

Abstract
Collisional activation of [M + H]+ parent ions from peptides of n amino acid residues may yield a rearrangement that involves loss of the C-terminal amino acid residue to produce (bn−1+H2O) daughters. We have studied this reaction by a retrospective examination of the m/z spectra of two collections of data. The first set comprised 398 peptides from coat protein digests of a number of plant viruses by various enzymes, where conditions in the tryptic digests were chosen so as to produce many missed cleavages. In this case a large effect was observed– 323 (bn−1+H2O) daughter ions (~81%), including 185 (~ 46%) “strong” decays with ratios (bn−1+H2O)/(bn−1)>1. The second set comprised 1200 peptides, all from tryptic digests, which were carried out under more stringent conditions, resulting in relatively few missed cleavages. Even here, 190 (bn−1+H2O) ions (~ 16 %) were observed, including 87 (> 7%) “strong” decays, so the effect is still appreciable. The results suggest that the tendency for (bn−1+H2O) ion formation is promoted by the protonated side chain of a non-C-terminal basic amino acid residue, in the order arginine ≫ lysine ≥ histidine, and that its (non-C-terminal) position is not critical. The results can be interpreted by a mechanism in which hydrogen bonding between the protonated side chain and the (n-1) carbonyl oxygen facilitates loss of the C-terminal amino acid residue to give a product ion having a carboxyl group at the new C-terminus.
INTRODUCTION
Gaskell and coworkers [1] were the first to report a decay mechanism of peptide [M + H]+ parent ions containing n amino acid residues, in which (bn−1 + H2O) daughter ions are formed – a fragmentation especially favored in peptides containing arginine. They also showed, by 18O isotopic labeling, that the loss of the C-terminal amino acid residue is accompanied by retention of one of the carboxyl oxygens at the new C-terminus, and that the structure of the daughter ion appears to be the same as that of the [M + H]+ ion of the peptide with one fewer amino acid residue. In a follow-up paper [2] they showed that partial or complete equilibration of oxygen exchange among the carboxyl oxygens and the (n−1) carbonyl oxygen occurs on collision induced dissociation (CID) of protonated RPPGF or TRKR, or sodium cationized YGGFL. Gaskell et al suggested a mechanism for the reaction [1], and other workers have subsequently proposed alternative mechanisms [3–5]. (See Refs. [6,7] for notation; species denoted by lower case letters carry an implied positive charge.)
Gaskell and coworkers [1] noted that “For the examples of bradykinin and related peptides, the rearrrangement is strongly promoted when arginine is the amino acid residue lost” and “is also favored by the presence of an arginine residue at or near the N-terminus”. In support of their observation that the decomposition was not observed for peptides with C-terminal amide or ester functions they proposed a mechanism (Scheme 1) initiated by interaction of the C-terminal hydroxyl group with the (n−1) carbonyl group, although they noted that not all of their observations were explained by this mechanism and stated that “The strong influence of amino acid composition … suggests mechanistic complexities that require further elucidation.”.
Scheme 1.

Formation of the (bn−1 + H2O) ion, as proposed in ref. 1.
Gonzales et al [3] carried out subsequent studies of metastable ion decomposition and CID of [M + H]+ ions of several small peptides (tetrapeptides to heptapeptides), and concluded that the (bn−1 + H2O) ion abundance is largest when an arginine residue is located at the (n−1) position, but absent when arginine is located at the C-terminus. The latter observation was ascribed to preferred formation of C-terminal-containing ions rather than N-terminal-containing ions, (such as b ions), when a highly basic residue is located at the C-terminus. When an arginine residue is located at the (n−1) position they proposed formation of a salt-bridge between the protonated guanidino group of the arginine residue and the deprotonated C-terminal carboxyl group (Scheme 2), with the location of the additional proton not being specified. (In accord with the “mobile proton” model [8] and the proposals of several groups [9–20] it is likely that several positions of the peptide backbone could serve as protonation sites, based on the observation of non-specific fragmentations of the peptide backbone.) Formation of the (bn−1 + H2O) ion is then initiated by interaction of the positively charged guanidino group with the carbonyl group of its own arginine residue. However, the presence of the rearrangement ion was not exclusive to this structural or compositional feature; it was also observed in pentapeptides when arginine was absent, but when histidine or lysine was present at the N-terminus, though at low abundance. The (bn−1 + H2O)/bn−1 abundance ratio increased with increasing basicity of the N-terminal amino acid.
Scheme 2.

Formation of the (bn−1 + H2O) ion for peptides having an n−1 arginine residue [3].
Vachet et al [4] made measurements of kinetic energy lost on decomposition of parent ions of a number of small arginine-containing peptides (leucine enkephalin and analogues, bradykin and Substance P). The (bn−1 + H2O) ion, in addition to formation from [M + H]+ ions of bradykinin (RPPGFSPFR) and des-Arg1-bradykinin (in the latter case supporting earlier observations [1] but contrary to others [3] on the activity of a C-terminal arginine residue), was also observed from [M + H]+ ions of YGRFL, where the arginine residue occupies the (n−2) position. These three peptides showed anomalously high energy losses in the formation of product ions involving a dissociation adjacent to an arginine residue. The authors argued, with support from computer modelling studies (molecular mechanics), that this was because the protonated peptides could adopt a conformation in which the proton is shared between the side chain of arginine and the carbonyl oxygen on the adjacent (n−1) amino acid residue. This led to the mechanism depicted in Scheme 3 for the formation of the (bn−1 + H2O) ion from the [M + H]+ ion of YGRFL. The increased positive charge on the carbonyl carbon, induced by the proton bridge, makes it more susceptible to attack by oxygen of the C-terminal hydroxy group.
Scheme 3.

Formation of the (bn−1 + H2O) ion for peptides having an n−2 arginine residue (based on Fig. 5 of ref. 4, after correction and modification).
Deery et al [5] studied a number of small peptides, all of which contained a C-terminal arginine residue and one other arginine residue somewhere in the peptide backbone, i.e. bradykinin, Lys-bradykinin (KRPPGFSPFR), Lys-Ala3-bradykinin (KRPAGFSPFR) and dynorphin A fragments 1–7 (YGGFLRR). Their [M + H]+ ions, after low energy CID, all yielded abundant (bn−1 + H2O) ions. The lowest energy conformation of the [M + H]+ ion of bradykinin was thought to involve a salt-bridge structure in which the two terminal arginine residues are protonated and the C-terminal hydroxyl group is deprotonated. Evidence in support of the salt-bridge structure from molecular modelling [17, 21] and experimental [22] studies was cited. Upon low energy CID the salt-bridge structure breaks down to give either of two isomeric forms; one isomer retained the ionizing proton at the guanidino group of C-terminal arginine (Fig. 1), while the other isomer retained the charge at the guanidino group of N-terminal arginine. They proposed that “When the charge is located at the C-terminal end of the peptide … the formation of the rearrangement ion is a very facile fragmentation pathway” and that when the ionizing proton is located on the guanidino group of N-terminal arginine, formation of the [M + H − 60]+ ion rather than of the (bn−1 + H2O) ion is observed. Evidence to support these proposals came from the non-observation of the (bn−1 + H2O) ion in the CID mass spectra of M+ ions of bradykinin derivatized to have a fixed positive charge (trimethylammonium), rather than a proton, at the N-terminus. Thus, a proton at the C-terminus (i.e. on the guanidino group) was considered necessary for formation of the (bn−1 + H2O) ion. However, they did not suggest a mechanism, and it is not obvious how the structure in Fig. 1 would give rise to the (bn−1 + H2O) ion. We will suggest that the fixed charge result does not rule out the possibility of involvement of a protonated guanidino group at the N-terminus in the case of decomposition of [M + H]+ ions of non-derivatized bradykinin, and that the results do not contradict those of Gonzales et al [3].
Figure 1.
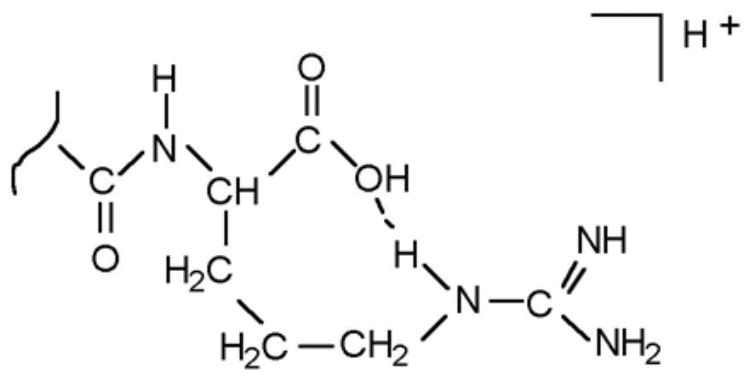
Structure proposed [5] for the bradykinin [M + H]+ ion, showing charge retention at the C-terminus.
A number of subsequent measurements on (bn−1 + H2O) ions have been reported. They include studies by Dikler et al [23], who reduced the basicity of arginine residues by derivatizing with acetylacetone, effectively converting an arginine residue to a pyrimidinyl ornithine residue, and also by Tsaprailis et al [24], who observed the effect in surface induced dissociation at 50 eV in a quadrupole mass spectrometer or by high energy CID (~8.9 – 10.0 keV, laboratory reference frame) in an EB sector mass spectrometer. However, the yield became zero with sustained off-resonance irradiation (18.7 eV) in the much longer time frame of a Fourier transform ion cyclotron resonance mass spectrometer.
According to all the proposals that have been made, the (bn−1 + H2O) ions have a structure with a C-terminal carboxyl group, as pointed out early on by Thorne and Gaskell [25], with supporting evidence given by their similar decompositions to (bn−2 + H2O) ions.
However, much of the evidence for this fragmentation has been obtained from a rather restricted group of compounds, bradykinin and its relatives, as well as a few others. These were obvious candidates in which to look for the effect, following its original discovery by Gaskell and coworkers [1], but they are not likely to provide a fair picture of its overall significance. In contrast, we present here a retrospective study of the m/z spectra of two fairly extensive collections of data: (i) 398 peptides from digests of coat proteins of a number of plant viruses by various enzymes; (ii) 1200 peptides from tryptic digests of 46 proteins. Both collections were gathered previously without regard to the presence of (bn−1+H2O)-type ions, so the samples are expected to be reasonably unbiased. These studies reveal a much larger and more varied group of peptide parent ions that decay in this way, so it is clear that the effect is much more widespread than originally believed. Thus our analysis provides a broader picture of the conditions required for the production of (bn−1+H2O)-type ions. It may consequently aid in their discovery and application, as well as shedding light on the production process.
Moreover, awareness of this mode of decay has practical applications, since it may cause unexpected errors in sequencing. Indeed, our interest in the topic was initially aroused in 1998 during sequencing of the JGMV coat proteins, when we misinterpreted a number of (bn−1+H2O) ions as y ions; two examples are given below. We were alerted to the problem when our initially deduced sequences disagreed with those predicted from the nucleotide data, and it seemed doubtful that they resulted from mutations. We eventually realized that the source of the disagreement must simply be the presence of (bn−1+H2O) ions, and finally became aware of the previous work in the field [1–5].
However, our mistake is an easy one to make, and the constraints provided by the nucleotide sequence are not always present– for example in de novo sequencing of an unknown protein– so it is important to be aware of this mode of decay, especially when non-C-terminal basic residues are present in the peptide being analyzed.
EXPERIMENTAL METHODS
Set 1– Plant virus coat proteins
Isolation and purification of plant virus coat proteins
All plant virus coat proteins were generously provided by Dr. S. Haber (Cereal Research Centre, Agriculture and Agrifood Canada, Winnipeg, MB), and Dr. D. L. Seifers (Agricultural Research Center, Kansas State University, Manhattan, KS). They included two strains of brome mosaic virus (BMV) [26–28], foxtail mosaic virus (FoMV) [29,30], four strains of wheat streak mosaic virus (WSMV) [31], four strains of Johnsongrass mosaic virus (JGMV) from Australia, Kansas, Nigeria and Israel [32–34], and high plains virus (HPV) from Kansas and Idaho [35]. We have previously described isolation and purification of the coat proteins from these viruses by ultracentrifugal precipitation through multiple sucrose density gradients. An additional centrifugal filtration through a 5,000 Da molecular mass cut-off membrane (Millipore Corporation, Bedford, MA) was used to remove residual impurities, such as SDS detergent and cesium salts introduced during earlier purification steps. The sequences of the coat proteins, as published in refs. [26–35] and given in Table S1 (supplemental), are based on those predicted by the cDNA sequences of the isolate types as listed in the SWISS-PROT/TrEMBL [36] or NCBI [37] databases, but they incorporate some modifications revealed by our previously reported mass spectral results.
Enzymatic digestions of plant virus coat proteins
The peptides examined were generated by partial or complete digestion of the virus coat proteins with various proteases. In order to make sequencing easier, conditions were chosen so as to favor the production of fairly long proteolytic fragments– for example, short (2–4 hr) tryptic digests in solution. This practice, together with the frequent use of non-tryptic enzymes, produced many fragments that contained non-C-terminal basic residues.
Trypsin was purchased from Sigma Chemical Company (St. Louis, MO). Chymotrypsin, as well as endoproteases Lys-C, Arg-C, Glu-C, and Asp-N were obtained from Roche Diagnostics (Indianapolis, IN). The lyophilized protein samples were dissolved (1 mg/mL) in aqueous 25mM ammonium bicarbonate (pH 7.8) for all digestions, except that a 10 mM Tris-HCl buffer solution (pH 7.6) was used for Asp-N digestions. Then the enzyme in the same buffer solution was added, with a 1:100 weight ratio of enzyme to protein, and the resulting mixture was incubated at 37°C for 2 to 24 hours. Finally, the digestion was terminated by freezing; the sample digest solution was then stored below −20°C.
Mass spectrometry
Mass spectra of whole digest mixtures were recorded with our Manitoba/SCIEX prototype tandem quadrupole/time-of-flight mass spectrometer (QqTOF) (PE/Sciex, Concord, ON, Canada), [38], which was coupled to a MALDI ion source for these experiments [39,40], as shown schematically in Fig. S1. The matrix was prepared from a 100 mg/mL solution of 2,5-dihydroxybenzoic acid (DHB) in acetone/water (1:1 v/v). 0.5 μL of sample digest solution, containing 0.1% trifluoroacetic acid, was mixed with an equal volume of the matrix solution, the mixture was deposited on a stainless steel target, and the solvent was allowed to evaporate. When dry, the target was placed on the sample stage of the instrument, and was subjected to 4 ns, 337 nm, controlled intensity pulses from a nitrogen laser (Model VSL-337ND, Laser Science, Inc, Franklin, MA). Ions desorbed from the analytes consisted primarily of [M + H]+ ions of the various peptides present. These were cooled in quadrupole q0, selected by m/z value in quadrupole Q1, and then subjected to collision-induced dissociation (CID) in quadrupole q2. The collision gas was nitrogen or argon at a pressure of ~10−2 Torr, with initial laboratory frame ion kinetic energies of 50 to 180 eV per charge (varied to adjust the extent of fragmentation). The daughter ions observed were compared to the corresponding calculated masses of the fragment ions using a commercially available program (ProMac 1.5.3.1, supplied by MDS Sciex), which was provided with the peptide sequences expected in the enzyme digests.
Set 2– 1200 tryptic peptides, generated for measurement of chromatographic retention times
Tryptic digests of 69 proteins (listed in Table S2)
The proteins were commercially-available proteins of known sequence, along with some others that we had previously sequenced, which had all been selected primarily for a study of their chromatographic behavior [41–43]. The proteins were prepared for mass spectrometry as described above and in ref. 42, i.e. they were individually reduced, alkylated with iodoacetamide, dialyzed, and digested overnight with trypsin (Sigma Chemical Company, St. Louis, MO). Mixtures of these digests were fractionated by micro-RP HPLC, and deposited on MALDI targets to provide a library of 1200 tryptic peptides. Daughter ions from CID of the peptides in the corresponding chromatographic fractions were then recorded with the QqTOF instrument.
These data were analyzed by computer using a set of scripts written in “Perl” for OS-X. The first program constructed mass-intensity tables for each peptide’s MS/MS spectrum. Another script computed the masses of the bn−1 and the (bn−1+18) ions from the known peptide sequences. If the mass entries in the peak table fell within 100 PPM of the calculated values, the program recorded the peak intensities and the ratio between them.
Chemical computations
The PC Spartan Pro set of programs (Wavefunction, Inc., Irvine, CA) were used to calculate structures and relative energies of protonated lysine, arginine and histidine. Molecular mechanics (MMFF94) calculations identified low energy conformations, which were then geometry optimized by the semi-empirical PM3 method; we found, in agreement with Hehre et al [44], that this method works better than the AM1 method for hydrogen bonded structures. The energies of the minimized structures were then computed by two methods: at the ab initio Hartree Fock 6–31G* level, and also by Density Functional Theory, using a perturbed Becke-Perdew functional and a DN** numerical basis set, pBP/DN**.
RESULTS AND DISCUSSION
Analysis of viral coat proteins
The enzymatic digests of the viral coat proteins yielded many peptide fragments. The 356 fragments that yielded (bn−1 + H2O) ions from CID of their [M + H]+ ions are collected in Table S3 (supplemental), and examples from two of the digests are shown in Table 1. The 75 fragments that did not give observed (bn−1 + H2O) ions are listed in Table S4. Since the extent of fragmentation of the [M + H]+ ions is dependent upon the CID conditions, the abundance of the (bn−1 + H2O) ion in a given spectrum has been denoted as strong, medium or weak relative to the abundance of the bn−1 ion in the same spectrum: [bn−1 + H2O]/bn−1 ratio: >1, S (strong); 0.5 to 1, M (medium); ~0.1 to 0.5, W (weak)]. Table 2 summarizes the data.
Table 1.
Peptides from BMV whose [M + H]+ ions yield [bn−1 + H2O] ions in their CID mass spectra
| Peptidea | Peptide sequenceb | [M + H]+ m/z | [bn−1 + H2O] abundancec |
|---|---|---|---|
| BMV-N and BMV-P (N-acetylated) | |||
| 2- 11 | @STSGTGKMTR | 1067.52 | M |
| 9- 14 | MTRVQR (BMV-N only) | 790.44 | S |
| 9- 15 | MTRVQRR (BMV-N only) | 946.57 | S |
| 12- 15 | VQRR (BMV-N only) | 558.35 | S |
| 15- 19 | RAAAR | 544.33 | S |
| 15- 20 | RAAARR | 700.43 | S |
| 20- 22 | RNR | 445.26 | S |
| 23- 35 | RTAGVQPVIVEPL (BMV-N only) | 1378.81 | S |
| 24- 41 | TAGVQPVIVEPLAAGQGK | 1734.98 | W |
| 27- 41 | VQPVIVEPLAAGQGK | 1505.87 | W |
| 42- 53 | AIKAIAGYSISK | 1536.84 | W |
| 42- 64 | AIKAIAGYSISKWEASSDAITAK | 2381.27 | M |
| 45- 64 | AIAGYSISKWEASSDAITAK | 2069.06 | M |
| 65- 81 | ATNAMSITLPHELSSEK | 1828.91 | M |
| 65- 86 | ATNAMSITLPHELSSEKNKELK | 2441.27 | S |
| 74- 81 | PHELSSEK | 926.46 | M |
| 74- 83 | PHELSSEKNK | 1168.60 | M |
| 82-102 | NKELKVGRVLLWLGLLPSVAG | 2262.37 | M |
| 84-102 | ELKVGRVLLWLGLLPSVAG | 2020.23 | M |
| 84-103 | ELKVGRVLLWLGLLPSVAGR | 2176.33 | S |
| 87-102 | VGRVLLWLGLLPSVAG | 1650.01 | M |
| 87-103 | VGRVLLWLGLLPSVAGR | 1806.11 | S |
| 90-105 | VLLWLGLLPSVAGRIK | 1735.10 | S |
| 112-130 | QAQAEAAFQVALAVADSSK | 1904.97 | W |
| 117-131 | AAFQVALAVADSSKE | 1506.78 | W |
| 131-142 | EVVAAMYTDAFR | 1372.66 | W |
| 166-188 | AVVVHLEVEHVRPTFDDFFTPVY | 2716.38 | S |
| 166-189 | AVVVHLEVEHVRPTFDDFFTPVYR | 2872.48 | S |
| 173-181 | VEHVRPTFD | 1099.55 | S |
| 175-188 | HVRPTFDDFFTPVY | 1740.84 | S |
Enzymatic fragments of the viral coat proteins: BMV-N and BMV-P
Basic residues are underlined.
[bn−1 + H2O]/bn−1 ratios: >1, S (strong); 0.5~1, M (medium); <0.5, W (weak)
Table 2.
Number of peptide examples containing non-C terminal arginine, lysine or histidine residues that give observable (bn−1 + H2O) ions.
| Abundancea | |||
|---|---|---|---|
| Strong (185) | Medium (91) | Weak (47) | Not detected (75) |
| 3R(1); 2R (9); R (58) | 2R (1); R (4) | 3K(2); 2K(2); K (13) | W (15) |
| 4K(4); 3K (4); 2K (12); K (12) | 4K(1); 3K(4); 2K (22); K (26) | 2H (1); H (13) | K (1) |
| 3R,H(1); 2R,3K,H (1); 2R,2K(1); | H (13) | 2R,H (1) | H (5) |
| 2R,K(4); 2R,H(2) | 2R,2K(1); R,4K(1); R,2K,H(2); | R, 2K(1) | K,H (1) |
| R,3K (6); R,2K (8); R,K,2H (4) | R, 3K(1); R,2K(2); R, K (2) | K,H (1) | H,W (3) |
| R,2K,H (2); R,K,H (6); | 2K,H(1); K,H (6) | None (13)c | None (50) |
| R,K (20); R,2H(5); R,H (21) | None (3)b | ||
| 2K,H (2); K, H(2) | |||
Abundance as defined in Table 1. Number of examples, in parentheses, of peptides containing the indicated number of arginine, lysine or histidine residues, and, for column 4, including the number of tryptophan residues. (Note: these numbers exclude the duplicate entries in Table S3.)
WSMV-EB3 96–107 (two threonines present) and JGMV-Aus 37–52 (three serines, three threonines present) contain C-terminal lysine; JGMV-IS 158–174 (one serine, two threonines present) does not contain a basic residue.
BMV 131–142 (one threonine present); FoMV-H93 150–167 (two serine, one threonine present); JGMV-Aus 137–146; JGMV-KS 125–133 (one threonine present), 138–146; JGMV-IS 268–284 (one serine present), 269–284 (one serine present), 273–284 (one serine present) contain C-terminal arginine. BMV 24–41 (one threonine present), 27–41 (one threonine present), 112–130 (two serines present); JGMV-IS 234–244 (one threonine present) contain C-terminal lysine. FoMV-H93 206–217 does not contain a basic residue.
About 95% of the 356 examples listed in Table S3 for which (bn−1 + H2O) ions were observed, contain at least one of the basic residues arginine, lysine or histidine, located internally or at the N-terminus; only 16 do not. There are no examples of a peptide [M + H]+ ion fragmenting to give a strong abundance (bn−1 + H2O) ion when only a single basic amino acid residue is located at its C-terminus. As indicated in footnote b of Table 2, there are two examples, with C-terminal lysine only, that give medium abundance (bn−1 + H2O) ions, and footnote c lists 8 peptide examples with C-terminal arginine only, and 4 peptides with C-terminal lysine only, that give weak abundance (bn−1 + H2O) ions. However, Table S4 lists 54 peptide fragments with C-terminal arginine, lysine, or histidine that do not yield detectable (bn−1 + H2O) ions, so the yield of this ion is not really significant when a basic residue is located only at the C-terminus. This is particularly noteworthy in view of the preponderance of peptide fragments of this type, a consequence of the enzymatic digestions with trypsin, Lys-C, and Arg-C. It is also apparent from Table S3 that, under the general purpose conditions for the digestions, many of these digestions are incomplete; the majority of the peptide fragments contain at least one of the basic residues arginine, lysine (or histidine) as a non-C-terminal residue; though only a few contain them as multiple residues.
Because a single basic peptide residue at the C-terminus seldom promotes formation of the (bn−1 + H2O) ion, we can now use the results in Tables 2 and S3 to assess the effectiveness of non-C-terminal basic residues in promoting its formation. This is most easily done for peptide fragments containing only a single basic residue. When single arginine residues only are considered, the abundance of the ion is strong in 58 examples, medium in 4 examples, and weak or not detected in no examples. When lysine only is considered, the abundance is strong in 12 examples, medium in 26 examples, weak in 13 examples, and not detected in one example. When histidine only is considered, the abundance is strong in no example, medium in 13 examples, weak in 13 examples, and not detected in 5 examples. It is therefore clear that the ability of the residues to promote (bn−1 + H2O) ion formation increases in the sequence histidine < lysine ≪ arginine. In general, the abundances are also strong or medium when multiple argine/lysine residues are present, but there are a few cases of weak abundance. Possibly, in the latter cases, proton bridging between two basic groups is favored over the necessary proton bridging to oxygen of the C-terminal CO group needed for (bn−1 + H2O) ion formation. There are also the 16 examples noted above, where the (bn−1 + H2O) ion is present, albeit at medium or weak abundance, when a non-C-terminal basic amino acid is absent. We shall return to this point later.
The tryptophan residue is also (weakly) basic (see below). However, non-C-terminal tryptophan is ineffective in promoting (bn−1 + H2O) ion formation. This is clear from Table S4, which includes 15 examples in which the effect of tryptophan is not swamped by other basic residues, and for which (bn−1 + H2O) ion formation is not detected.
Table 3 lists peptides having a single non-C-terminal arginine, lysine or histidine residue, arranged according to the distance of the basic residue from the C-terminus, so the influence of each residue can be assessed in the absence of competition from other basic residues. In the case of arginine, the (bn−1 + H2O) ion abundance is not detected or weak when it is located at the C-terminus and strong in 17 of the 18 examples listed (medium in the remaining example) when it is located away from the C-terminus (both with and without a C-terminal arginine being present). This is true even when it is located as far from the C-terminus as the 22nd residue. The (bn−1 + H2O) ion abundance is uniformly strong in the 9 arginine-containing examples in which the normally weakly producing histidine is also present. It is strong in all 5 remaining examples, where non-C-terminal lysine residues are also present; (histidine is present in three of these cases, though here the abundance is, no doubt, enhanced by the lysine residue). In the case of lysine the abundance can be strong or medium with little apparent dependence on the position of the residue. A strong abundance is observed even when lysine is 12 residues from the C-terminus. In the case of histidine there appears to be little relationship between abundance of the (bn−1 + H2O) ion, and residue position; a weak abundance is observed even when histidine is 18 residues from the C-terminus.
Table 3.
Dependence of the abundance of the (bn−1 + H2O) ion on the position of an arginine, lysine or histidine residue.a
| Sequence | Position of underlined basic residueb | (bn−1 + H2O) abundance | Peptide source | |
|---|---|---|---|---|
| Peptides containing a single arginine residue (lysine and histidine absent): | ||||
| ATQEEFNR | 1 | ND | JGMV-IS | 121-128 |
| NLTDFNLAR | 1 | ND | JGMV-IS | 225-233 |
| GNVGESQENTER | 1 | W | JGMV-IS | 273-284 |
| FGLDGNVGESQENTER | 1 | W | JGMV-IS | 269-284 |
| MFGLDGNVGESQENTER | 1 | W | JGMV-IS | 268-284 |
| NLTDFNLARY | 2 | S | JGMV-IS | 225-234 |
| ATQEEFNRWY | 3 | S | JGMV-IS | 121-130 |
| FNRWYN | 4 | S | JGMV-IS | 126-131 |
| ATQEEFNRWYN | 4 | S | JGMV-IS | 121-131 |
| FNLARYAFD | 5 | S | JGMV-IS | 229-237 |
| SLARYAFDFY | 7 | S | JGMV-NI | 221-230 |
| NLTDFNLARYAFDFYE | 8 | S | JGMV-IS | 225-240 |
| DEQMRILMNGLMVWCIENGTSP | 18 | M | JGMV-NI | 132-153 |
| GRLNGAPALPNNGQYFIEAPQ | 20 | S | FoMV | 197-217 |
| Peptides containing a single non-C-terminal arginine in addition to just a C-terminal lysine residue: | ||||
| WYNRIK | 3 | S | JGMV-Aus | 130-135 |
| WYNRIK | 3 | S | JGMV-NI | 120-125 |
| FWYNRIK | 3 | S | JGMV-NI | 119-125 |
| @SGNEDAGRQK | 3 | S | JGMV-Aus | 1- 10 |
| HTQFQFWYNRIK | 3 | S | JGMV-NI | 114-125 |
| PIIPRGFDK | 5 | S | WSMV-IHC | 200-208 |
| RQVNEIFK | 8 | S | FoMV | 167-174 |
| MRLPMVSNK | 8 | S | JGMV-NI | 81- 89 |
| RGLTPAAFVQAAIIFTMESMDK | 22 | S | FoMV | 51- 72 |
| Peptides containing single arginine plus one or more non-C-terminal histidine residues only: | ||||
| ATHTQFQFWYNRIK | 3 | S | JGMV-Aus | 122-135 |
| NVHTYRGAK | 4 | S | JGMV-NI | 284-292 |
| HHFSDAAEAYIEYRNSK | 4 | S | JGMV-IS | 197-213 |
| DGNVGESSENTERHTAA | 5 | S | JGMV-NI | 263-279 |
| AAAIRGSTNHMF | 8 | S | JGMV-Aus | 259-270 |
| AREAHAQMK | 8 | S | JGMV-NI | 240-248 |
| AAAIRGSTNHMFGL | 10 | S | JGMV-NI | 249-262 |
| NASPTLRQIMHHFSDAAE | 12 | S | JGMV-IS | 187-204 |
| AVVVHLEVEHVRPTFDDFFTPVY | 12 | S | BMV | 166-188 |
| Peptides containing single arginine plus one or two non-C-terminal lysine residues only: | ||||
| FWYNRIKK | 4 | S | JGMV-NI | 119-126 |
| ATHTQFQFWYNRIKK | 4 | S | JGMV-NI | 112-126 |
| Peptides containing single arginine plus one or more non-C-terminal lysine or histidine residues | ||||
| NVHTYRGAKI | 5 | S | JGMV-NI | 284-293 |
| ATHTQFQFWYNRIKKEY | 6 | S | JGMV-NI | 112-128 |
| FNRWYNAIKKE | 9 | S | JGMV-IS | 126-136 |
| Peptides containing one or more non-C-terminal lysines plus a C-terminal arginine residue: | ||||
| NLNDKSLAR | 5 | M | JGMV-Aus | 226-234 |
| YTKPAYANR | 7 | M | FoMV | 109-117 |
| FYTKPAYANR | 7 | M | FoMV | 108-117 |
| TMMDGETQVTYPLKPVVENASPTLR | 12 | S | JGMV-IS | 169-193 |
| GETQVTYPLKPVVENASPTLR | 12 | S | JGMV-IS | 173-193 |
| EFPLKPIVENAKPTLR | 5,12 | M | JGMV-NI | 169-184 |
| SEFPLKPIVENAKPTLR | 5,12 | M | JGMV-NI | 168-184 |
| DQSEFPLKPIVENAKPTLR | 5,12 | S | JGMV-Aus | 176-194 |
| GNNQSEFPLKPIVENAKPTLR | 5,12 | M | JGMV-NI | 164-184 |
| IKKEYDVDDEQMR | 11,12 | M | JGMV-Aus | 134-146 |
| Peptides containing one or more non-C-terminal histidines plus a C-terminal arginine residue: | ||||
| HFSDAAEAYIEMR | 12 | S | JGMV-NI | 189-201 |
| QCMMHFSDAAEAYIEMR | 12 | W | JGMV-NI | 185-201 |
| GSTNHMFGLDGNVGESSENTER | 17 | W | JGMV-NI | 254-275 |
| QIMHHFSDAAEAYIEYR | 18,19 | W | JGMV-IS | 194-210 |
| Peptides containing non-C-terminal lysine and histidine plus a C-terminal arginine residue | ||||
| HLIQYKPDQR | 5,10 | M | JGMV-Aus | 106-115 |
| AILNLDHLIQYKPDQR | 5,10 | M | JGMV-Aus | 100-115 |
| Arginine-less peptides containing one or more lysine residues: | ||||
| YAFDFYEITSK | 1 | W | JGMV-IS | 234-244 |
| VSANDQSEFPLKPI | 3 | W | JGMV-Aus | 172-185 |
| WTMVSANDQSEFPLKPI | 3 | S | JGMV-Aus | 169-185 |
| SATPAENQPASADGKPAQTTATS | 9 | W | JGMV-KS | 11- 33 |
| DVDVGSTGTFVIPKLK | 1,3 | M | JGMV-NI | 60- 75 |
| DQSEFPLKPIVENAK | 1,8 | S | JGMV-Aus | 176-190 |
| AIKAIAGYSISK | 1,10 | W | BMV | 42- 53 |
| VVNNAGKDNEQQLEFK | 1,10 | M | WSMV-IHC | 149-164 |
| SATPAANQTASGDGKPAQTTATAENK | 1,12 | M | JGMV-Aus | 11- 36 |
| DKDVDVGSTGTFVIPK | 1,15 | M | JGMV-Aus | 68- 83 |
| SQTESQDKETGESVNKDK | 1,3,11 | S | JGMV-IS | 11- 28 |
| VVNNAGKDNEQQLEFKIEPMYK | 1,7,16 | S | WSMV-IHC | 149-170 |
| DKIKPEMINNMIK | 1,10,12 | M | WSMV-IHC | 72- 84 |
| ASTATKDKDVDVGSTGTFVIPK | 1,15,17 | S | JGMV-KS | 62- 83 |
| SGGTKASTATKDKDVDVGSTGTFVIPK | 1,15,17,23 | S | JGMV-KS | 57- 83 |
| Arginine-less peptides containing one or more histidine residues: | ||||
| GSTNHMFGL | 5 | W | JGMV-NI | 254-262 |
| GSTNHMFGLDGNVGE | 11 | W | JGMV-NI | 254-268 |
| NMHSLLGVQQSH | 1,10 | M | JGMV-IS | 293-304 |
| Arginine-less peptides containing lysine and histidine residues: | ||||
| DHLIQYKP | 2,7 | W | JGMV-NI | 95-102 |
| PHELSSEK | 1,7 | M | BMV | 74- 81 |
| PHELSSEKNK | 1,3,9 | M | BMV | 74- 83 |
ND = not detected
Basic residues are underlined. Sequences containing more than one arginine or no basic residues are not included.
Position of residue counting from the C-terminus
Table S3 also yields information on the longest and shortest peptide fragment for which the (bn−1 + H2O) ion was observed. The longest peptide we found had 27 residues and the shortest had three. We note also that Farrugia and O’Hair found that the (bn−1 + H2O) ion was insignificant in the CID mass spectrum of the [M + H]+ ion of the dipeptide arg.gly, where the (b1 + H2O) ion should be observed at m/z 175; a small peak at this value was attributed to the isobaric y1 ion of gly.arg [45]. In that case a gas phase rearrangement reaction was proposed that led to identical tandem mass spectra for arg.gly and gly.arg via a mixed anhydride intermediate. We suggest that this alternative reaction pathway, not available for longer peptides because it requires an adjacent terminal amino group, switches off the pathway leading to the (b1 + H2O) ion.
Mistakes in sequencing
As mentioned above, we first made contact with (bn−1+H2O) ions when sequencing some of the JGMV peptides. In a number of cases, our interpretation of prominent ions in the spectra as y ions led to sequences that disagreed with those deduced from the nucleotide data in the databases. Two examples are given here:
RMNLDEPYMPR vs MRNLDEPYMPR– Here a straightforward interpretation of the mass spectrum (Fig. S2) led to the first sequence, assuming that a prominent ion at 1265.57 Da was interpreted as y10. The second sequence, the one predicted by the nucleotide data, was in apparent disagreement. The definitive low mass ions were not observed, but re-interpretation of the 1265.57 Da ion as a (b10+H2O) ion reconciled the observed spectrum with the nucleotide prediction (see Table S5). Note the presence of non-C terminal arginine. Also note that in this case the predicted masses in the two sequences are identical so higher mass accuracy is of no help.
YHEEFNRWY vs ATQEEFNRWY– Again, a straightforward interpretation of the mass spectrum (Fig. S3) led to the first sequence, with a prominent ion at 1180.52 Da interpreted as y8, but re-interpretation of this ion as a (b9+H2O) ion reconciled the observed spectrum with the second sequence, the nucleotide prediction (see Table S6). Note again the presence of non-C-terminal arginine. In this case there is a mass difference of ~ 22 mDa between the two possible sequences, so higher mass accuracy could have distinguished between them.
Chemical computations
As noted above, amino acid basicities have often been invoked in discussing the fragmentation of protonated peptides. The gas phase basicities and proton affinities (PAs) of amino acids and peptides have been reviewed [46]; the highest PAs were observed for arginine (244.8), lysine (235.6), histidine (231.5) and tryptophan (223.9 kcal/mol) (these values closely parallel their relative basicities). In the cases of free arginine, histidine and tryptophan, their side chains are intrinsically more basic than their terminal amino groups. In the case of lysine, the basicities of the side chain and terminal amino group are similar (if the ~2 kcal/mol reduction in basicity of the latter, see below, is neglected). The basicities of free lysine and arginine are enhanced by ~13 kcal/mol compared to the basicities of their side chains, while those of histidine and tryptophan are increased by ~4 and ~2 kcal/mol, respectively. In the case of lysine the enhancement was ascribed to proton bridging between the side chain and the amino terminus. The net result is that the order of basicities is tryptophan ≪ histidine ≤ lysine ≪ arginine.
Of more concern to the present discussion are the relative basicities of the amino acid residues in a peptide, where their amino groups have been converted to the much less basic amide groups of the peptide bond, and are less effective in their H-bond interactions with the protonated side chain. However, proton bridging between a protonated side chain and a carbonyl group is still possible. To assess the importance of the bridging contribution, the relative stabilities of various protonated structures of lysine, arginine and histidine were computed, as described in the experimental section. The results are presented in Figs. 2–4. For each amino acid, the hydrogen bond lengths, bond angles at hydrogen, and the computed energies relative to the most stable structure are shown. Because entropy differences among the charged structures of a given amino acid are similar, the energy differences in Figs. 2–4, when combined with PA data for the amino acids can be directly related to either relative basicities or relative PAs of the amino acids, as utilized in the ensuing discussion.
Figure 2.

Structures of protonated lysine showing H-bonding interactions: protonated N-terminus to side-chain NH2 group (structure 1); protonated side-chain to C=O group (structure 2) and to terminal NH2 group (structure 3). Hydrogen bonds are denoted by dashed lines. The indicated bond lengths and bond angles were optimized by the semi-empirical PM3 method. Relative single point energies (kcal/mol) for the optimized geometries were computed by the HF/6–31G*//PM3 and DFT/pBP/DN**//PM3 methods
Figure 4.

Structures of protonated histidine showing H-bonding interactions: protonated imidazole ring to terminal NH2 group (structure 8) and to the C=O group (structure 9). See Fig. 3 for explanation of notations.
In the case of lysine, Fig. 2, the flexible side chain permits nearly ideal H-bonding interactions. The H-bond lengths of ~1.74 Å in each structure are as expected, and the bond angles at hydrogen are not significantly reduced from the ideal 180° angle. Both computational methods slightly favor structure 3 in which the side chain protonated amino group is H-bonded to the terminal amino group. Structure 2, having the protonated side chain amino group H-bonded to the carbonyl oxygen, is less stable than this by 1.2 kcal/mol (HF/6–31G*//PM3) or by 2.4 kcal/mol (DFT/pBP/DN**//PM3). Structure 1, having the protonated terminal amino group H-bonded to the side chain amino group, is less stable than 3 by 5.1 kcal/mol (HF/6–31G*//PM3) or by 1.7 kcal/mol (DFT/pBP/DN**//PM3). (It is a moot point whether 1 and 3 are different structures; they differ only by the position of the proton in an asymmetric double potential energy well). Hehre et al. [44] also found, at the (HF/6–31G*//PM3) theory level, that protonation at the side chain amino group is favored over protonation of the amino group at the N-terminus, but the structure proton-bridged to carbonyl oxygen 2 was slightly favored. The small preference for side chain protonation is consistent with the observation that the basicity of the terminal amino group is reduced by ~2 kcal/mol by the inductive effect of the COOH group [44]. In summary, a lysine residue in a peptide appears to be no more than ~2 kcal/mol less basic than free lysine.
In the case of arginine, Fig. 3, only structures involving protonation at the highly basic guanidino function need to be considered. The protonated guanidino function has two sites that can potentially undergo H-bonding interactions with either the amino terminus or with the carbonyl oxygen to yield the four proton-bridged structures shown. The relative energies computed are in good agreement for either computational method for all four structures. The energy differences among the structures are small. Structures 6 and 7, involving proton bridging to the NH2 terminus, are favored. Structures 5 and 6, which differ in energy by 2.8 (HF) or 4.5 (DFT) kcal/mol represent the more stable of the amino- and carbonyl-bridged structures, respectively. Thus an arginine residue in a peptide appears to be no more than ~ 4 kcal/mole less basic than free arginine.
Figure 3.

Structures of protonated arginine showing H-bonding interactions: protonated guanido group (NH) to C=O group (structure 4) and to terminal NH2 group (structure 6); protonated guanido group (NH2) to C=O group (structure 5) and to terminal NH2 group (structure 7). See Fig. 3 for explanation of notations.
In the case of histidine, Fig. 4, protonation at nitrogen of the imidazole ring has been assumed because this site is significantly more basic than the amino function. Two structures are shown, one involving proton bridging to carbonyl oxygen, the other involving proton bridging to the amino function. The energies of the two structures are virtually the same, the two computational methods disagreeing on whether 8 or 9 is more stable. Structurally, the H-bonding interactions appear favorable, yet the basicity enhancement of free histidine over its side chain, noted in Harrison’s review [46], is only ~4 kcal/mol.
The remaining common amino acid that should be considered is tryptophan. Although it is less strongly basic than those already discussed, its side-chain is still a favored protonation site compared to others, such as amide linkages. In this case, molecular models show that it is not possible for the protonated site of the indole group to approach either the amino or carbonyl function closely enough to undergo H-bonding interactions, so that the basicity of tryptophan is not enhanced by these interactions, and remains relatively low when compared to lysine, arginine and histidine. Moreover, and certainly of critical significance here, a protonated tryptophan side chain is unable to transport a proton to a carbonyl group, whether or not it is basic enough to do so.
We can now assess the relative basicities of the amino acid residues in a peptide. As noted above, the relatively high basicity of free lysine has been ascribed to proton bridging between the basic side chain and the NH2 terminus. However, the computations reveal that there is little energy difference between H-bonding to the amino group or to the carbonyl oxygen. (We have ignored the possibility of H-bonding to the amide nitrogen. High level ab initio calculations have shown the oxygen atom to be the most basic centre in simple amides [47,48].) Indeed, arginine, lysine and histidine residues are all expected to have high basicity in a peptide owing to favorable H-bonding interactions of their protonated side-chains with adjacent carbonyl groups, either their own or that of the preceding residue (counting from the N-terminus). In the case of arginine, the interaction of the side chain and the carbonyl oxygen of an adjacent amino acid was estimated to result in a stabilization of 24–30 kcal/mol [4], a value significantly higher than expected from the experimental value of ~13 kcal/mol noted in Harrison’s review [46]. Tryptophan is unable to undergo H-bonding interactions or to transport a proton to the carbonyl function.
Of the mechanisms proposed for formation of the (bn−1 + H2O) ion, the arguments presented here favor a modified form of the mechanism shown in Scheme 3. Here it has has been generalized (Scheme 4) to accommodate the observation that the (bn−1 + H2O) ion can be formed when the protonated side chain is remote from the C-terminus. The essential feature is the transport of a proton to the n−1 carbonyl group by a protonated side chain to give a proton-bridged structure in which increased positive charge on the carbonyl carbon makes it more susceptible to nucleophilic attack by the C-terminal hydroxyl group, and concomitant decomposition by concerted electron shifts to give stable products, as shown. (The proposal of a neutral carboxyl group is consistent with the preference for the neutral bridged structure rather than a “zwitterion” in guanidino-carboxylic acid systems [49,50].) Support for the 5-membered cyclic intermediate shown in Schemes 3 and 4 is provided by the 18O labelling results of Ballard and Gaskell [2], which confirm oxygen exchange between the n−1 carbonyl and the C-terminal carboxyl group through cyclic intermediates, and is consistent with kinetic energy loss experiments [6]. Moreover, it is analogous to one of the mechanisms proposed for decomposition of sodiated peptides [51,52] (although we realize that an alternative mechanism has also been proposed for [M + Na]+ decompositions [53]).
Scheme 4.

Formation of the (bn−1 + H2O) ion for peptides containing a non-C-terminal arginine residue.
It has been noted that protonation at carbonyl oxygen strengthens the peptide bond whereas protonation at the less basic amide nitrogen weakens it [13,54,55]. However, to compensate, nucleophilic attack by hydroxyl oxygen at the carbonyl carbon would weaken the peptide bond and facilitate the decompositions depicted in Schemes 3 and 4. It should be born in mind that the population of this reactively suitable conformation would, in general, decrease as the protonated side chain becomes more remote, and as competition from other carbonyl oxygen potential H+-binding sites increases. In the collision region q2 of our QqTOF mass spectrometer, the ions are progressively activated by a sequence of low energy collisions so that, initially, the small population of conformationally suitable ions reacts to give (bn−1 + H2O). As the reactive population is depleted it is replenished from the bulk population by re-equilibration among the conformations.
We consider this mechanism to be more easily reconciled with the body of experimental evidence discussed here than alternatives presented earlier, including decomposition of activated precursor ions by a charge-remote mechanism, or one involving energetically less-favorable H-bonding to the less basic amide nitrogen. The first alternative is weakened by the experiment in which a fixed charge is placed on the N-terminus, whereupon the (bn−1 + H2O) ion is not observed [2]. As well, although the side chain of tryptophan is intrinsically basic, its protonated side chain is unable to transport a proton to carbonyl oxygens, so the (bn−1 + H2O) ion is not observed. The second alternative would weaken the peptide bond, and it has been emphasized [13,24] that protonation of backbone amide nitrogens promotes formation of bn and yn ions, which are suppressed when the (bn−1 + H2O) ion is enhanced. Sequestering of the proton by an arginine side chain reduces the populations of random amide N-protonated species.
The formation of (bk + H2O) ions from [M + H]+ ions of n-residue peptides in which the (k+1)th residue is serine or threonine has also been observed [56]. In these cases, the decomposition was believed to be assisted by the hydroxyl group of serine or threonine H-bonding to the kth carbonyl oxygen. The exceptions listed in footnotes b and c of Table 2 do not conform to the general observations on the requirement for the presence of a non-C-terminal basic residue, and it is intriguing that all but three of these peptides contain serine or threonine. While the mechanism proposed [56] for formation of the (bk + H2O) ion is inadequate to explain formation of the (bn−1 + H2O) ion here, perhaps H-bonding interactions of the hydroxyl groups with the n−1 carbonyl groups can facilitate its formation. Peptide conformation may play a significant role here, but mechanisms involving the protonated N-terminus may also need to be invoked.
CID of a collection of tryptic peptides that had been generated for measurement of chromatographic retention times
This collection contained 1200 peptides, from digests of 46 proteins. It differed from the collection of viral coat proteins discussed above in that these digests all employed trypsin, and the digest conditions were considerably more drastic (typically overnight). Thus these conditions are more typical of those commonly used in sequencing applications. As might be expected from the conditions used, the fraction of spectra exhibiting missed cleavages, and the fraction of non-C-terminal basic residues in the daughter ions observed, were both much smaller than in the first set. Consequently, the percentage of bn−1+18 ions observed was also considerably smaller. Table S7 lists the 190 cases of bn−1+18 decay observed (~ 16%), [strong 87, medium 60 weak 43]; an example of the results from the digestion of one of the proteins (bovine serum albumin) is given in Table 4.
Table 4.
Peptides from Bovine Serum Albumin (BSA) whose [M+H]+ ions yield [bn−1+H2O] ions in their CID mass spectra
| Peptide | Peptide Sequence | [M+H]+ m/z | [bn−1+H2O]Δ/[bn−1] | abundance* |
|---|---|---|---|---|
| 29-34 | SEIAHR | 712.370 | 27/47 | M |
| 123-130 | NECFLSHK | 1034.470 | 49/58 | S |
| 310-318 | SHCIAEVEK | 1072.508 | 32/55 | M |
| 460-468 | CCTKPESER | 1166.488 | 25/36 | M |
| 35-44 | FKDLGEEHFK | 1249.617 | 114/287 | W |
| 402-412 | HLVDEPQNLIK | 1305.723 | 17/95 | W |
| 89-100 | SLHTLFGDELCK | 1419.683 | 35/64 | M |
| 360-371 | RHPEYAVSVLLR | 1439.794 | 1467/173 | S |
| 76-88 | TCVADESHAGCEK | 1463.591 | 30/36 | M |
| 298-309 | LKECCDKPLLEK | 1532.795 | 29/42 | M |
| 437-451 | KVPQVSTPTLVEVSR | 1639.937 | 86/64 | S |
| 267-280 | ECCHGDLLECADDR | 1749.657 | 30/157 | W |
| 508-523 | RPCFSALTPDETYVPK | 1880.927 | 184/40 | S |
| 529-544 | LFTFHADICTLPDTEK | 1907.915 | 15/29 | M |
| 168-183 | RHPYFYAPELLYYANK | 2045.057 | 23/15 | S |
>1, S (strong); 0.5–1, M (medium); <0.5, W (weak)
ratio of counts
A significant difference from the first set can be noted. Since the percentage of ions containing non-C-terminal arginines or lysines is greatly reduced, the percentage of (bn−1 + H2O) decays caused by histidine is much larger (~54%), [32 strong, 39 medium and 31 weak], supporting our interpretation of non-C-terminal histidine as a significant cause of this decay mode. We note also that there appears to be no particular requirement for the non-C-terminal histidine to be close to the C-terminus; Table S7 shows that it can be as much as 17 residues away (in Rabbit Aldolase 154–173).
A second difference is the much larger number of ions observed in this set that contained tryptophan as the sole non-C-terminal basic residue (114), listed in Table S8. In this case, (bn−1 + H2O) ions were observed in only 3 cases (two of which contained serine), confirming the ineffectiveness of tryptophan in producing (bn−1 + H2O) ions.
Thus the results of this analysis give additional support to the general picture of (bn−1 + H2O) ion production given by the viral coat protein measurements.
CONCLUSIONS
Peptide sequencing by mass spectrometry is most readily achieved in those peptides in which protonation occurs randomly at the backbone amide nitrogen atoms (the “mobile proton” model), to give significant yields of sequence-specific b–series and y–series ions. In cases where the peptide contains highly basic residues, i.e. histidine, lysine, and especially arginine, (but not tryptophan), the populations of the backbone-protonated species are reduced, while populations having the protons localized on a basic side chain are enhanced to the point at which a rearrangement leading to a prominent (bn−1 + H2O) product ion by loss of the C-terminal amino acid occurs. The fragmentation is not specific to the residue position (except that it cannot be located at the C-terminus). These observations should be useful in assisting peptide sequence determinations, especially when typical sequence information ions are of low abundance.
Supplementary Material
Figure S1. Schematic diagram of the orthogonal MALDI QqTOF instrument.
Figure S2. CID spectrum of a peptide at m/z 1421.67 from a Glu-C digest of the JGMV coat protein.
Figure S3. CID spectrum of a peptide at m/z 1343.62 from a Lys-C digest of the JGMV coat protein.
Acknowledgments
We thank Dr. S. Haber (Cereal Research Centre, Agriculture and Agrifood Canada, Winnipeg, MB) and Dr. D. L. Seifers (Agricultural Research Center, Kansas State University, Manhattan, KS) for providing the virus coat protein samples. We are grateful to J. McNabb for technical support. The work was supported by grants from the U. S. National Institutes of Health (GM59240), and from the Natural Sciences and Engineering Research Council (NSERC) of Canada.
Footnotes
Preliminary accounts of some of these measurements were given at the 12th Lake Louise Workshop on Tandem Mass Spectrometry (1999), and at the 52nd Annual Conference of the American Society for Mass Spectrometry (2004).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thorne GC, Ballard KD, Gaskell SJ. Metastable Decomposition of Peptide [M + H]+ Ions via Rearrangement Involving Loss of the C-Terminal Amino Acid Residue. J Am Soc Mass Spectrom. 1990;1:249–257. [Google Scholar]
- 2.Ballard KD, Gaskell SJ. Intramolecular [O-18] Isotopic Exchange in the Gas-Phase Observed During the Tandem Mass-Spectrometric Analysis of Peptides. J Am Chem Soc. 1992;114:64–71. [Google Scholar]
- 3.Gonzalez J, Besada V, Garay H, Reyes O, Padron G, Tambara Y, Takao T, Shimonishi Y. Effect of the Position of a Basic Amino Acid on C-Terminal Rearrangement of Protonated Peptides Upon Collision-Induced Dissociation. J Mass Spectrom. 1996;31:150–158. doi: 10.1002/(SICI)1096-9888(199602)31:2<150::AID-JMS287>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 4.Vachet RW, Asam MR, Glish GL. Secondary Interactions Affecting the Dissociation Patterns of Arginine-Containing Peptide Ions. J Am Chem Soc. 1996;118:6252–6256. [Google Scholar]
- 5.Deery MJ, Summerfield SG, Buzy A, Jennings KR. A Mechanism for the Loss of 60 u from Peptides Containing an Arginine Residue at the C-Terminus. J Am Soc Mass Spectrom. 1997;8:253–261. [Google Scholar]
- 6.Roepstorff P, Fohlman J. Proposal for a Common Nomenclature for Sequence Ions in Mass Spectra of Peptides. Biomed Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 7.Biemann K. Contributions of Mass Spectrometry to Peptide and Protein Structure. Biomed Environ Mass Spectrom. 1988;16:99–111. doi: 10.1002/bms.1200160119. [DOI] [PubMed] [Google Scholar]
- 8.Dongré AR, Somogyi Á, Wysocki VH. Surface-Induced Dissociation: An Effective Tool to Probe Structure, Energetics and Fragmentation Mechanisms of Protonated Peptides. J Mass Spectrom. 1996;31:339–350. doi: 10.1002/(SICI)1096-9888(199604)31:4<339::AID-JMS322>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 9.Hunt DF, Yates JR, III, Shabanowitz J, Winston S, Hauer CR. Protein Sequencing by Tandem Mass Spectrometry. Proc Natl Acad Sci USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biemann K, Martin SA. Mass Spectrometric Determination of the Amino Acid Sequence of Peptides and Proteins. Mass Spectrom Rev. 1987;6:1–76. [Google Scholar]
- 11.Mueller DR, Eckersley M, Richter WJ. Hydrogen Transfer Reactions in the Formation of “y + 2” Sequence Ions from Protonated Peptides. Org Mass Spectrom. 1988;23:217–222. [Google Scholar]
- 12.Kenny PT, Nomoto K, Orlando R. Fragmentation Studies of Peptides: The Formation of y Ions. Rapid Commun Mass Spectrom. 1992;6:95–97. doi: 10.1002/rcm.1290060205. [DOI] [PubMed] [Google Scholar]
- 13.Burlet O, Orkiszewski RS, Ballard KD, Gaskell SJ. Charge Promotion of Low-Energy Fragmentations of Peptide Ions. Rapid Commun Mass Spectrom. 1992;6:658–662. doi: 10.1002/rcm.1290061106. [DOI] [PubMed] [Google Scholar]
- 14.Burlet O, Yang CY, Gaskell SJ. Influence of Cysteine to Cysteic Acid Oxidation on the Collision-Activated Decomposition of Protonated Peptides –Evidence for Intraionic Interactions. J Am Soc Mass Spectrom. 1992;3:337–344. doi: 10.1016/1044-0305(92)87061-3. [DOI] [PubMed] [Google Scholar]
- 15.Tang XJ, Thibault P, Boyd RK. Fragmentation Reactions of Multiply-Protonated Peptides and Implications for Sequencing by Tandem Mass Spectrometry With Low-Energy Collision-Induced Dissociation. Anal Chem. 1993;65:2824–2834. doi: 10.1021/ac00068a020. [DOI] [PubMed] [Google Scholar]
- 16.Jones JL, Dongré AR, Somogyi Á, Wysocki VH. Sequence Dependence of Peptide Fragmentation Efficiency Determined by Electrospray Ionization/Surface/Induced Dissociation Mass Spectrometry. J Am Chem Soc. 1994;116:8368–8369. [Google Scholar]
- 17.Cox KA, Gaskell SJ, Morris M, Whiting A. Role of the Site of Protonation in the Low-Energy Decompositions of Gas-Phase Peptide Ions. J Am Soc Mass Spectrom. 1996;7:522–531. doi: 10.1016/1044-0305(96)00019-0. [DOI] [PubMed] [Google Scholar]
- 18.Summerfield SG, Cox KA, Gaskell SJ. The Promotion of d-Type Ions During the Low Energy Collision-Induced Dissociation of Some Cysteic Acid-Containing Peptides. J Am Soc Mass Spectrom. 1997;8:25–31. [Google Scholar]
- 19.Summerfield SG, Gaskell SJ. Fragmentation Efficiencies of Peptide Ions Following Low Energy Collisional Activation. Int J Mass Spectrom Ion Proc. 1997;165:509–521. [Google Scholar]
- 20.Sharp JS, Tomer KB. Formation of [(b(n−1) + OH + H]+ Ion Structural Analogs by Solution-Phase Chemistry. J Am Soc Mass Spectrom. 2005;16:607–621. doi: 10.1016/j.jasms.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Schnier PD, Price WD, Jockusch RA, Williams ER. Blackbody Infrared Radiative Dissociation of Bradykinin and Its Analogues: Energetics, Dynamics, and Evidence for Salt-Bridge Structures in the Gas Phase. J Am Chem Soc. 1996;118:7178–7189. doi: 10.1021/ja9609157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell S, Rodgers MT, Marzluff EM, Beauchamp JL. Deuterium Exchange Reactions as a Probe of Biomolecule Structure. Fundamental Studies of Gas Phase H/D Exchange Reactions of Protonated Glycine Oligomers with D2O, CD3OD, CD3CO2D, and ND3. J Am Chem Soc. 1995;117:12840–12854. [Google Scholar]
- 23.Dikler S, Kelly JW, Russell DH. Improving Mass Spectrometric Sequencing of Arginine-Containing Peptides by Derivatization With Acetylacetone. J Mass Spectrom. 1997;32:1337–1349. doi: 10.1002/(SICI)1096-9888(199712)32:12<1337::AID-JMS599>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 24.Tsaprailis G, Nair H, Somogyi Á, Wysocki VH, Zhong W, Futrell JH, Summerfield SG, Gaskell SJ. Influence of Secondary Structure on the Fragmentation of Protonated Peptides. J Am Chem Soc. 1999;121:5142–5154. [Google Scholar]
- 25.Thorne GC, Gaskell SJ. Elucidation of Some Fragmentations of Small Peptides Using Sequential Mass Spectrometry on a Hybrid Instrument. Rapid Commun Mass Spectrom. 1989;3:217–221. doi: 10.1002/rcm.1290030704. [DOI] [PubMed] [Google Scholar]
- 26.Lane LC, Kaesburg P. Multiple Genetic Components in Bromegrass Mosaic Virus. Nature New Biol. 1971;232:40–43. doi: 10.1038/newbio232040a0. [DOI] [PubMed] [Google Scholar]
- 27.Haber S, Hamilton RI. Brome Mosaic Virus Isolated in Manitoba, Canada. Plant Dis. 1989;73:195–199. [Google Scholar]
- 28.She YM, Haber S, Seifers DL, Loboda A, Chernushevich I, Perreault H, Ens W, Standing KG. Determination of the Complete Amino Acid Sequence for the Coat Protein of Brome Mosaic Virus by Time-of-Flight Mass Spectrometry. Evidence for Mutations Associated with Change of Propagation Host. J Biol Chem. 2001;276:20039–20047. doi: 10.1074/jbc.M100189200. [DOI] [PubMed] [Google Scholar]
- 29.Seifers DL, Harvey TL, Haber S, She YM, Chernushevich I, Ens W, Standing KG. Natural Infection of Sorghum by Foxtail Mosaic Virus in Kansas. Plant Dis. 1999;83:905–912. doi: 10.1094/PDIS.1999.83.10.905. [DOI] [PubMed] [Google Scholar]
- 30.Bancroft JB, Rouleau M, Johnston R, Prins L, Mackie GA. The Entire Nucleotide Sequence of Foxtail Mosaic Virus RNA. J Gen Virology. 1991;72:2173–2181. doi: 10.1099/0022-1317-72-9-2173. [DOI] [PubMed] [Google Scholar]
- 31.Stenger DC, Seifers DL, French R. Patterns of Polymorphism in Wheat Streak Mosaic Virus: Sequence Space Explored by a Clade of Closely Related Viral Genotypes that Rivals that between the Most Divergent Strains. Virology. 2002;302:58–70. doi: 10.1006/viro.2001.1569. [DOI] [PubMed] [Google Scholar]
- 32.Seifers DL. Partial Characterization of a Colorado Isolate of Agropyron Mosaic Virus. Plant Dis. 1992;76:564–569. [Google Scholar]
- 33.Seifers DL, Haber S, Ens She Y-M, Standing KG, Salomon R. Characterization of a Distinct Johnsongrass mosaic virus Strain Isolated from Sorghum in Nigeria. Arch Virol. 2005;150:557–576. doi: 10.1007/s00705-004-0411-y. [DOI] [PubMed] [Google Scholar]
- 34.Seifers DL, Salomon R, Marie-Jeanne V, Alliot B, Signoret P, Haber S, Loboda A, Ens W, She YM, Standing KG. Characterization of a Novel Potyvirus Isolated from Maize in Israel. Phytopathology. 2000;90:505–513. doi: 10.1094/PHYTO.2000.90.5.505. [DOI] [PubMed] [Google Scholar]
- 35.She YM, Seifers DL, Haber S, Ens W, Standing KG. Characterization of the Agent of High Plains Disease: Mass Spectrometry Determines the Sequence of the Disease-Specific Protein. J Biol Chem. 2004;279:488–494. doi: 10.1074/jbc.M308506200. [DOI] [PubMed] [Google Scholar]
- 36.Swiss Prot/TrEMBL databases. Swiss Institute of Bioinformatics; Geneva, Switzerland: [(last accessed April 2004)]. http://www.expasy.ch. [Google Scholar]
- 37.National Center for Biotechnology Information. [(last accessed April 2004)]; http://www.ncbi.nlmnih.gov:80/entrez.
- 38.Shevchenko A, Chernushevich IV, Ens W, Standing KG, Thomson B, Wilm M, Mann M. Rapid ‘de Novo’ Peptide Sequencing by a Combination of Nanoelectrospray, Isotopic Labeling, a, Quadrupole/Time-of-Flight Mass Spectrometer. Rapid Commun Mass Spectrom. 1997;11:1015–1024. doi: 10.1002/(SICI)1097-0231(19970615)11:9<1015::AID-RCM958>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 39.Chernushevich IV, Ens W, Standing KG. Orthogonal-Injection TOFMS for Analyzing Biomolecules. Anal Chem. 1999;71:452A–461A. doi: 10.1021/ac990524l. [DOI] [PubMed] [Google Scholar]
- 40.Loboda AV, Krutchinsky AN, Bromirski M, Ens W, Standing KG. A Tandem Quadrupole/Time-of-Flight Mass Spectrometer with a Matrix-Assisted Laser Desorption/Ionization Source: Design and Performance. Rapid Commun Mass Spectrum. 2000;14:1047–1057. doi: 10.1002/1097-0231(20000630)14:12<1047::AID-RCM990>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 41.Krokhin OV, Craig R, Spicer V, Ens W, Standing KG, Beavis RC, Wilkins JA. An improved model for prediction of retention times of tryptic peptides in ion-pair reverse-phase HPLC; its application to protein peptide mapping by off-line HPLC-MALDI MS. Mol Cell Proteomics. 2004;3:908–919. doi: 10.1074/mcp.M400031-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Krokhin OV, Ying S, Cortens JP, Ghosh D, Spicer V, Ens W, Standing KG, Beavis RC, Wilkins JA. Use of peptide retention time prediction for protein identification by off-line reversed-phase HPLC-MALDI MS/MS. Anal Chem. 2006;78:6265–6269. doi: 10.1021/ac060251b. [DOI] [PubMed] [Google Scholar]
- 43.Krokhin OV, Cheng K, Sousa S, Ens W, Standing KG, Wilkins JA. Mass Spectrometric Based Mapping of the Disulphide Bonding Patterns of Integrin α Chains. Biochemistry. 2003;42:12950–12959. doi: 10.1021/bi034726u. [DOI] [PubMed] [Google Scholar]
- 44.Hehre WJ, Yu J, Klunzinger PE, Lou L. A Brief Guide to Molecular Mechanics and Quantum Chemical Calculations. Wavefunction, Inc; Irvine, CA: 1998. [Google Scholar]
- 45.Farrugia JM, O’Hair RAJ. Involvement of Salt Bridges in a Novel Gas Phase Rearrangement of Protonated Arginine-Containing Dipeptides which precedes Fragmentation. Int J Mass Spectrom. 2003;222:229–242. [Google Scholar]
- 46.Harrison AG. The Gas-Phase Basicities and Proton Affinities of Amino Acids and Peptides. Mass Spectrom Rev. 1997;16:201–217. [Google Scholar]
- 47.Herreros M, Gal JF, Maria PC, Decouzon M. Gas-Phase Basicity of Simple Amides Toward Proton and Lithium Cation: An Experimental and Theoretical Study. Eur Mass Spectrom. 1999;5:259–265. [Google Scholar]
- 48.Tortajada J, Léon E, Morizur JP, Luna A, Mó O, Yáñez M. Potential Energy Surface of Protonated Formamide and of Formamide-X+ (X = Li, Na, Mg, and Al) Complexes. J Phys Chem. 1995;99:13890–13898. [Google Scholar]
- 49.Melo A, Ramos MJ. Proton Transfer in Arginine-Carboxylate Interactions. Chem Phys Lett. 1995;245:498–502. [Google Scholar]
- 50.Zheng YJ, Ornstein RL. What Happens to Salt-Bridges in Nonaqueous Environments: Insights from Quantum Mechanics Calculations. J Am Chem Soc. 1996;118:11237–11243. [Google Scholar]
- 51.Tang X, Ens W, Standing KG, Westmore JB. Daughter Ion Mass Spectra from Cationized Molecules of Small Oligopeptides in a Reflecting Time-of-Flight Mass Spectrometer. Anal Chem. 1988;60:1791–1799. doi: 10.1021/ac00168a029. [DOI] [PubMed] [Google Scholar]
- 52.Teesch LM, Orlando RC, Adams J. Location of the Alkali Metal Atom in Gas-Phase Peptide Complexes. J Am Chem Soc. 1991;113:3668–3675. [Google Scholar]
- 53.Grese RP, Cerny RL, Gross ML. Metal Ion-Peptide Interactions in the Gas Phase: A Tandem Mass Spectrometry Study of Alkali Metal Cationized Peptides. J Am Chem Soc. 1989;111:2835–2842. [Google Scholar]
- 54.McCormack AL, Somogyi Á, Dongré AR, Wysocki VH. Fragmentation of Protonated Peptides: Surface-Induced Dissociation in Conjunction with a Quantum Mechanical Approach. Anal Chem. 1993;65:2859–2872. doi: 10.1021/ac00068a024. [DOI] [PubMed] [Google Scholar]
- 55.Somogyi Á, Wysocki VH, Mayer I. The Effect of Protonation Site on Bond Strengths in Simple Peptides: Application of ab initio and Modified Neglect of Differential Overlap Bond Order and Modified Neglect of Differential Overlap Energy Partitioning. J Am Soc Mass Spectrom. 1994;5:704–717. doi: 10.1016/1044-0305(94)80002-2. [DOI] [PubMed] [Google Scholar]
- 56.Fang S, Takao T, Satomi Y, Mo W, Shimonishi Y. Novel Rearranged Ions Observed for Protonated Peptides via Metastable Decomposition in Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. J Am Soc Mass Spectrom. 2000;11:345–351. doi: 10.1016/s1044-0305(99)00153-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic diagram of the orthogonal MALDI QqTOF instrument.
Figure S2. CID spectrum of a peptide at m/z 1421.67 from a Glu-C digest of the JGMV coat protein.
Figure S3. CID spectrum of a peptide at m/z 1343.62 from a Lys-C digest of the JGMV coat protein.