

Abstract
Previously, we showed that oxidant exposure in renal proximal tubular cells (RPTC) induces mitochondrial dysfunction mediated by PKC-ε. This study examined the role of ERK1/2 in mitochondrial dysfunction induced by oxidant injury and whether PKC-ε mediates its effects on mitochondrial function through the Raf-MEK1/2-ERK1/2 pathway. Sublethal injury produced by tert-butylhydroperoxide (TBHP) resulted in three- to fivefold increase in phosphorylation of ERK1/2 and p38 but not JNK. This was followed by decreases in basal and uncoupled respirations (41%), state 3 respiration and ATP production coupled to complex I (46%), and complex I activity (42%). Oxidant exposure decreased aconitase activity 30% but not pyruvate, α-ketoglutarate, and malate dehydrogenase activities. Inhibition of ERK1/2 restored basal and state 3 respirations, ΔΨm, ATP production, and complex I activity but not aconitase activity. In contrast, activation of ERK1/2 by expression of constitutively active MEK1 suppressed basal, uncoupled, and state 3 respirations in noninjured RPTC to the levels observed in TBHP-injured RPTC. MEK1/2 inhibition did not change Akt or p38 phosphorylation, demonstrating that the protective effect of MEK1/2 inhibitor was not due to activation of Akt or inhibition of p38 pathway. Inhibition of PKC-ε did not block TBHP-induced ERK1/2 phosphorylation in whole RPTC or in mitochondria. We conclude that 1) oxidant-induced activation of ERK1/2 but not p38 or JNK reduces mitochondrial respiration and ATP production by decreasing complex I activity and substrate oxidation through complex I, 2) citric acid cycle dehydrogenases are not under control of the ERK1/2 pathway in oxidant-injured RPTC, 3) the protective effects of ERK1/2 inhibition are not due to activation of Akt, and 4) ERK1/2 and PKC-ε mediate oxidant-induced mitochondrial dysfunction through independent pathways.
Keywords: renal proximal tubular cells, mitochondria, respiratory complex, ATP, extracellular signal-regulated kinases, injury, repair
The kidney is a major target of toxicant-induced damage as it receives a large percentage of the cardiac output and concentrates numerous chemicals through its diverse transporters. Renal proximal tubular cells (RPTC) are the primary target of nephrotoxicants within the kidney. RPTC selectively accumulate many chemicals, which results in an increased intracellular concentration of these compounds and leads to toxic injury. Mitochondrial dysfunction is a common result of nephrotoxicant exposure in RPTC and an important mechanism of toxicity for a number of drugs and toxicants. Many nephrotoxicants act as mitochondrial inhibitors or uncouplers inducing necrosis or apoptosis in RPTC (1, 38, 40). RPTC are highly dependent on oxidative metabolism for ATP synthesis and have little capacity for generating energy through glycolysis when oxidative metabolism is impaired (7, 11). The decrease in cellular ATP level that is often associated with mitochondrial dysfunction results in the reduction of ATP-dependent processes including Na+-K+-ATPase activity and active Na+ transport, which, in turn, disrupts Na+-dependent transports and ion homeostasis in RPTC. Therefore, the results of energy depletion in RPTC are dissolution of cellular structure and function. Prolonged mitochondrial energy deficits in nephrotoxicant-injured RPTC limit restoration of RPTC transport functions and contribute to the lack of overall cellular repair and development of acute renal failure.
Oxidative stress plays a key role in renal dysfunction associated with exposure to numerous drugs and environmental nephrotoxicants (9, 16, 17, 29). Oxidant damage denatures proteins causing the loss of their function and increasing their susceptibility to proteolysis. Tert-butylhydroperoxide (TBHP) is commonly used as a model compound to induce oxidant injury in in vitro models of RPTC (12, 33, 38). Oxidant exposure in RPTC also inappropriately activates some proteins, including important elements of signaling cascades such as protein kinase C (PKC) isozymes and members of mitogen-activated protein kinase (MAPK) family (21, 35). Activation of some of these signaling proteins leads to mitochondrial dys-function and ATP depletion (35). Extracellular signal-regulated kinases (ERK1 and ERK2) have been implicated in the regulation of a variety of functions in different cell types. ERK1/2 are involved in the regulation of proliferation, differentiation, contraction, transport, injury, and apoptosis through the regulation of transcription factors and direct phosphorylation of some proteins. Regulation of mitosis and proliferation by ERK1/2-dependent pathway has been documented in many cell types including renal epithelial cells (14, 16, 27). ERK1/2 are activated by numerous toxicants and have been implicated in cell injury in different cell types. Both protection and potentiation of ischemia-reperfusion- or toxicant-induced injury by ERK1/2 have been reported in different cell types, including RPTC (4, 5, 19, 21, 24-26, 41, 46). It has been suggested that the balance between the activation of ERK1/2 and the JNK may be a major factor that determines the outcome of cell injury following oxidant injury in RPTC (4). It was proposed that survival following oxidant injury is associated with the activation of both ERK1/2 and JNK, whereas oxidant-induced death was associated with activation of JNK only (5).
Although a connection between ERK1/2 and specific mitochondrial functions has not been reported yet, the presence of ERK1/2 protein has been shown recently in mitochondria isolated from different tissues and cell types, including cardiomyocytes and brain cells (3, 8, 48). In the cardiomyocyte mitochondria, ERK1/2 form complexes with PKC-ε and appear to play a role in PKC-ε-mediated cardioprotection against ischemia-reperfusion injury (8). In the brain, ERK1/2 are present in the mitochondrial outer membrane and/or the inter-membrane space (3). It has been established that mitochondrial integrity and function directly participate in the signaling pathway initiated by the release of cytochrome c from mitochondria and culminating in apoptosis. ERK1/2 protect against the cytochrome c-mediated apoptosis in some cell types by 1) phosphorylating mitochondrial Bad on serine 112 and inhibiting its proapoptotic function, maintaining the integrity of mitochondria, and preventing the release of cytochrome c, and/or 2) inhibiting caspase 9 by direct phosphorylation on threonine 125 (2, 20).
The targets of the deleterious effects of ERK1/2-mediated phosphorylation are unknown. Our previous study showed that activation of ERK1/2 is involved in the mitochondrial dysfunction induced by cisplatin in RPTC (30). Oxidant stress-induced activation of ERK1/2 decreases growth factor-dependent survival of RPTC (42). Inhibition of the MEK1/2-ERK1/2 pathway is as potent as epidermal growth factor in maintaining viability in this cell type (42). Because ERK1/2 are involved in the regulation of functions associated with survival, it is likely that the MEK1/2-ERK1/2 pathway regulates mitochondrial activities associated with cell life and death in renal proximal tubules, including functions related to ATP production. Therefore, the aim of the present study was to examine whether ERK1/2 pathway plays a role in mitochondrial dysfunction induced by an oxidant and in the repair of mitochondrial function following oxidant injury in RPTC and examine putative mitochondrial targets of ERK1/2 in oxidant-injured RPTC. The second goal of this study was to determine whether the MEK1/2-ERK1/2 pathway is a downstream effector of PKC-ε in oxidant-induced mitochondrial dysfunction in RPTC.
MATERIALS AND METHODS
Materials
Female New Zealand White rabbits (2.0–2.5 kg) were purchased from Myrtle's Rabbitry (Thompson Station, TN). The cell culture media were purchased from MediaTech Cellgro (Herndon, VA). U0126 [1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)-butadiene] and U0124 [1,4-diamino-2,3-dicyano-1,4-bis(methylthio)-butadiene] were supplied by LC Laboratories (Woburn, MA) and Calbiochem (La Jolla, CA), respectively. Protease inhibitors and ATP Bioluminescence Assay Kit HS II were obtained from Roche (Mannheim, Germany); N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD), isocitrate dehydrogenase, and phosphatase inhibitors cocktail were from Sigma (St. Louis, MO); and MitoTracker Red 580 was supplied by Invitrogen/Molecular Probes (Eugene, OR). Phospho-ERK1/2, phosphoMEK1/2, MEK1/2, phospho-Akt(Ser471), Akt and phospho-p38 antibodies were purchased from Cell Signaling Technology (Beverly, MA). ERK1/2, phospho-JNK, JNK, p38, and fluorescein-conjugated phosphoERK1/2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary IgGs coupled to horseradish peroxidase were supplied by Kirkegaard and Perry Laboratory (Gaithersburg, MD) and Chemicon (Temecula, CA), respectively. Myristoylated PKC-εV1–2 inhibitor (N-Myr-Glu-Ala-Val-Ser-Leu-Lys-Pro-Thr) was supplied by Biomol (Plymouth Meeting, PA). U0126 and SL327 were purchased from LC Laboratories and Tocris Cookson (Ellisville, MO), respectively. U0124 and PD98059 were supplied by Calbiochem. All other reagents have been purchased from Sigma or as described previously (30-32, 35).
Isolation and culture of RPTC
Renal proximal tubules were isolated from rabbit kidneys by the iron oxide perfusion method and cultured in 35-mm culture dishes in improved conditions as previously described (31, 32). The culture medium was a 50:50 mixture of DMEM and Ham's F-12 nutrient mix without phenol red, pyruvate, and glucose, supplemented with 15 mM NaHCO3, 15 mM HEPES, and 6 mM lactate (pH 7.4, 295 mosmol/kgH2O). Human transferrin (5 μg/ml), selenium (5 ng/ml), hydrocortisone (50 nM), bovine insulin (10 nM), and l-ascorbic acid-2-phosphate (50 μM) were added to the media immediately before daily media change (2 ml/dish).
TBHP treatment of RPTC monolayer
RPTC monolayers reached confluence within 6 days and were treated with TBHP (350 μM, 45 min) on day 7 of culture. Controls were treated with the diluent (dimethyl sulfoxide, 0.1%) (DMSO). Following TBHP exposure, the monolayer was washed with fresh, warm (37°C) medium and cultured for an additional 4 days. In experiments using the selective inhibitor of MEK1/2, RPTC were treated with 10 μM U0126 for 0.5 h before TBHP treatment and daily starting with the media change immediately following TBHP exposure. U0124 was used as a negative control for MEK1/2 inhibitor, U0126. In addition, two other MEK1/2 inhibitors, SL327 and PD98059, were used in some experiments. RPTC samples were taken at various time points after TBHP exposure for measurements of different mitochondrial functions and immunoblotting.
For the assessment of the role of PKC-ε in ERK1/2 activation by TBHP, 20 μM PKC-εV1–2 inhibitor was added 1 h before TBHP exposure and every day with the daily media change.
Assessment of RPTC viability
Cell viability was assessed using the annexin V/propidium iodide binding assay as described previously (34). Annexin V binding to RPTC plasma membrane was used as a marker of apoptosis, whereas propidium iodide binding to DNA served as a marker of necrosis. RPTC monolayers were washed twice with a binding buffer consisting of 10 mM HEPES (pH 7.4), 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 1.8 mM CaCl2 followed by incubation (15 min on ice) in the binding buffer containing propidium iodide (2 μg/ml). Monolayers were washed three times with the binding buffer and incubated in the presence of annexin V-FITC (125 ng/ml) for 10 min at room temperature. RPTC were washed three times with the binding buffer and processed for flow cytometry. Propidium iodide and annexin V-FITC fluorescence was quantified by flow cytometry (FACSCalibur, BD Biosciences) using excitation at 488 nm and emission at 590 nm and 530 nm for propidium iodide and annexin V-FITC, respectively. For each sample, 10,000 events were counted. Cells positive for annexin V and negative for propidium iodide were considered apoptotic. Cells negative for annexin V and positive for propidium iodide were considered necrotic.
Active MEK1 expression
Adenoviral vector carrying cDNA coding the constitutively active MEK1 (Ser 218 and Ser 22 replaced by Glu) was constructed by Dr. S. Tanaka as described earlier (28). The adenovirus was amplified in AD293 cells as described previously by Arany and colleagues (5) and generously provided by Dr. I. Arany. Adenovirus carrying an empty pShuttle vector was obtained from BD Biosciences Clontech (Palo Alto, CA) and was amplified in HEK293 cells. To increase levels of active ERK1/2, adenoviral infection was carried out in confluent RPTC 48 h before TBHP exposure. RPTC were incubated with adenoviral vectors (50 MOI) for 24 h, culture media were replaced, and cells were cultured for another 24 h before TBHP exposure. Infection with adenovirus carrying an empty vector was used as negative control.
Oxygen consumption
RPTC monolayers were gently detached from the dishes using a rubber policeman and transferred to the oxygen consumption (Qo2) measurement chamber. Qo2 was measured polarographically using a Clark type electrode as described previously (30-32). Basal Qo2 was used as a marker of overall mitochondrial function in RPTC. Uncoupled Qo2 was used as a marker of electron transfer rate and was measured in the presence of carbonyl cyanide p-(trifluoro-methoxy)phenylhydrazone (FCCP; 2 μM). State 3 respiration supported by electron donors linked to the respiratory complex I, II, or IV was measured in whole RPTC in 2 ml warm (37°C) solution resembling an intracellular electrolyte milieu (120 mM KCl, 5mMKH2PO4, 10 mM HEPES, 1 mM MgSO4, and 2 mM EGTA, pH 7.4) containing digitonin (0.1 mg/ml), 5 mM glutamate, and 5 mM malate (electron donors to complex I), 10 mM succinate (electron donor to complex II) + 0.1 μM rotenone, or 1 mM ascorbate + 1mM TMPD (electron donors to complex IV). RPTC were suspended in the above buffer containing one of the substrates, transferred to the Qo2 chamber, and state 3 respiration was initiated by addition of 0.4 mM ADP.
ATP production rate
The assessment of ATP production at state 3 respiration (the maximum rate of ATP synthesis) was carried out by a modified method of Borkan et al. (12). In brief, the culture media were aspirated and replaced with 1 ml of a buffer solution resembling an intracellular electrolyte milieu (120 mM KCl, 5 mM KH2PO4, 10 mM HEPES, 1 mM MgSO4, and 2 mM EGTA, adjusted to pH 7.4 with KOH), containing digitonin (0.1 mg/ml) and 5 mM glutamate + 5 mM malate or 10 mM succinate + 0.1 μM rotenone as the substrates. The reaction was initiated by adding excess ADP (2 mM final concentration) and carried out for 5 min at 37°C. Initial experiments determined that ATP production in these conditions was linear for 10 min. The reaction was terminated by adding an aliquot of ice-cold perchloric acid (3% final concentration), and the suspension was snap-frozen in liquid nitrogen. Following thawing, the suspension was spun down at 15,000 g for 1 min at 4°C. The supernatant was neutralized to pH 7.5 and centrifuged again 15,000 g for 10 min at 4°C. The final supernatant was analyzed for ATP content by the luciferase method using an ATP Bioluminescence Assay Kit HS II (Roche) and the manufacturer's protocol. The initial pellet was assayed for protein content following solubilization in a buffer containing 100 mM Tris·HCl (pH 7.5), 150 mM NaCl, and 0.05% Triton X-100.
Mitochondrial membrane potential
Mitochondrial membrane potential (ΔΨm) was assessed as described previously (30) using JC-1, a cationic dye that exhibits potential-dependent accumulation and formation of red fluorescent J-aggregates in mitochondria, which is indicated by fluorescence emission shift from green (525 nm) to red (590 nm). At different time points of the recovery period, RPTC monolayers were loaded with 5 μM JC-1 for 30 min at 37°C. After being loaded, the monolayers were washed with ice-cold PBS (pH 7.4), scraped off culture dishes, washed again, and resuspended in PBS. Fluorescence was determined by flow cytometry (FACSCalibur, BD Biosciences) using excitation by a 488-nm argon-ion laser. The JC-1 monomer (green) and the J-aggregates (red) were detected separately in FL1 (emission, 525 nm) and FL2 (emission, 590 nm) channels, respectively. Ten-thousand events were counted for each sample. ΔΨm Is presented as the JC-1 red/green fluorescence intensity ratio.
Isolation of RPTC mitochondria
RPTC were homogenized in the ice-cold isolation buffer [225 mM mannitol, 10 mM HEPES (adjusted to pH 7.4 with KOH), 75 mM sucrose, 0.1 mM EGTA, and 0.1% BSA (fatty acid free)] using a Dounce homogenizer and centrifuged at 1,000 g for 5 min at 4°C. Supernatant was recentrifuged at 15,000 g for 15 min at 4°C. The pellet containing RPTC mitochondria was washed twice in the washing buffer (395 mM sucrose, 10 mM HEPES, 0.1 mM EGTA, adjusted to pH 7.4 with KOH) and spun down again at 15,000 g for 15 min at 4°C. The final mitochondrial pellet was resuspended in the Laemmli sample buffer (23) and used for immunoblot analysis or in 25 mM potassium phosphate buffer containing 5 mM MgCl2, pH 7.2, for direct assessment of the activities of the respiratory complexes.
Activity of mitochondrial respiratory complexes
Activities of respiratory complexes I and II were measured in isolated mitochondria according to the method of Birch-Machin et al. (10). Immediately following isolation, mitochondria were suspended in the hypotonic assay buffer (25 mM potassium phosphate buffer containing 5 mM MgCl2, pH 7.2) and freeze-thawed in liquid nitrogen. Complex I (NADH:Ubiquinone Oxidoreductase) activity was assayed spectrophotometrically by following the oxidation of NADH (0.25 mM) at 340 nm at 30°C in the assay buffer and in the presence of 62.5 μM ubiquinone, 0.25% BSA, antimycin A (2 μg/ml), and mitochondria. The decrease in absorbance (NADH oxidation) was recorded for 3 min, rotenone (10 μg/ml) was added, and the absorbance was recorded for another 2 min. Complex I activity was calculated as the rotenone-sensitive NADH:ubiquinone oxidoreductase activity. Complex II (succinate:ubiquinone oxidoreductase) activity was assayed spectrophotometrically at 590 nm by following the reduction of dichlorophenolindophenol (0.25 mM) at 30°C in the assay buffer and in the presence of succinate (20 mM), antimycin A (2 μg/ml), rotenone (10 μg/ml), 0.25% BSA, and ubiquinone (62.5 μM). The increase in absorbance (dichlorophenolindophenol oxidation) was recorded for 3 min. The results were corrected for changes in the absorbance in blank samples (containing no mitochondria) in all assays.
Pyruvate dehydrogenase complex activity was measured spectrophotometrically using a modified method of Hinman and Blass (18) by following the formation of NADH at 340 nm at 37°C. Isolated mitochondria were resuspended in the assay buffer (50 mM phosphate buffer, pH 7.4, containing 1 mM MgCl2, 0.2 mM EGTA, and 0.1% Triton X-100). The reaction mixture contained the assay buffer, 5 mM pyruvate, 2 mM NAD+, 0.2 mM thiamine pyrophosphate, 0.2 mM coenzyme A, 0.3 mM dithiotreitol, and mitochondria. Blank samples containing no pyruvate were included in all assays. The activity of the pyruvate dehydrogenase complex was expressed as nanomoles of NADH produced per minute per milligram of mitochondrial protein.
Aconitase activity was assessed by the method of Drapier and Hibbs (15). The mitochondrial pellet was resuspended in 50 mM Tris·HCl, pH 7.4, containing 0.2% Triton X-100, incubated for 5 min on ice, and centrifuged at 10,000 g for 5 min at 4°C. Aconitase activity was assayed immediately in 100 mM Tris·HCl buffer, pH 7.4, containing 1 mM MgCl2, 1 mM NADP, and 1 mM potassium citrate using the supernatant resulting from this centrifugation. The reaction was started by adding isocitrate dehydrogenase (2 U/ml), carried out at 37°C, and the absorbance (340 nm) was followed for 3 min. Blank samples containing no isocitrate dehydrogenase were included in all assays. The activity of aconitase was expressed as nanomoles of NADPH formed per minute per milligram of protein.
α-Ketoglutarate dehydrogenase complex activity was measured in isolated mitochondria according to the method of Tretter and Adam-Vizi (43) by following the formation of NADH at 340 nm at 37°C. Mitochondrial aliquots were added to a 50 mM phosphate buffer (pH 7.4) containing 1 mM MgCl2, 0.2 mM EGTA, 0.1% Triton X-100, 0.2 mM thiamine pyrophosphate, 2 mM NAD+, 1 mM α-ketoglutarate, and 0.4 mM ADP. The reaction was initiated by the addition of 0.2 mM coenzyme A. Blank samples containing no α-ketoglutarate were included in all assays. The activity of α-ketoglutarate dehydrogenase complex was expressed as nanomoles of NADH produced per minute per milligram of mitochondrial protein.
Malate dehydrogenase activity was measured in isolated mitochondria by following the oxidation of NADH at 340 nm at 30°C. The mitochondrial pellet was resuspended in 100 mM potassium phosphate buffer, pH 7.4, containing 0.14 mM NADH. The reaction was started by adding 0.3 mM oxaloacetate and the absorbance was followed for 3 min at 340 nm at 30°C. Blank samples containing no oxaloacetate were included in all assays. The activity of malate dehydrogenase was expressed as nanomoles of NADH oxidized per minute per milligram of mitochondrial protein.
Immunoblotting
Immunoblot analysis was used to determine the total and phosphorylated forms of ERK1/2 in RPTC homogenates and mitochondria. RPTC homogenates were lysed in the modified radio-immune precipitation assay buffer (50 mM Tris·HCl, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, 1 mM Na3VO4, 1 mM NaF, and the protease and phosphatase inhibitor cocktails; pH 7.4), incubated on ice for 10 min, and spun down at 100,000 g for 15 min at 4°C to pellet the detergent-insoluble fraction. The supernatant was combined with Laemmli sample buffer (23) and boiled as described previously (30). Proteins (20 μg/lane) were separated by SDS-PAGE and transferred electrophoretically to a nitrocellulose membrane. Blots were blocked for1hin Tris-buffered saline buffer containing 0.5% casein and 0.1% Tween 20 (blocking buffer) and incubated overnight at 4°C in the presence of primary antibodies diluted in the blocking buffer. Following being washed, the membranes were incubated with secondary IgG coupled to horseradish peroxidase and washed again. The super-signal chemiluminescent system was used for protein detection. Quantification of the results was performed using scanning densitometry.
Immunocytochemistry
RPTC mitochondria were visualized by staining alive RPTC monolayers (for 30 min at 37°C with shaking) with 75 nM MitoTracker Red 580 dissolved in culture media. Following staining, the monolayers were washed three times with ice-cold Tris-buffered saline (TBS), fixed with 3.7% formaldehyde, and immunocytochemical localization of phosphorylated-ERK1/2 in RPTC was performed. Fixed monolayers were permeabilized for 10 min using TBS containing 0.1% Triton X-100 and nonspecific binding was blocked for 1 h in TBS containing 10% horse serum. RPTC were washed with TBS and incubated with the fluorescein-conjugated anti-phospho ERK1/2 polyclonal antibody (5 μg/ml in TBS containing 1.5% horse serum) for 1 h at room temperature. Monolayers were washed three times with TBS, mounting media were added, and the monolayers were examined using a Zeiss LSM410 confocal laser-scanning microscope at a magnification of ×400.
Protein assay
Protein concentration was determined using the bicinchoninic acid assay with bovine serum albumin as the standard.
Statistical analysis
Data are presented as means ± SE and were analyzed for significance by ANOVA. Multiple means were compared using Fisher's protected least significance difference test with a level of significance of P < 0.05. RPTC isolated from an individual rabbit represented one experiment (n = 1) consisting of data obtained from two to three culture plates.
RESULTS
Effect of TBHP-induced injury on RPTC viability
Figure 1 shows that the 45-min treatment of RPTC with TBHP did not produce any significant amount of cell death. The number of apoptotic and necrotic cells in TBHP-treated monolayers was not different from controls (Fig. 1, A and B). These data show that TBHP exposure produced sublethal injury in RPTC. Inhibition of the ERK1/2 pathway had no effect on the viability of control and TBHP-treated cells at 4 h after injury. However, the presence of U0126 for 4 days in RPTC cultures decreased the levels of apoptosis in both controls and TBHP-treated cells (Fig. 1A). U0126 had no effect on RPTC necrosis (Fig. 1B). These data suggest that ERK1/2 is involved in basal apoptosis during normal cell turnover in our model.
Fig. 1.
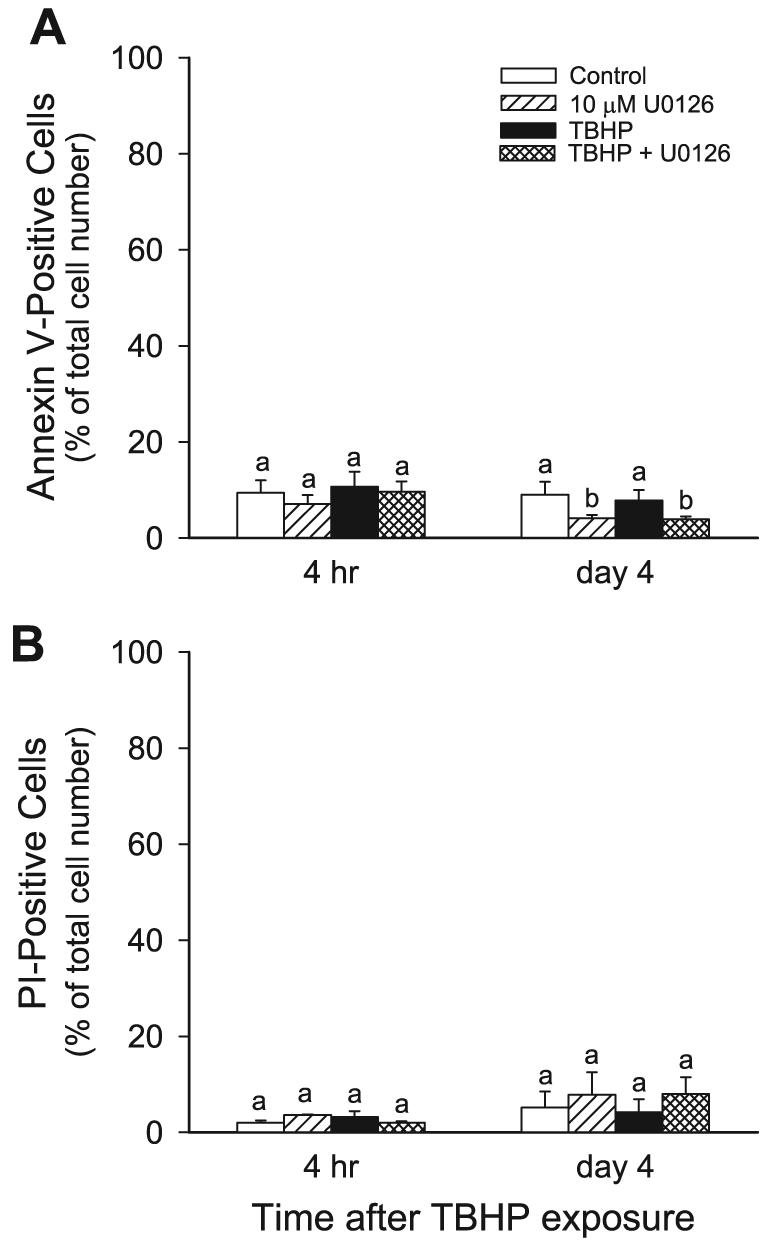
Effects of tert-butylhydroperoxide (TBHP)-induced injury and the inhibition of ERK1/2 activation on renal proximal tubular cells (RPTC) viability. The graphs show the percentage of annexin V-FITC-positive/propidium iodide (PI)-negative (apoptotic; A) and PI-positive/annexin V-FITC-negative (oncotic; B) RPTC as determined by flow cytometry. RPTC monolayers were incubated in the presence of 2 μg PI/ml and 125 ng annexin V-FITC/ml as described in materials and methods. PI and annexin V-FITC fluorescence was quantified using excitation at 488 nm and emission at 590 nm and 530 nm for PI and annexin V-FITC, respectively. For each sample, 10,000 events were counted. Results are average ± SE of 3 independent experiments (RPTC isolations). Values with dissimilar superscripts on a given day are significantly different (P < 0.05) from each other.
Effect of TBHP-induced injury on MEK1/2-ERK1/2 pathway in RPTC
As shown in Fig. 2A, treatment of RPTC with TBHP resulted in ERK1/2 phosphorylation (indicative of activation) within 10 min of the exposure. The protein levels of phosphor-ylated ERK1/2 increased at 10 min of TBHP exposure (Fig. 2A) and remained increased during the first 8 h of the recovery period (Fig. 2B). The ratio of phosphorylated ERK1/2 to total ERK1/2 protein in RPTC homogenates reached the maximum at 20 and 30 min of TBHP exposure (20-fold increase compared with controls) and returned to control levels at 24 h of the recovery period. The treatment of RPTC with U0126, a specific inhibitor of MEK1/2 (kinases upstream of ERK1/2), blocked the increases in ERK1/2 phosphorylation during and following TBHP exposure, whereas U0124, a negative control for U0126, had no effect on ERK1/2 phosphorylation in TBHP-injured RPTC (Fig. 2B).
Fig. 2.

A: protein levels of ERK1/2 and phosphorylated ERK in RPTC homogenates during the exposure to TBHP (0.35 mM). Confluent monolayers of RPTC were treated with TBHP and samples were taken at different time points during the oxidant exposure for immunoblot analysis. Controls were treated with the diluent (DMSO, 0.01%). No changes in ERK1/2 phosphorylation in control RPTC were observed during the experiment. B: protein levels of ERK1/2 and phosphorylated ERK in RPTC homogenates during the recovery following TBHP-induced injury. Some RPTC were treated with 10 μM U0126 or 10 μM U0124 before TBHP exposure and after each daily media change starting from the media change to remove TBHP. Controls were treated with the diluent (DMSO, 0.01%). No changes in ERK1/2 phosphorylation in control RPTC were observed during the experiment. C: protein levels of ERK1/2 and phosphorylated ERK1/2 in homogenates of noninjured (lanes 1-3) and TBHP-injured (lanes 4-5) RPTC infected with adenoviral vector carrying cDNA coding for constitutively active (Ca) MEK1. TBHP (0.35 mM) exposure and collection of RPTC samples were carried out at 48 h following adenoviral infection. The blots presented in Fig. 2 are representative of 3–4 independent experiments.
To activate ERK1/2 in RPTC, the cells were infected with adenoviral cDNA carrying cDNA coding for constitutively active MEK1, the kinase upstream of ERK1/2. This experimental approach increased the levels of phosphorylated (active) ERK1/2 in RPTC without any effect on total protein levels of ERK1/2 (Fig. 2). ERK1/2 phosphorylation in cells treated with TBHP and cells transfected with constitutively active MEK1 and treated with TBHP was similar indicating that TBHP exposure resulted in phosphorylation of most ERK1/2 present in RPTC. However, activation of ERK1/2 by TBHP was transient and subsided 8 h after the exposure, whereas activation of ERK1/2 by expression of constitutively active MEK1 was continuous.
Thus these data suggest that ERK1/2 is activated during TBHP exposure and the early recovery period following TBHP exposure in RPTC. The results also show that ERK1/2 is activated in the MEK1/2-dependent pathway.
Mitochondrial MEK1/2-ERK1/2 pathway during RPTC repair following TBHP-induced injury
Our previous reports demonstrated that PKC-α and PKC-ε are present in RPTC mitochondria (25, 30, 35). Using both immunoblotting and immunocytochemistry, we show here that ERK1/2 is localized and phosphorylated in RPTC mitochondria following TBHP exposure (Figs. 3 and 4). Figures 2 and 4 show low levels of phosphorylated ERK1/2 in control RPTC and RPTC treated with 10 μM U0126. Exposure of RPTC to TBHP increased the levels of phosphorylated ERK1/2 (Figs. 2 and 4). Dual staining of RPTC using red fluorescing dye MitoTracker Red 580 that accumulates specifically in the mitochondria and the antibody specific for phosphorylated ERK1/2 show that some of phosphorylated ERK1/2 in these cells colocalized in mitochondria resulting in a yellow and orange fluorescent signals (Fig. 4C). These data are consistent with the results obtained using immunoblotting that show ERK1/2 presence in RPTC mitochondria and phosphorylation of ERK1/2 in mitochondria of TBHP-injured RPTC (Fig. 3). Colocalization of phosphoERK1/2 and mitochondria was particularly prominent in the perinuclear area of TBHP-injured RPTC (Fig. 4C). The phosphorylation of ERK1/2 in mitochondria of oxidant-treated RPTC was blocked by MEK1/2 inhibitor U0126 (Fig. 4D).
Fig. 3.

Protein levels and phosphorylation of MEK1/2, ERK1/2, and p38 in RPTC mitochondria during the recovery following TBHP-induced injury. Some RPTC were treated with 10 μM U0126 before TBHP exposure and after each daily media change starting from the media change to remove TBHP. Controls were treated with the diluent (DMSO, 0.01%). No changes in ERK1/2 phosphorylation in control RPTC mitochondria were observed during the experiment (data not shown). The blots presented in Fig. 3 are representative of 3–4 independent experiments.
Fig. 4.

Subcellular localization of phosphorylated ERK1/2 in RPTC. RPTC mitochondria were visualized by staining with MitoTracker Red 580 (red fluorescence) and followed by immunocytochemistry using phospho-ERK1/2 antibody conjugated to fluorescein (green fluorescence). Yellow/orange fluorescence (overlay of red and green) indicates mitochondrial localization of phosphorylated ERK1/2 in RPTC. A: control RPTC. B: RPTC treated with 10 μM U0126. C: RPTC treated with 0.35 mM TBHP (end of the exposure). D: RPTC treated with 10 μM U0126 and 0.35 mM TBHP (end of the exposure). The images are representative of 3 independent experiments.
This is the first report demonstrating the presence of ERK1/2 in RPTC mitochondria. The levels of phosphorylated ERK1/2 in RPTC mitochondria increased starting at 1 h of the recovery following TBHP exposure and reached a maximum at 8 h of the recovery period. (Figs. 3 and 4). Because the levels of the total ERK1/2 protein did not change in RPTC mitochondria, the phosphorylation (indicative of activation) of ERK1/2 in mitochondria was not due to the translocation of active ERK1/2 from the cytosol (Fig. 3). These events were preceded by phosphorylation (indicative of activation) of mitochondrial MEK1/2 (within 10 min) and p38 (within 5 min) of TBHP exposure (Fig. 3). Treatment of RPTC with U0126 before TBHP exposure blocked mitochondrial ERK1/2 but not p38 phosphorylation (Fig. 3). U0124 had no effect on ERK1/2 phosphorylation in RPTC mitochondria (data not shown). Thus these results show that 1) MEK1/2, ERK1/2, and p38 are present in RPTC mitochondria, 2) oxidant exposure activates mitochondrial MEK1/2, ERK1/2, and p38, and 3) ERK1/2 but not p38 is phosphorylated (activated) in RTPC mitochondria in the MEK1/2-dependent pathway.
Qo2
Basal Qo2 was used as a marker of the overall respiration in RPTC. Basal Qo2 decreased to 53% of control levels at the end of TBHP exposure (45 min), remained at similar level at 4 h, and increased to 81% of controls on day 4 following the injury (Figs. 5 and 6A). TBHP-induced decreases in basal Qo2 followed ERK1/2 phosphorylation, which was indicative of ERK1/2 activation (Figs. 2 and 3). MEK1/2 inhibitor, U0126, was used to test whether the ERK1/2 pathway plays a role in oxidant-induced decreases in RPTC respiration. Inhibition of ERK1/2 activation by U0126 reduced the decreases in basal Qo2 at 4 h to 81% of the control level and promoted full recovery of basal Qo2 in TBHP-injured RPTC on day 4 (Fig. 6A). Treatment of RPTC with U0124, a negative control for U0126, had no effect on the recovery of basal Qo2 in TBHP-injured RPTC (data not shown). Inhibition of ERK1/2 activation using other pharmacological MEK1/2 inhibitors (25 μM PD98059 and 5 μM SL237) also protected against the decreases in basal Qo2 (Fig. 6B). However, these inhibitors were not as effective as U0126 (Fig. 6A). In contrast, activation of ERK1/2 by expressing constitutively active MEK1 in RPTC decreased basal Qo2 by 78% at 48 h following the transfection (Fig. 6C). Furthermore, expression of constitutively active MEK1 exacerbated TBHP-induced decreases in basal Qo2 (Fig. 6C). Basal Qo2 was not altered in RPTC infected with adenovirus carrying an empty vector (18.8 ± 2.6 vs. 17.7 ± 0.5 nmol O2·mg protein−1·min−1 in controls vs. RPTC transfected with the empty vector, respectively). Infection with adenovirus carrying an empty vector had no effect on TBHP-induced decreases in basal Qo2 (data not shown).
Fig. 5.
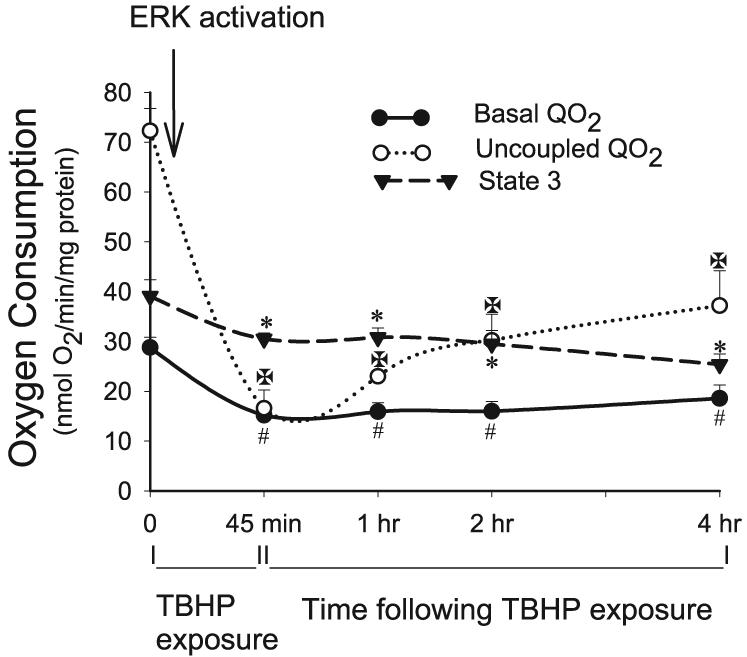
Basal oxygen consumption (Qo2), a marker of overall mitochondrial function (depicted by ● and solid line), uncoupled Qo2, a marker of electron transport rate through the respiratory chain (depicted by ○ and dotted line), and state 3 respiration, the maximum respiratory activity of mitochondria (depicted by ▼ and dashed line), at the end (45 min) of TBHP (0.35 mM) exposure and during 4 h following TBHP removal. Uncoupled Qo2 was measured in the presence of carbonyl cyanide p-(trifluoro-methoxy)phenylhydrazone (FCCP; 2 μM). State 3 respiration was measured in a buffer (120 mM KCl, 5 mM KH2PO4, 10 mM HEPES, 1 mM MgSO4, and 2 mM EGTA, pH 7.4) containing digitonin (0.1 mg/ml), 5 mM glutamate + 5 mM malate (electron donors to complex I), and in the presence of 0.4 mM ADP. Results are average ± SE of 6 independent experiments. #,*Significantly different (P < 0.05) from controls (time 0).
Fig. 6.

Effect of inhibition and activation of the MEK1/2-ERK1/2 pathway on the recovery of Qo2 following TBHP-induced injury in RPTC. A and B: effect of inhibition of MEK1/2-ERK1/2 pathway on the recovery of basal Qo2. C: effect of activation of MEK1/2-ERK1/2 pathway on basal Qo2 in noninjured and TBHP-injured RPTC. D and E: effect of inhibition of MEK1/2-ERK1/2 pathway on the recovery of uncoupled Qo2. F: effect of activation of MEK1/2-ERK1/2 pathway on uncoupled Qo2 in noninjured and TBHP-injured RPTC. To inhibit ERK1/2 activation, RPTC were treated with MEK1/2 inhibitors: 10 μM U0126 (A and D), 25 μM PD98059 (B and E), and 5 μM SL237 (B and E) before TBHP exposure and after each daily media change starting from the media change to remove TBHP. To increase ERK1/2 activation, RPTC were infected with adenovirus carrying constitutively active MEK1 2 days before TBHP exposure (C and F). Results are average ± SE of 3–8 independent experiments (RPTC isolations). Values with dissimilar superscripts at a given time point are significantly different (P < 0.05) from each other.
Uncoupled Qo2 was used as a marker of the mitochondrial electron transfer rate and the integrity of respiratory complexes. Uncoupled Qo2 decreased to 23% of control levels at the end of TBHP exposure in RPTC, increased to 51% of controls at 4 h following the exposure, and returned to control levels on day 4 of the recovery period (Figs. 5 and 6D). The inhibition of ERK1/2 activation by U0126 reduced the decreases in uncoupled Qo2 at 4 h to 73% of controls (Fig. 6D). Treatment of RPTC with U0124, a negative control for U0126, had no effect on uncoupled Qo2 and recovery of mitochondrial function in TBHP-injured RPTC (data not shown). Inhibition of MEK1/2 and ERK1/2 activation by PD98059 and 5 μM SL237 reduced the decreases in uncoupled Qo2 (Fig. 6E), but these inhibitors were less effective than U0126 (Fig. 6D). Activation of ERK1/2 by expressing constitutively active MEK1 decreased uncoupled Qo2 in RPTC by 92% at 48 h following the transfection (Fig. 6F). TBHP had no further effect on uncoupled Qo2 in RPTC expressing active ERK1/2 (Fig. 6F). Uncoupled Qo2 was not altered in RPTC infected with adenovirus carrying an empty vector (47.5 ± 3.6 vs. 44.5 ± 2.1 nmol O2·mg protein−1·min−1 in controls vs. RPTC transfected with the empty vector, respectively). Infection with adenovirus carrying an empty vector had no effect on TBHP-induced decreases in uncoupled Qo2 (data not shown).
These results show that the activation of ERK1/2 plays a role in mitochondrial dysfunction induced by TBHP-induced injury in RPTC. The inhibition of ERK1/2 activation decreases mitochondrial dysfunction and promotes the recovery of mitochondrial function following the injury. In contrast, constitutive activation of ERK1/2 results in a marked decrease in RPTC Qo2 and potentiates TBHP-induced decreases in RPTC respiration. Furthermore, these data suggest that the electron transport chain is a target for ERK1/2.
State 3 respiration
Because the data suggested that a component(s) of the respiratory chain is the ultimate target of the ERK1/2 pathway, we examined whether state 3 respiration coupled to respiratory complexes I, II, and/or IV is under control of ERK1/2. Complex I-coupled state 3 respiration (using glutamate + malate as electron donors) gradually decreased after TBHP exposure and was 43% lower than controls at 4 h following TBHP-induced injury (Figs. 5 and 7A). Complex I-coupled respiration recovered on day 4 following the injury (Fig. 7A). Inhibition of ERK1/2 (using 3 different MEK1/2 inhibitors: U0126, PD98059, and SL237) had no effect on complex I-coupled state 3 respiration in noninjured RPTC but totally prevented the decreases in complex I-coupled state 3 respiration due to TBHP injury (Fig. 7, A and B). Similar results were obtained using other electron donors (citrate, α-ketoglutarate) to the respiratory complex I (Table 1). Treatment with U0124 (that does not block the MEK1/2) did not affect TBHP-induced changes in state 3 respiration supported by substrates coupled to complex I (data not shown). In contrast, ERK1/2 activation decreased complex I-coupled state 3 respiration in noninjured RPTC by 56% at 48 h following infection but had no further effect on TBHP-induced decreases in complex I-coupled state 3 respiration (Fig. 7C). Complex I-coupled state 3 respiration was not changed by infection of RPTC with adenovirus carrying an empty vector (33.1 ± 2.7 vs. 34.7 ± 2.6 nmol O2·mg protein−1·min−1 in controls vs. RPTC transfected with the empty vector, respectively). Likewise, infection with adenovirus carrying an empty vector had no effect on TBHP-induced decreases in complex I-coupled state 3 respiration (data not shown).
Fig. 7.

Effect of inhibition and activation of the MEK1/2-ERK1/2 pathway on the recovery of state 3 respiration following TBHP-induced injury in RPTC. RPTC were treated with TBHP (0.35 mM, 45 min) and samples were taken at different time points of the recovery period to assess state 3 respiration. Controls were treated with the diluent (DMSO, 0.01%). To inhibit ERK1/2 activation, RPTC were treated with MEK1/2 inhibitors: 10 μM U0126 (A, D, G), 25 μM PD98059 (B and E), and 5 μM SL237 (B and E) before TBHP exposure and after each daily media change starting from the media change to remove TBHP. To activate ERK1/2, RPTC were infected with adenovirus carrying constitutively active MEK1 2 days before TBHP exposure (C and F). A, B, C: state 3 respiration coupled to complex I. D, E, F: state 3 respiration coupled to complex II. G: state 3 respiration coupled to complex IV. Results are average ± SE of 3–6 independent experiments (RPTC isolations). Values with dissimilar superscripts at a given time point are significantly different (P < 0.05) from each other.
Table 1.
Effect of inhibition of ERK1/2 activation on state 3 respiration during recovery period following TBHP-induced injury in RPTC
| Respiratory Substrate |
||||
|---|---|---|---|---|
| α-Ketoglutarate |
Citrate |
|||
| Treatment | 4 h | day 4 | 4 h | day 4 |
| Control | 26.1±2.1 | 26.6±0.2 | 26.5±3.3 | 24.7±4.3 |
| U0126 | 26.2±2.1 | 28.5±2.4 | 24.9±1.9 | 26.3±3.6 |
| TBHP | 18.2±2.4* | 27.2±4.2 | 18.4±1.7* | 22.8±3.4 |
| TBHP + U0126 | 26.1±5.2 | 25.0±3.7 | 25.2±2.0 | 26.5±2.3 |
Results are average ± SE of 7–8 independent experiments.
Significantly different (P < 0.05) from controls (time 0). Units measured as nmol O2·min−1·mg protein−1. Renal proximal tubular cells (RPTC) were cultured until confluent, treated with 0.35 mM TBHP for 45 min in the absence or presence of U0126 (MEK 1/2 inhibitor, 10 μM), washed with fresh, warm culture medium to remove the oxidant. U0126 was present in the media during 4 days of the recovery period. Controls were treated with the diluent (DMSO). Samples were taken at 4 h and 4 days following TBHP exposure to assess state 3 respiration (the maximum respiratory activity of mitochondria in the presence of excess ADP and substrates). State 3 respiration was measured in a buffer (120 mM KCl, 5 mM KH2PO4, 10 mM HEPES, 1 mM MgSO2, and 2 mM EGTA, pH 7.4) containing digitonin (0.1 mg/ml), 10 mM α-ketoglutarate, or 10 mM citrate (electron donors to complex I), and 0.4 mM ADP.
TBHP-induced injury had no effect on state 3 respiration coupled to the complexes II and IV (Fig. 7, D, E, and G). Furthermore, inhibition of the ERK1/2 by U0126 had no effect on state 3 respiration coupled to complexes II and IV (Fig. 7, D, E, and G). However, continuous activation of ERK1/2 by transfecting RPTC with constitutively active MEK1 decreased complex II-coupled state 3 respiration by 34% in noninjured and 61% in TBHP-injured RPTC (Fig. 7F). Complex II-coupled state 3 respiration was not changed by infection of RPTC with adenovirus carrying an empty vector (59.8 ± 7.7 vs. 54.2 ± 6.3 nmol O2·mg protein−1·min−1 in controls vs. RPTC transfected with the empty vector, respectively). Likewise, infection with adenovirus carrying an empty vector had no effect on TBHP-induced decreases in complex I-coupled state 3 respiration (data not shown).
These results demonstrate that transient activation of the ERK1/2 pathway by TBHP decreases state 3 respiration coupled to the respiratory complex I and that the inhibition of this pathway restores state 3 respiration in TBHP-injured RPTC. Continuous activation of ERK1/2 results in significant reduction of state 3 respiration coupled to both complex I and complex II resulting in decreased flow of electrons through all complexes of the respiratory chain.
ATP production
ATP synthesis is the fundamental function of the mitochondria and provides the energy for reabsorptive processes in RPTC. The rate of ATP synthesis was measured during state 3 (the maximal capacity of mitochondria to generate ATP) in digitonin-permeabilized RPTC incubated in the presence of excess ADP and metabolic substrates linked to the respiratory complex I (glutamate + malate) and the respiratory complex II (succinate). In these conditions, ATP was not utilized by active transports driven by the plasma membrane-associated ATPases. The rate of ATP production in the presence of electron donors linked to complex I was 40.0 ± 2.6 nmol·mg protein−1·min−1 in control RPTC. TBHP exposure decreased complex I-linked ATP production to 60% of the control levels (Fig. 8A). Inhibition of TBHP-induced activation of ERK1/2 prevented decreases in ATP production (Fig. 8A). Complex I-linked ATP synthesis returned on day 4 following the oxidant exposure regardless of the presence or absence of U0126. TBHP-induced injury had no effect on ATP synthesis supported by complex II-linked substrate (succinate) and the inhibition of ERK1/2 had no effect on this function (Fig. 8B). U0124 did not affect ATP production in TBHP-injured RPTC regardless of the substrate used as the electron donor (data not shown). These results demonstrate that ERK1/2 mediates TBHP-induced decreases in ATP production from oxidative substrates coupled to the respiratory complex I. Furthermore, inhibition of ERK1/2 activation protects against oxidant-induced decreases in ATP production.
Fig. 8.
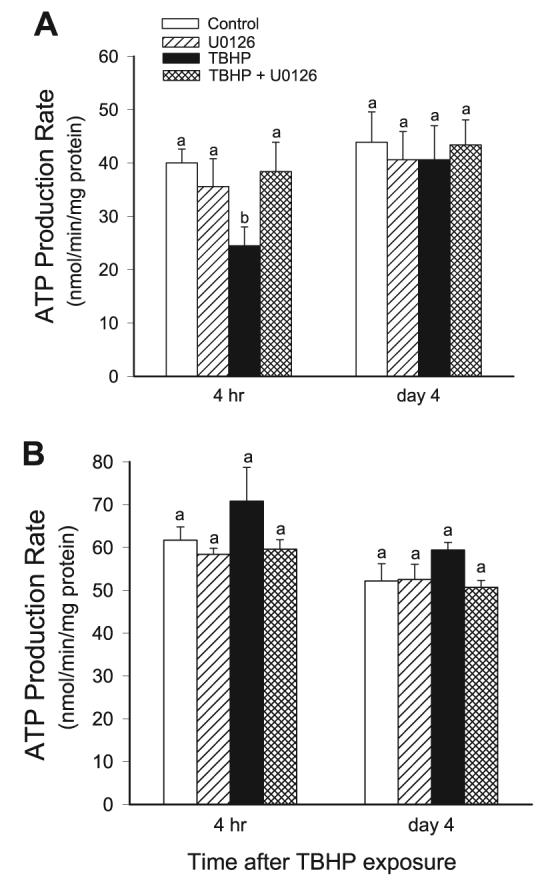
Effect of inhibition of the MEK-ERK1/2 pathway on the recovery of ATP production rate following TBHP-induced injury in RPTC. RPTC were treated with TBHP (0.35 mM, 45 min), and samples were taken at different time points of the recovery period to assess ATP production rate. Controls were treated with the diluent (DMSO, 0.01%). To inhibit ERK1/2 activation, RPTC were treated with 10 μM U0126 before TBHP exposure and after each daily media change starting from the media change to remove TBHP. ATP production rate was measured at 37°C in a buffer (120 mM KCl, 5 mM KH2PO4, 10 mM HEPES, 1 mM MgSO4, and 2 mM EGTA, pH 7.4) containing digitonin (0.1 mg/ml), 5 mM glutamate, and 5 mM malate (electron donors to complex I) or 10 mM succinate (the electron donor to complex II) + 0.1 μM rotenone, and 2.0 mM ADP. A: ATP production coupled to complex I (using glutamate and malate as substrates). B: ATP production coupled to complex II (using succinate as a substrate). Results are average ± SE of 5 independent experiments (RPTC isolations). Values with dissimilar superscripts at a given time point are significantly different (P < 0.05) from each other.
Activities of the respiratory complexes
The activities of NADH:ubiquinone oxidoreductase (the respiratory complex I) and succinate:ubiquinone oxidoreductase (the respiratory complex II) in mitochondria isolated from RPTC were examined to determine whether ERK1/2 mediates TBHP-induced mitochondrial dysfunction by targeting either of these complexes. Figure 9A shows that TBHP-induced injury decreased the activity of complex I to 58% of control levels at 4 h following the exposure. The activity of complex I returned on day 4 of the recovery period (Fig. 9A). In contrast, the activity of complex II was unaffected by TBHP-induced injury (Fig. 9B). Inhibition of ERK1/2 activation by U0126 prevented TBHP-induced decreases in complex I activity and had no effect on the activity of complex II (Fig. 9, A and B). These results suggest that the activation of ERK1/2 mediates the dysfunction of complex I in oxidant-injured RPTC and that the inhibition of ERK1/2 activation maintains the function of complex I in RPTC mitochondria.
Fig. 9.
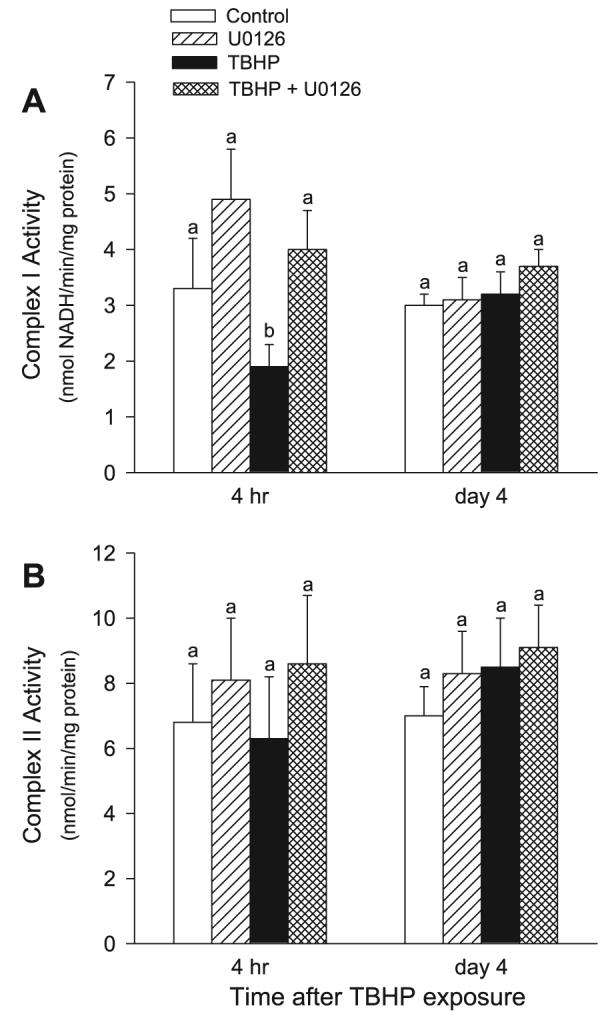
Effect of inhibition of the MEK1/2-ERK1/2 pathway on the recovery of activities of the NADH:Ubiquinone Oxidoreductase (the respiratory complex I; A) and the Succinate:Ubiquione Oxidoreductase (the respiratory complex II; B) in isolated mitochondria following TBHP-induced injury in RPTC. RPTC were treated with TBHP (0.35 mM, 45 min) and samples were taken at different time points of the recovery period to assess the activities of the respiratory complex I and complex II. Controls were treated with the diluent (DMSO, 0.01%). To inhibit ERK1/2 activation, RPTC were treated with 10 μM U0126 before TBHP exposure and after each daily media change starting from the media change to remove TBHP. Mitochondria were isolated from RPTC at different time points following TBHP-induced injury and the activities of complex I and complex II were determined spectrophotometrically at 30°C as described in materials and methods. Results are average ± SE of 5 independent experiments (RPTC isolations). Values with dissimilar superscripts at a given time point are significantly different (P < 0.05) from each other.
Activities of the citric acid cycle dehydrogenases
The rate of electron transport through the respiratory chain is determined by the integrity and activity of the respiratory complexes and the availability of mobile electron carriers (ubiquinone and cytochrome c) and electron donors (NADH and FADH2). The citric acid cycle is a major source of NADH and FADH2 in RPTC, which generate most of their ATP by mitochondrial oxidative phosphorylation. The decrease in state 3 respiration and ATP production coupled to the respiratory complex I in TBHP-injured RPTC could be due to both, decreased activity of complex I and reduced activity of the dehydrogenases of the citric acid cycle. Therefore, in addition to testing the activity of respiratory complexes I and II, we examined the activity of the enzymes that catalyze generation of NADH (the substrate for respiratory complex I) by the oxidative decarboxylation of pyruvate and the citric acid cycle. In addition, we tested the activity of aconitase, a known target of oxidative stress within the citric acid cycle. Figure 10, A, C, and D, shows that the sublethal injury induced by TBHP had no effect on the activities of the pyruvate dehydrogenase complex, the α-ketoglutarate dehydrogenase complex, and the malate dehydrogenase. Inhibition of ERK1/2 activation by U0126 had no effect on the activity of these enzymes in injured and noninjured RPTC (Fig. 10, A, C, and D). In contrast, aconitase activity was decreased by 30% in TBHP-injured RPTC (Fig. 10B). However, inhibition of ERK1/2 activation by U0126 did not prevent this decrease (Fig. 10B). These data show that ERK1/2 does not induce mitochondrial dysfunction in oxidant-injured RPTC through decreasing activities of the major regulatory enzymes of the citric acid cycle and reducing the production of electron donors to mitochondrial respiratory complexes.
Fig. 10.

Effect of TBHP-induced injury and inhibition of the MEK1/2-ERK1/2 pathway on the activities of pyruvate dehydrogenase (A), aconitase (B), α-ketoglutarate dehydrogenase (C), and malate dehydrogenase (D) in RPTC mitochondria at 4 h following the injury. RPTC were treated with TBHP (0.35 mM, 45 min), and samples were taken at 4 h of the recovery period to assess activities of pyruvate dehydrogenase, aconitase, α-ketoglutarate dehydrogenase, and malate dehydrogenase. Controls were treated with the diluent (DMSO, 0.01%). To inhibit ERK1/2 activation, some RPTC were treated with 10 μM U0126 before TBHP exposure and after each daily media change starting from the media change to remove TBHP. Mitochondria were isolated from RPTC at 4 h following TBHP-induced injury and enzymatic activities were determined spectrophotometrically as described in materials and methods. Results are average ± SE of 5 independent experiments (RPTC isolations). Values with dissimilar superscripts at a given time point are significantly different (P < 0.05) from each other.
Δ ψ m
Electron transport through the respiratory chain results in the generation of proton and pH gradients across the inner mitochondrial membrane. These reactions produce the transmembrane potential (Δψm), which represents most of the energy of the proton gradient. Δψm in RPTC was assessed by the measurement of changes in the J-aggregate/JC-1 monomer ratio. The J-aggregate/JC-1 monomer ratio in control RPTC was 1.30 ± 0.25. The J-aggregate/ JC-1 monomer ratio was not altered at 2 h (0.93 ± 0.28 vs. 1.03 ± 0.09 in TBHP-injured and control RPTC, respectively) and 4 h following TBHP injury (Fig. 11). These data suggest that the sublethal injury due to TBHP exposure did not induce the mitochondrial membrane permeability transition in RPTC until 24 h following TBHP exposure. At 24 and 48 h after the injury, the J-aggregate/JC-1 monomer ratio decreased to 33 and 26% of control levels (Fig. 11, data not shown). On day 4 of the recovery period, Δψm returned to the control levels (Fig. 11). Inhibition of ERK1/2 activation in TBHP-injured RPTC using U0126 and PD98059 reduced the decreases in Δψm at 24 after the oxidant exposure, whereas U0124 had no effect on TBHP-induced decreases in Δψm (Fig. 11, data not shown). Thus these data show that ERK1/2 activation mediates oxidant-induced decreases in the mitochondrial membrane potential in RPTC and that the inhibition of ERK1/2 activation maintains Δψm in oxidant-injured RPTC. The delay between decreases in the activity of complex I, state 3 respiration coupled to complex I, and the decreases in Δψm suggest that 1) the decrease in Δψm in sublethally injured RPTC was due to decreased activity of the respiratory chain but not the mitochondrial permeability transition and 2) the decreases in Δψm due to reduced activity of the respiratory chain develop with a delay.
Fig. 11.

Effect of inhibition of the MEK1/2-ERK1/2 pathway on the recovery of the mitochondrial membrane potential (Δψm) following TBHP-induced injury in RPTC. RPTC were treated with TBHP (0.35 mM, 45 min) and samples were taken at different time points of the recovery period to assess Δψm. Controls were treated with the diluent (DMSO, 0.01%). To inhibit ERK1/2 activation, RPTC were treated with MEK1/2 inhibitors, 10 μM U0126 (A) or 25 μM PD98059 (B) before TBHP exposure and after each daily media change starting from the media change to remove TBHP. Δψm Was assessed using 5 μM JC-1 as described in materials and methods. Fluorescence was determined by flow cytometry using excitation by a 488-nm argon-ion laser. The JC-1 monomer (green) and the J-aggregates (red) were detected separately in FL1 (emission, 525 nm) and FL2 (emission, 590 nm) channels, respectively. Ten thousand events were counted for each sample. Δψm Is presented as the JC-1 red/green fluorescence intensity ratio. Results are average ± SE of 4 independent experiments (RPTC isolations). Values with dissimilar superscripts at a given time point are significantly different (P < 0.05) from each other.
Does oxidant injury activate ERK1/2 in the PKC-ε-dependent pathway?
Previously, we showed that TBHP activates PKC-ε in RPTC and that PKC-ε mediates mitochondrial dysfunction induced by this toxicant. Furthermore, our data suggested that PKC-ε targets the function of mitochondrial complex I- and complex I-coupled ATP production. Because present data show that complex I also is a target of ERK1/2 in oxidant-injured RPTC, we tested whether ERK1/2 is a downstream target of PKC-ε. Figures 2, 3, and 12A show that ERK1/2 activation in TBHP-treated RPTC and in mitochondria occurred before the activation of PKC-ε (2 h following TBHP exposure). Furthermore, PKC-εV1–2 inhibitor (20 μM) blocked the translocation of PKC-ε to mitochondria in TBHP-injured RPTC but did not prevent the activation of ERK1/2 in the whole RPTC and in mitochondria (Fig. 12A). Therefore, ERK1/2 activation in TBHP-injured RPTC occurred in PKC-ε-independent manner.
Fig. 12.

A: phosphorylation of PKC-ε and ERK1/2 in RPTC mitochondria during (0.5 and 1 h) and following (1, 2, and 4 h) TBHP exposure (left). Lack of effect of PKC-ε inhibition on ERK1/2 activation in RPTC mitochondria during (0.5 and 1 h) and following (1, 2, and 4 h) TBHP exposure (right). PKC-εV1–2 inhibitor was added 1 h before TBHP exposure and every day with the daily media change. B: phosphorylation of protein kinase B/Akt during recovery following TBHP (0.35 mM) exposure in the absence (left) and the presence of MEK1/2 inhibitor 10 μM U0126 (right). The blots are representative of 3–5 independent experiments.
Is Akt involved in protective effects of MEK1/2 inhibitor?
Protein kinase B/Akt is a serine/threonine kinase involved in cell survival in many cell types. We tested whether protective effects of U0126 and the inhibition of ERK1/2 pathway against oxidant-induced injury were associated with increased activation of Akt. Figure 12B shows that TBHP exposure increased phosphorylation of Akt, which suggested Akt activation during and following oxidant-induced injury. Inhibition of ERK1/2 activation by U0126 neither increased nor decreased Akt phosphorylation, suggesting that the protective effects of MEK1/2 inhibitor were not due to modulation of Akt activation.
Finally, we tested whether more severe oxidant injury (500 μM TBHP) alters the balance between different kinases of the MAPK family. Exposure to 0.5 mM TBHP resulted in 70% decrease in RPTC respiration and extensive necrosis (∼60%; data not shown). Mitochondrial function did not recover following injury induced by 0.5 mM TBHP (data not shown). The presence of U0126 had no significant effect on mitochondrial dysfunction and cell death induced by 0.5 mM TBHP (data not shown). Figure 13 shows that in contrast to sublethal injury caused by 0.35 mM TBHP, the exposure of RPTC to 0.5 mM TBHP resulted in phosphorylation (indicative of activation) of JNK (54 kDa only). Furthermore, in contrast to sublethal injury which induced Akt phosphorylation, severe oxidant injury blocked Akt phosphorylation (Fig. 13). Phosphorylation of p38 kinase was equivalent in both models of oxidant injury (Fig. 13). U0126 had no effect on p38 phosphorylation and did not restore Akt phosphorylation but reduced JNK phosphorylation in RPTC treated with 0.5 mM TBHP (Fig. 13). These data suggest that severe oxidant injury in RPTC activates protein kinases involved in stress signaling (JNK) and inhibits activation of protein kinases involved in cell survival (Akt).
Fig. 13.

Phosphorylation of ERK1/2, JNK1/2, p38, and protein kinase B/Akt during TBHP exposure and recovery of RPTC following TBHP exposure. Left: sublethal injury (mitochondrial dysfunction without cell death) induced by 0.35 mM TBHP. Middle: lethal injury (mitochondrial dysfunction followed by 70% cell death) induced by 0.50 mM TBHP. Right: effect of MEK1/2 inhibitor, 10 μM U0126, on ERK1/2, JNK1/2, p38, and protein kinase B/Akt phosphorylation following exposure to 0.5 mM TBHP. TBHP exposure had no effect on total protein levels of protein kinases shown in this figure. The blots are representative of 3 independent experiments.
DISCUSSION
Our study suggests that an oxidant exposure resulting in sublethal injury activates the MEK1/2-ERK1/2 pathway in RPTC and that the activation of this pathway precedes mitochondrial dysfunction (decreases in basal, uncoupled and state 3 respirations, and ATP production). In contrast to studies carried out in proximal tubular cell lines grown in the presence of glucose (5), sublethal oxidant injury in RPTC in primary culture is not associated with activation of JNK. Subsiding ERK1/2 activation is followed by the recovery of mitochondrial functions. Our data also show that oxidant exposure activates ERK1/2 in RPTC mitochondria. The present study is the first report demonstrating the presence of ERK1/2 in the renal proximal tubular mitochondria and activation of mitochondrial ERK1/2 by an oxidant-induced injury. Furthermore, our data show that MEK1/2 is present in RPTC mitochondria and is phosphorylated (hence activated) before ERK1/2 phosphorylation. Previously, we reported mitochondrial activation of PKC-ε in TBHP-injured RPTC, which was due to the translocation of phosphorylated PKC-ε to RPTC mitochondria (35). In contrast to PKC-ε, the mitochondrial activation of ERK1/2 in TBHP-injured RPTC is not due to the translocation of active ERK1/2 but phosphorylation of ERK1/2 within the mitochondria. This suggests that mitochondrial MEK1/2 activates mitochondrial ERK1/2.
The role of ERK1/2 in the regulation of mitochondrial function and in mediating toxicant effects on mitochondrial function has not been explored. In cardiomyocytes, phosphorylation of ERK1/2 is the signaling event downstream of the generation of mitochondrial reactive oxygen species (45). Mitochondria are capable of signaling the impairment of electron transport through the respiratory chain, particularly through complex I, by the modules of the MAPK pathway such as Raf-1 and MEKK-1 (13). However, in our study, the activation of ERK1/2 preceded mitochondrial dysfunction in TBHP-injured RPTC. Therefore, the activation of MEK1/2-ERK1/2 pathway appears to be downstream of the oxidative stress but upstream of mitochondrial dysfunction. The decreases in state 3 respiration demonstrating dysfunction of the respiratory chain did not occur until 2 h following oxidant removal and followed both the early activation of ERK1/2 in the whole RPTC (within 10 min of TBHP exposure) and later activation of ERK1/2 in the mitochondrial (within 1 h following oxidant removal). Furthermore, the mitochondrial membrane potential did not change during the first 4 h after oxidant exposure. Therefore, it is unlikely that the activation of ERK1/2 in RPTC was due to impairment of the respiratory chain or the mitochondrial permeability transition and generation of the reactive oxygen species by mitochondria. Inhibition of ERK1/2 activation by U0126 reduced TBHP-induced decreases in basal and uncoupled respiration and prevented decreases in state 3 respiration and ΔΨm. Thus we conclude that the initial early activation of ERK1/2 was not caused by reactive oxygen species generated by mitochondria. However, a secondary role of the mitochondrial reactive oxygen species in maintaining ERK1/2 in the active state at later time points following injury cannot be ruled out.
Elucidating a function of a specific kinase or a signaling pathway requires selective tools to inhibit the kinase under study. To inhibit the activation of ERK1/2 in TBHP-injured RPTC, we used U0126, one of the most specific and widely used inhibitors of MEK1/2, a kinase immediately upstream of ERK1/2. U0126 had no adverse effects on any tested mitochondrial function in control RPTC. To test whether the protective effects of U0126 were due to nonspecific effects unrelated to MEK1/2 inhibition, we used U0124, a negative control for U0126 that does not inhibit MEK1/2 and subsequent ERK1/2 activation. In contrast to U0126, U0124 did not affect the decreases in basal respiration, electron transport rate, complex I-coupled respiration, and ATP production following oxidant injury. Furthermore, two other MEK1/2 inhibitors (PD98059 and SL237) also reduced the oxidant-induced decreases in mitochondrial function in RPTC. Therefore, we concluded that protective effects of U0126 were due to the inhibition of the ERK1/2 pathway and not a nonspecific effect of this inhibitor. Because U0126 directly inhibits MEK1/2 and, indirectly, the activation of ERK1/2, the effects of TBHP-induced injury on mitochondrial function may be mediated by either of these two kinases, or another pathway activated by MEK1/2. However, because ERK1/2 is the only kinase known to be phosphorylated by MEK1/2, the effects of TBHP-injury in our model are most likely mediated by ERK1/2.
To activate ERK1/2, we transfected RPTC with adenovirus carrying constitutively active MEK1 cDNA. Continuous activation of ERK1/2 in noninjured RPTC resulted in a suppression of mitochondrial respiration indicating mitochondrial dysfunction. Oxidant injury in RPTC expressing active ERK1/2 had no further inhibitory effect on mitochondrial respiration indicating that continuous ERK1/2 activation is sufficient to suppress mitochondrial respiration to the levels observed in oxidant-injured RPTC. Furthermore, in contrast the recovery of TBHP-injured noninfected RPTC, mitochondrial function did not recover in TBHP-injured RPTC infected with adenovirus carrying constitutively active MEK1 cDNA. Both injured and noninjured RPTC expressing active ERK1/2 progressively died over 4 days following TBHP-induced injury and measurement of mitochondrial function was impossible due to cell loss. These events were not caused by adenovirus carrying empty vector demonstrating that these effects were specific to either MEK1 or ERK1/2 activation.
Our data show that the activation of ERK1/2 is involved in the decreases in the respiratory function of mitochondria and ATP production in TBHP-injured RPTC. Inhibition of MEK1/2 (using 3 different pharmacological inhibitors: U0126, PD98050, and SL0237) blocked ERK1/2 activation in the whole RPTC and in the mitochondria, reduced the decreases in basal respiration, electron transport rate, and ATP production, and promoted recovery of mitochondrial function in TBHP-injured RPTC. These results suggested that the respiratory chain is a target for ERK1/2. To look further into the mechanism of ERK1/2-mediated decreases in mitochondrial function, we tested whether the respiratory complexes were the targets of ERK1/2. Different oxidative substrates were used as electron donors to differentiate between respiration linked to the generation of NADH and complex I, FADH2 production and complex II, or the function of complex IV. The measurements of state 3 respiration (maximum respiration in the presence of excess ADP and respiratory substrates) and ATP production using electron donors (respiratory substrates) to different respiratory complexes demonstrated that the inhibition of ERK1/2 abolished oxidant-induced decreases in state 3 respiration and ATP synthesis linked to complex I but not to complexes II or IV. In contrast, ERK1/2 activation markedly decreased state 3 respiration linked to complex I and somewhat reduced state 3 respiration coupled to complex II. These results suggest that transient activation of ERK1/2 decreases oxidation of substrates through complex I, whereas continuous ERK1/2 activation decreases oxidation of substrates through both complexes I and II.
Because state 3 respiration linked to complex II was unaffected by TBHP-induced injury, we concluded that complex III was not a target of TBHP and ERK1/2. Our data also suggest that the availability of ubiquinone, the carrier of electrons between complexes I or II and complex III, was adequate to support the electron transport. Therefore, we concluded that sublethal oxidant induced injury and ERK1/2 target respiratory complex I and/or NADH-producing dehydrogenases upstream of complex I but have no apparent effects on complexes II, III, and IV. The choice of respiratory substrates used in this study precluded the involvement of dehydrogenases upstream of the citric acid cycle. Thus we tested whether the NADH-producing dehydrogenases of the citric acid cycle and/or the pyruvate dehydrogenase complex are targeted by TBHP-induced injury and ERK1/2. The results show that none of the dehydrogenases tested (pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase) was a target of TBHP and/or ERK1/2. In addition, we tested the activity of aconitase, an important regulatory step of the citric acid cycle, known to be very sensitive to oxidative stress (15, 36, 39). Our data show that aconitase is a target of the oxidant but the decrease in its activity is not mediated by ERK1/2. In contrast, direct measurements of activities of complex I and II showed that TBHP decreased the activity of complex I but not complex II and that the decrease in the activity of complex I was mediated by ERK1/2. Thus our results show that the respiratory function and ATP synthesis in TBHP-injured RPTC are under the control of ERK1/2. TBHP-induced injury decreases mitochondrial respiration through reducing the activity of complex I and this effect is mediated by the activation of ERK1/2.
Numerous studies have shown that the activation of the Ras-ERK1/2 pathway plays a pivotal role in mediating survival of different cell types in dissimilar experimental models (24, 26, 41). Activation of this pathway is thought to mediate the protective effects of many growth factors including nerve growth factor, hepatocyte growth factor, and epidermal growth factor (25, 46). ERK1/2 activation appears to be a survival signal in cell line models (4, 5, 19). ERK1/2 activation ameliorates cell injury caused by oxidative stress induced by H2O2 in cell lines derived from RPTC (TKPTS and LLC-PK1) (5, 19). However, our previous and present data show that, in contrast to its established role in the survival of other cell types, activation of ERK1/2 in renal proximal tubules mediates injury induced by nephrotoxicant cisplatin and the oxidant TBHP (30). Oxidant-induced activation of the MEK1/2-ERK1/2 pathway is detrimental to respiratory functions and ATP production in RPTC and the inhibition of ERK1/2 activation offers protection against the decrease in these functions. Our data are in agreement with previous reports suggesting that ERK1/2 contributes to the injury and cell death in renal cortical cells, brain tissue, and lung epithelium caused by an exposure to toxicants (cisplatin, cephaloridine), ischemia-reperfusion, and oxidative stress (5, 22, 37, 44, 47). The protective effects of ERK1/2 activation in cell lines may be due to different mechanisms of cell damage and death in this in vitro model. While oncosis is the major mechanism of toxicant-induced injury in vivo and in primary cultures, apoptosis predominates in different types of cell lines. Based on data from the literature and the data from our laboratory, we speculate that ERK1/2 may play protective role in apoptosis but detrimental role in those mechanisms of cell injury that culminate in necrosis. The data presented in Fig. 13 and unpublished data from our laboratory suggest that the activation of protein kinase B/Akt is the major prosurvival factor in injured renal proximal tubules in primary culture.
Our previous study showed that PKC-ε is activated by oxidant-induced injury and suggested that the respiratory complex I is also a target for PKC-ε (35). Because PKC-ε can phosphorylate and activate Raf-1 in some cell types, we tested whether the MEK1/2-ERK1/2 pathway is activated by PKC-ε in oxidant-injured RPTC. However, both the temporal relationship between PKC-ε activation (2 h after TBHP injury) and ERK1/2 activation (10 min of TBHP exposure) and the lack of inhibition of ERK1/2 activation by TBHP in the presence of PKC-ε inhibitor demonstrated that the MEK1/2-ERK1/2 pathway is not activated by PKC-ε in oxidant-injured RPTC. Thus the oxidant-induced decreases in function of the respiratory complex I are mediated by PKC-ε and ERK1/2 through the independent pathways.
In conclusion, ERK1/2, but not JNK or p38, plays an important role in the sublethal injury and the early recovery of mitochondrial function after oxidant injury in RPTC. ERK1/2 activation mediates the effects of oxidant on mitochondrial function by decreasing the activity of the respiratory complex I, thus decreasing the electron transport through complex I and ATP production coupled to complex I. Furthermore, continuous activation of ERK1/2 in noninjured RPTC results in significant reduction of respiratory functions, predominantly due to suppression of complex I-coupled respiration and substrate oxidation. Inhibition of ERK1/2 activation in renal proximal tubules represents a means to diminish oxidant-induced mitochondrial dysfunction and oxidant-induced injury in RPTC and promote the repair of mitochondrial function. The protective effects of ERK1/2 inhibition against oxidant-induced mitochondrial dysfunction are not due to activation of Akt pathway. In contrast, JNK is not activated in sublethally injured RPTC and activation of p38 by oxidant does not mediate mitochondrial dysfunction. Finally, ERK1/2 is not a downstream target of PKC-ε in mediating mitochondrial dysfunction in oxidant-injured RPTC.
ACKNOWLEDGMENTS
We thank Dr. I. Arany (Department of Internal Medicine, Division of Nephrology, University of Arkansas for Medical Sciences, Little Rock, AR) for the generous gift of the adenovirus carrying constitutively active MEK1cDNA. The use of the facilities in the University of Arkansas for Medical Sciences, Digital and Confocal Microscopy Laboratory, supported by National Institutes of Health Grant 2 P20 RR-16460 (INBRE) is also acknowledged.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R-01-DK-59558.
REFERENCES
- 1.Aleo MD, Wyatt RD, Schnellmann RG. Mitochondrial dysfunction is an early event in ochratoxin A but not oosporein toxicity to rat renal proximal tubules. Toxicol Appl Pharmacol. 1991;107:73–80. doi: 10.1016/0041-008x(91)90332-9. [DOI] [PubMed] [Google Scholar]
- 2.Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr125 by ERK MAPK. Nat Cell Biol. 2003;5:647–655. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- 3.Alonso M, Melani M, Converso D, Jaitovich A, Paz C, Carreras MC, Medina JH, Poderoso JJ. Mitochondrial extracellular signal-regulated kinases 1/2 (ERK1/2) are modulated during brain development. J Neurochem. 2004;89:248–256. doi: 10.1111/j.1471-4159.2004.02323.x. [DOI] [PubMed] [Google Scholar]
- 4.Arany I, Megyesi JK, Kaneto H, Price P, Safirstein RL. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am J Physiol Renal Physiol. 2004;287:F543–F549. doi: 10.1152/ajprenal.00112.2004. [DOI] [PubMed] [Google Scholar]
- 5.Arany I, Megyesi JK, Kaneto H, Tanaka S, Safirstein RL. Activation of ERK or inhibition of JNK ameliorates H2O2 cytotoxicity in mouse renal proximal tubule cells. Kidney Int. 2004;65:1231–1239. doi: 10.1111/j.1523-1755.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 6.Awazu M, Omori S, Hida M. MAP kinase in renal development. Nephrol Dial Transplant. 2002;17:5–7. doi: 10.1093/ndt/17.suppl_9.5. [DOI] [PubMed] [Google Scholar]
- 7.Bagnasco S, Good D, Balaban R, Burg M. Lactate production in isolated segments of the rat nephron. Am J Physiol Renal Fluid Electrolyte Physiol. 1985;248:F522–F526. doi: 10.1152/ajprenal.1985.248.4.F522. [DOI] [PubMed] [Google Scholar]
- 8.Baines CP, Zhang J, Wang G, Zheng Y, Xiu JX, Cardwell EM, Bolli R, Ping P. Mitochondrial PKCε and MAPK form signaling modules in the murine heart. Enhanced mitochondrial PKCε-MAPK interactions and differential MAPK activation in PKCε-induced cardioprotection. Circ Res. 2002;90:390–397. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 9.Baliga R, Ueda N, Walker PD, Shah SV. Oxidant mechanisms in toxic acute renal failure. Am J Kidney Dis. 1997;29:465–477. doi: 10.1016/s0272-6386(97)90212-2. [DOI] [PubMed] [Google Scholar]
- 10.Birch-Machin M, Jackson S, Kler RS, Turnbull DM. Study of skeletal muscle mitochondrial dysfunction. In: Lash LH, Jones DP, editors. Methods in Toxicology. Academic; New York: 1993. [Google Scholar]
- 11.Bonventre JV, Brezis M, Siegel N, Rosen S, Portilla D, Venkatachalam MA. Acute renal failure. I. Relative importance of proximal vs distal tubular injury. Am J Physiol Renal Physiol. 1998;275:F623–F631. doi: 10.1152/ajprenal.1998.275.5.F623. [DOI] [PubMed] [Google Scholar]
- 12.Borkan SC, Emami A, Schwartz JH. Heat stress protein-associated cytoprotection of inner medullary collecting duct cells from rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol. 1993;265:F333–F341. doi: 10.1152/ajprenal.1993.265.3.F333. [DOI] [PubMed] [Google Scholar]
- 13.Cassarino DS, Halvorsen EM, Swerdlow RH, Abramova NN, Parker WD, Jr, Sturgill TW, Bennet JP., Jr Interaction among mitochondria, mitogen-activated protein kinases, and nuclear factor-κB in cellular models of Parkinson's disease. J Neurochem. 2000;74:1384–1392. doi: 10.1046/j.1471-4159.2000.0741384.x. [DOI] [PubMed] [Google Scholar]
- 14.Dmitrieva RI, Doris PA. Ouabain is a potent promoter of growth and activator of ERK1/2 in ouabain-resistant rat renal epithelial cells. J Biol Chem. 2003;278:28160–28166. doi: 10.1074/jbc.M303768200. [DOI] [PubMed] [Google Scholar]
- 15.Drapier JC, Hibbs JB. Aconitases: a class of metalloproteins highly sensitive to nitric oxide synthesis. Methods Enzymol. 1996;269:26–36. doi: 10.1016/s0076-6879(96)69006-5. [DOI] [PubMed] [Google Scholar]
- 16.Groves CE, Lock EA, Schnellmann RG. Role of lipid peroxidation in renal proximal tubule cell death induced by haloalkene cysteine conjugates. Toxicol Appl Pharmacol. 1991;10:54–62. doi: 10.1016/0041-008x(91)90330-h. [DOI] [PubMed] [Google Scholar]
- 17.Hannemann J, Baumann K. Nephrotoxicity of cisplatin, carboplatin and transplatin. A comparative in vitro study. Arch Toxicol. 1990;64:393–400. doi: 10.1007/BF01973462. [DOI] [PubMed] [Google Scholar]
- 18.Hinman LM, Blass JP. An NADH-linked spectrophotometric assay for pyruvate dehydrogenase complex in crude tissue homogenates. J Biol Chem. 1981;256:6583–6586. [PubMed] [Google Scholar]
- 19.Hung CC, Ichimura T, Stevens JL, Bonventre JV. Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by ERK1/2 activation. J Biol Chem. 2003;278:29317–29326. doi: 10.1074/jbc.M302368200. [DOI] [PubMed] [Google Scholar]
- 20.Jin K, Mao XO, Zhu Y, Greenberg DA. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem. 2002;80:119–125. doi: 10.1046/j.0022-3042.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- 21.Kohda Y, Gemba M. Enhancement of protein kinase C activity and chemiluminescence intensity in mitochondria isolated from the kidney cortex of rats treated with cephaloridine. Biochem Pharmacol. 2002;64:543–549. doi: 10.1016/s0006-2952(02)01216-9. [DOI] [PubMed] [Google Scholar]
- 22.Kohda Y, Hiramatsu J, Gemba M. Involvement of MEK/ERK pathway in cephaloridine-induced injury in rat renal cortical slices. Toxicol Lett. 2003;143:185–194. doi: 10.1016/s0378-4274(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 24.Lee YJ, Kang IJ, Bunger R, Kang YH. Enhanced survival effect of pyruvate correlates MAPK and NF-κB activation in hydrogen peroxide-treated human endothelial cells. J Appl Physiol. 2004;96:793–801. doi: 10.1152/japplphysiol.00797.2003. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Godwin ML, Nowak G. Protein kinase C-α inhibits the repair of oxidative phosphorylation after S-(1,2-dichlorovinyl)-l-cysteine injury in renal cells. Am J Physiol Renal Physiol. 2004;287:F64–F73. doi: 10.1152/ajprenal.00216.2003. [DOI] [PubMed] [Google Scholar]
- 26.Liu ZX, Nickel CH, Cantlay LG. HGF promotes adhesion of ATP-depleted renal tubular epithelial in a MAPK-dependent manner. Am J Physiol Renal Physiol. 2001;281:F62–F70. doi: 10.1152/ajprenal.2001.281.1.F62. [DOI] [PubMed] [Google Scholar]
- 27.Masaki T, Foti R, Hill PA, Ikezumi Y, Atkins RC, Nikolic-Paterson DJ. Activation of the ERK pathway precedes tubular proliferation in the obstructed rat kidney. Kidney Int. 2003;63:1256–1264. doi: 10.1046/j.1523-1755.2003.00874.x. [DOI] [PubMed] [Google Scholar]
- 28.Miyazaki T, Katagiri H, Kanegae Y, Takayanagi H, Sawada Y, Yamamoto A, Pando MP, Asano T, Verma IM, Oda H, Nakamura K, Tanaka S. Reciprocal role of ERK and NF-κB pathways in survival and activation of osteoclasts. J Cell Biol. 2000;148:333–342. doi: 10.1083/jcb.148.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nath KA, Croatt AJ, Likely S, Behrens TW, Warden D. Renal oxidant injury and oxidant response induced by mercury. Kidney Int. 1994;50:1032–1043. doi: 10.1038/ki.1996.406. [DOI] [PubMed] [Google Scholar]
- 30.Nowak G. Protein kinase C-α and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem. 2002;277:43377–43388. doi: 10.1074/jbc.M206373200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nowak G, Schnellmann RG. Improved culture conditions stimulate gluconeogenesis in primary cultures of renal proximal tubule cells. Am J Physiol Cell Physiol. 1995;268:C1053–C1061. doi: 10.1152/ajpcell.1995.268.4.C1053. [DOI] [PubMed] [Google Scholar]
- 32.Nowak G, Schnellmann RG. l-Ascorbic acid regulates growth and metabolism of renal cells: improvements in cell culture. Am J Physiol Cell Physiol. 1996;271:C2072–C2080. doi: 10.1152/ajpcell.1996.271.6.C2072. [DOI] [PubMed] [Google Scholar]
- 33.Nowak G, Aleo MD, Morgan JA, Schnellmann RG. Recovery of cellular functions following oxidant injury. Am J Physiol Renal Physiol. 1998;274:F509–F515. doi: 10.1152/ajprenal.1998.274.3.F509. [DOI] [PubMed] [Google Scholar]
- 34.Nowak G, Price PM, Schnellmann RG. Lack of functional p21WAF1/CIP1 gene accelerates caspase-independent apoptosis induced by cisplatin in renal cells. Am J Physiol Renal Physiol. 2003;285:F440–F450. doi: 10.1152/ajprenal.00233.2002. [DOI] [PubMed] [Google Scholar]
- 35.Nowak G, Bakajsova D, Clifton GL. Protein kinase C-ε modulates mitochondrial function and active Na+ transport after oxidant injury in renal cells. Am J Physiol Renal Physiol. 2004;286:F307–F316. doi: 10.1152/ajprenal.00275.2003. [DOI] [PubMed] [Google Scholar]
- 36.Powell CS, Jackson RM. Mitochondrial complex I, aconitase, and succinate dehydrogenase during hypoxia-reoxygenation: modulation of enzyme activities by MnSOD. Am J Physiol Lung Cell Mol Physiol. 2003;285:L189–L198. doi: 10.1152/ajplung.00253.2002. [DOI] [PubMed] [Google Scholar]
- 37.Ramachandiran S, Huang Q, Dong J, Lau SS, Monks TJ. Mitogen-activated protein kinases contribute to reactive oxygen species-induced cell death in renal proximal tubule epithelial cells. Chem Res Toxicol. 2002;15:1635–1642. doi: 10.1021/tx0200663. [DOI] [PubMed] [Google Scholar]
- 38.Schnellmann RG. Mechanisms of t-butyl hydroperoxide-induced toxicity to rabbit renal proximal tubules. Am J Physiol Cell Physiol. 1988;255:C28–C33. doi: 10.1152/ajpcell.1988.255.1.C28. [DOI] [PubMed] [Google Scholar]
- 39.Schnellmann RG. The cellular effects of a unique pestide sulfuramid (N-ethylperfluoroctane sulphonamide) on rabbit renal proximal tubules. Toxicol In Vitro. 1990;4:71–74. doi: 10.1016/0887-2333(90)90012-i. [DOI] [PubMed] [Google Scholar]
- 40.Schnellmann RG, Manning RO. Perfluoroctane sulfonamide: a structurally novel uncoupler of oxidative phosphorylation. Biochim Biophys Acta. 1990;1016:344–348. doi: 10.1016/0005-2728(90)90167-3. [DOI] [PubMed] [Google Scholar]
- 41.Shonai T, Adachi M, Sakata K, Takekawa M, Endo T, Imai K, Hareyama M. MEK/ERK pathway protects ionizing radiation-induced loss of mitochondrial membrane potential and cell death in lymphocytic leukemia cells. Cell Death Differ. 2002;9:963–971. doi: 10.1038/sj.cdd.4401050. [DOI] [PubMed] [Google Scholar]
- 42.Sinha D, Bannergee S, Schwartz JH, Lieberthal W, Levine JS. Inhibition of ligand-independent ERK1/2 activity in kidney proximal tubular cells deprived of soluble survival factors up-regulates Akt and prevents apoptosis. J Biol Chem. 2004;279:10962–10972. doi: 10.1074/jbc.M312048200. [DOI] [PubMed] [Google Scholar]
- 43.Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of α-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, Trzaskos JM, Feuerstein GZ. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther. 2003;304:172–178. doi: 10.1124/jpet.102.040246. [DOI] [PubMed] [Google Scholar]
- 45.Xu Z, Ji X, Boysen PG. Exogenous nitric oxide generates ROS and induces cardioprotection: involvement of PKG, mitochondrial KATP channels, and ERK. Am J Physiol Heart Circ Physiol. 2004;286:H1433–H1440. doi: 10.1152/ajpheart.00882.2003. [DOI] [PubMed] [Google Scholar]
- 46.Xue L, Murray JH, Tolkovsky AM. The Ras/Phosphatidylinositol 3-Kinase and Ras/ERK pathways function as independent survival modules each of which inhibits a distinct apoptotic signaling pathway in sympathetic neurons. J Biol Chem. 2002;12:8817–8824. doi: 10.1074/jbc.275.12.8817. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X, Shan P, Sasidhar M, Chupp GL, Flavell RA, Choi AM, Lee PJ. Reactive oxygen species and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am J Respir Cell Mol Biol. 2003;28:305–315. doi: 10.1165/rcmb.2002-0156OC. [DOI] [PubMed] [Google Scholar]
- 48.Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in lewy body diseases. Brain Pathol. 2003;13:473–481. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]