RA, Harden TK. Agonist action of adenosine triphosphates at the human P2Y1 receptor. Mol Pharmacol. 1998;54:1118–1123. [PubMed] [Google Scholar]
- Schachter JB, Li Q, Boyer JL, Nicholas RA, Harden TK. Second messenger cascade specificity and pharmacological selectivity of the human P2Y1-purinoceptor. Br J Pharmacol. 1996;118:167–173. doi: 10.1111/j.1476-5381.1996.tb15381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulapurkar ME, Laubinger W, Nahum V, Fischer B, Reiser G. Subtype specific internalization of P2Y1 and P2Y2 receptors induced by novel adenosine 5′-O-(1-boranotriphosphate) derivatives. Br J Pharmacol. 2004;142:869–878. doi: 10.1038/sj.bjp.0705859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulapurkar ME, Schäfer R, Hanck T, Flores RV, Weisman GA, Gonzalez FA, et al. Endocytosis mechanism of P2Y2 nucleotide receptor tagged with green fluorescent protein: clathrin and actin cytoskeleton dependence. Cell Mol Life Sci. 2005;62:1388–1399. doi: 10.1007/s00018-005-5052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubl JJ, Vöhringer C, Reiser G. Co-existence of two types of [Ca2+]i-inducing protease-activated receptors (PAR-1 and PAR-2) in rat astrocytes and C6 glioma cells. Neuroscience. 1998;86:597–609. doi: 10.1016/s0306-4522(97)00686-6. [DOI] [PubMed] [Google Scholar]
- Vigne P, Hechler B, Gachet C, Breittmayer JP, Frelin C. Benzoyl ATP is an antagonist of rat and human P2Y1 receptors and of platelet aggregation. Biochem Biophys Res Commun. 1999;256:94–97. doi: 10.1006/bbrc.1999.9558. [DOI] [PubMed] [Google Scholar]
- Vöhringer C, Schäfer R, Reiser G. A chimeric rat brain P2Y1 receptor tagged with green-fluorescent protein: high-affinity ligand recognition of adenosine diphosphates and triphosphates and selectivity identical to that of the wild-type receptor. Biochem Pharmacol. 2000;59:791–800. doi: 10.1016/s0006-2952(99)00390-1. [DOI] [PubMed] [Google Scholar]
- White PJ, Webb TE, Boarder MR. Characterization of a Ca2+ response to both UTP and ATP at human P2Y11 receptors: evidence for agonist-specific signaling. Mol Pharmacol. 2003;63:1356–1363. doi: 10.1124/mol.63.6.1356. [DOI] [PubMed] [Google Scholar]
- Wilkin F, Duhant X, Bruyns C, Suarez-Huerta N, Boeynaems JM, Robaye B. The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic cells. J Immunol. 2001;166:7172–7177. doi: 10.4049/jimmunol.166.12.7172. [DOI] [PubMed] [Google Scholar]