

Abstract
Previous studies showed that the gp120 envelope protein of HIV-1 is able to crosslink membrane IgM on normal human B cells and to induce their activation in a VH3 immunoglobulin gene-family-specific manner. Because this VH gene family is the largest in the human repertoire, this superantigen (SAg) property is thought to have deleterious consequences for the host, including a progressive decline of B cells with progression of the HIV-1-induced disease. Here, we have identified the sequence motifs on gp120 involved in SAg binding to normal Igs. We show that this SAg-binding activity is present in gp120s from highly divergent isolates of HIV-1 belonging to clades derived from various geographical origins, and that carbohydrate residues are not essential for its expression. The SAg-binding site is formed by protein sequences from two regions of the gp120 molecule. The core motif is a discontinuous epitope spanning the V4 variable domain and the amino-terminal region flanking the C4 constant domain. The most critical residues appear to be Leu395–Asp397 and Ile425–Gln427. Residues from the C2 constant domain (positions 252–272) also seem to play an accessory role in SAg binding of gp120 to normal human Igs. These findings are important in the design of a successful gp120-based vaccine against HIV-1.
Keywords: human antibodies, immunoglobulin variable genes
After HIV infection, strong humoral and cellular immune responses are developed to various viral antigens, including the envelope glycoprotein gp120. The sequence of this molecule can be subdivided into five variable regions (V1–V5) with 25% or fewer conserved residues, and five constant regions (C1–C5) with greater than 75% conservation (1). By virtue of its accessibility to immune effectors, this glycoprotein has been shown to elicit virus-neutralizing antibodies in a variety of experimental systems and to bind a portion of the neutralizing antibodies present in sera from HIV-1-infected subjects (2). Remarkably, the important immunological determinants of gp120 are present on both its constant and variable regions. Because of its strong immunogenicity and its binding to CD4, the viral coreceptor, gp120 is considered a potential subunit vaccine candidate against this virus (3).
However, in addition to its role in triggering potentially protective responses, gp120 is known to participate in responses detrimental to the host, for example, by triggering autoimmune responses (4), by inhibiting natural killer activity (5), by enhancing HIV infectivity (6), and by exhibiting superantigen (SAg)-like properties for B cells (7). More specifically, gp120 binds a subpopulation of B cells from normal individuals by means of immunoglobulins of the VH3 gene family and selectively induces Ig secretion by VH3+ B cells (7). This SAg-like property is thought to be responsible for the increase in VH3+ B cells during early stages of the infection followed by their progressive decline during advanced stages of the disease (8, 9). Given the severity of this B cell depletion, we have characterized the epitopes on the gp120 molecule engaged in B cell SAg interactions.
MATERIALS AND METHODS
Recombinant Proteins.
Recombinant gp120s from different HIV-1 isolates were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health from MicrogeneSys (West Haven, CT) (gp120CM, gp120LAV, gp120MN) and from K. Steimer (Chiron) (gp120SF2). Recombinant gp160 proteins were provided by M.-P. Kieny (Transgene, Strasbourg, France). Their characteristics are summarized in Table 1.
Table 1.
Recombinant HIV envelope proteins used
| Recombinant envelope protein | gp120 isolate | Clade | Geographic origin | Expression system |
|---|---|---|---|---|
| gp120SF2 | SF2 | B | U.S.A. | CHO cells |
| gp120SF2 | SF2 | B | U.S.A. | Yeast (strain 2150) |
| gp120LAV | LAI | B | France | Insect cells |
| gp120MN | MN | B | U.S.A. | Insect cells |
| gp120CM | CM | E | Thailand | Insect cells |
| gp160BRU | LAI | B | France | BHK cells |
| gp160MN/LAI | MN | B | U.S.A. | BHK cells |
| gp160RF/LAI | RF | B | U.S.A. | BHK cells |
| gp160ELI/LAI | ELI | D | Zaire | BHK cells |
| gp1604023/LAI | 4023 | D | Central Africa | BHK cells |
Synthetic Peptides.
Peptides covering the entire sequence of gp120 were obtained from the Agence Nationale de la Recherche sur le SIDA (Paris), and through the AIDS Research and Reference Reagent Program. All peptides were soluble at neutral pH, except peptides SP9045, SP89247, and SP89049, which had to be solubilized in an acidic buffer. Their sequences are presented in Table 2.
Table 2.
Sequences of the synthetic peptides used
| Peptide | Amino acid positions | gp120 domain | Isolate | Sequence |
|---|---|---|---|---|
| SP89334 | 1-10 | C1 | LAI | MRVKEKYQHL |
| 1919 | 1-20 | C1 | MN | MRVKGIRRNYQHWWGWGTML |
| 1920 | 23-30 | C1 | MN | LLMICSAT |
| 1921 | 27-35 | C1 | MN | CSATEKLWV |
| SP94437 | 37-46 | C1 | LAI | TVYYGVPVWK |
| SP89259 | 49-64 | C1 | LAI | TTTLFCASDAKAYDTE |
| P90405P90405 | 34-56 | C1 | LAI | LWVTVYYGVPVWKEATTTLVCAS |
| 8911486 | 67-85 | C1 | LAI | NVWATHACVPTDPNPQEV |
| 1927 | 84-96 | C1 | MN | ELVNVTENFNMWK |
| SP89246 | 88-98 | C1 | LAI | NVTENFNMWKN |
| SP89363 | 97-118 | V1 | LAI | CKNDMVEQMHEDIISLWDQSLQ |
| SP89243 | 115-131 | V1-V2 | LAI | SLKPCVKLTPLCVSLKC |
| SP89244 | 120-135 | V2 | LAI | VKLTPLCVSLKCTDLG |
| 1933 | 141-160 | V2 | MN | NSTANNNSNSEGTIKGGEMK |
| 1934 | 151-170 | V2-C2 | MN | EGTIKGGEMKNCSFNITTSI |
| SP93212 | 171-183 | C2 | LAI | RGKVQKEYAFFYK |
| 1937 | 181-200 | C2 | MN | LYKLDIVSIDNDSTSYRLIS |
| 1956 | 201-220 | C2 | MN | CNTSVITQACPKISFEPIPI |
| SP93872 | 193-207 | C2 | LAI | TTSYTLTSCNTSVIT |
| SP90404 | 202-219 | C2 | LAI | NTSVITQACPKVSFEPIP |
| 1958 | 221-240 | C2 | MN | HYCAPAGFAILKCNDKKFSG |
| SP93877 | 228-243 | C2 | LAI | FAILKCNNKTFNGTGP |
| SP93873 | 238-251 | C2 | LAI | FNGTGPCTLVSTVQ |
| 1960 | 241-260 | C2 | MN | KGSCKNVSTVQCTHGIRPVV |
| SP89045 | 252-272 | C2 | LAI | CTHGIRPVVSTQLLLNGSLAE |
| SP89247 | 270-285 | C2 | LAI | LAEEEVVIRSANFTDN |
| 1964 | 281-300 | C2 | MN | NFTDNAKTIIVHLNESVQIN |
| SP92371 | 301-333 | V3 | MN | CTRPNYNKRKRIHIGPGRAFYTTKNIIGTIRQA |
| SP89367 | 307-327 | V3-C3 | MN | CNKRKRIHIGPGRAFYTTKM |
| SP93853 | 330-346 | C3 | LAI | NMRQAHCNISRAKTNAT |
| SP89048 | 346-359 | C3 | LAI | TLKQIASKLREQFG |
| 1587 | 357-385 | C3 | IIIB | REQFGNNKTIIFKQSSGGDPEIVTHSFNC |
| SP93608 | 381-394 | C3-V4 | LAI | FNCGGEFFYCNSTQ |
| SP93855 | 392-414 | V4 | LAI | STQLFNSTWFNSTWSTEGSNNTE |
| SP89261 | 414-434 | C4 | LAI | EGSDTITLPCRIKQFINMWQE |
| SP89052 | 432-448 | C4 | LAI | WQEVGKAMYAPPISGQI |
| SP89053 | 449-464 | C4 | LAI | RCSSNITGLLLTRDGG |
| SP89248 | 460-474 | V5 | LAI | TRDGGNNNNGSEIFR |
| SP89057 | 465-479 | C5 | LAI | NNNNGSEIFRPGGGD |
| SP89054 | 474-489 | C5 | LAI | RPGGGDMRDNWRSELY |
| SP89258 | 487-509 | C5 | LAI | ELYKYRVVKIEPLGVAPTKAKRR |
| SP90385 | 491-516 | C5-gp41 | LAI | YKVVKIEPLGVAPTKAKRRVVQREKR |
Numbering is according to Ratner et al. (10). Domain assignment is according to ref. 2. Each amino acid is represented by a single-letter code that is the first letter of its name, except for arginine (R), asparagine (N), glutamine (Q), glutamic acid (E), lysine (K), phenylalanine (F), tryptophan (W), and tyrosine (Y).
Human Monoclonal Igs.
Human monoclonal IgG and IgM expressing variable regions of the VH3 gene family were purified from the serum of patients with multiple myeloma or Waldenström macroglobulinemia, as described previously (11).
Binding of Igs to HIV-1 Envelope Proteins.
Direct binding was assessed by enzyme-linked immunosorbent assay (ELISA). Polystyrene microtiter plates (Maxisorp F96, Nunc) were incubated overnight at +4°C with 100 μl of either rgp120 or rgp160 antigens (50 ng per well) diluted in borate-buffered saline (pH 8.4). Plates were washed with PBS (pH 7.2) containing 0.1% Tween-20 (Prolabo, Paris), and nonspecific sites of the wells were blocked with PBS containing 1% (wt/vol) BSA for 2 h at 37°C. A 100-μl volume of the test antibody diluted in PBS/BSA was added to each well and, after 2 h of incubation at 37°C, the plates were washed again with PBS/Tween. Bound antibodies were revealed with an alkaline phosphatase anti-immunoglobulin conjugate diluted (1:1000) in PBS/BSA. After a further incubation for 1 h at 37°C, the plates were washed with PBS/Tween and a volume of 100 μl per well of the alkaline phosphatase substrate p-nitrophenyl phosphate (Sigma) (1 mg/ml) diluted in 0.05 M carbonate buffer, pH 9.5, containing 2 mM MgCl2, was added. Absorbance was recorded at 405 nm with a multiscan automatic plate reader (MR 5000, Dynatech).
Competitive Immunoassays.
Plates were coated overnight at 4°C with recombinant proteins, and, after several washings with PBS/Tween, they were blocked with PBS/BSA. The test Ig at a dilution that achieved 50% of its maximal binding to the coated protein was mixed with various concentrations of the competitor peptide solution. After an overnight incubation at 4°C, the mixtures were transferred to protein-coated wells. The ELISA then was continued as described above. The percent inhibition of Ig binding to its target protein by the competitor peptide was calculated according to the formula
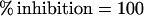 |
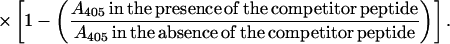 |
RESULTS
Recombinant gp120s from Divergent HIV-1 Isolates Display SAg-Like Activity.
The gp120 molecule is a SAg for normal B cells, and it interacts, through this SAg-binding site, with VH3+ Igs from normal subjects (7). To obtain further information on the gp120 SAg-binding site, we used two monoclonal Igs (IgM LAN and IgG SAU) isolated from two individuals who have not been exposed to HIV-1. First, we performed direct binding studies with rgp120 and rgp160 from various isolates. Fig. 1 shows the dose-dependent binding of monoclonal IgG SAU to divergent rgp120s. It also demonstrates that glycosylated and unglycosylated forms of gp120 from different clades exhibit the SAg-binding property, implying that the activity does not depend on glycosylation. Similar results were obtained with IgM LAN (not shown). To probe the effect of protein denaturation on the SAg-binding site, gp120SF2 was heated at 100°C in 0.1% sodium dodecyl sulfate. This treatment did not affect the SAg-binding activity. These data suggest that the SAg site is present on gp120s from sequence-divergent isolates and that it is not particularly sensitive to gp120 conformation, in that it survives heat denaturation.
Figure 1.

SAg binding of IgG SAU to rgp120s from different HIV-1 isolates. Microtiter wells were coated with 50 ng of rgp120s, and Ig binding was revealed by an alkaline phosphatase-labeled anti-IgG conjugate. The results are from one representative experiment. gp120-SF2 ng refers to an unglycosylated form of gp120SF2 that was heat inactivated.
The B Cell SAg-Binding Site Maps to Distant Motifs of gp120.
Next, we undertook a series of inhibition experiments aimed at precisely mapping the Ig-binding site on gp120. For this purpose, we tested the inhibitory capacity of a panel of synthetic peptides encompassing the whole sequence of the gp120 molecule. When tested at equimolar concentrations in a competitive assay using monoclonal IgM LAN, only peptides SP93855, SP89261, and to a lesser degree, SP89045, had an inhibitory activity (Fig. 2). All other peptides completely failed to induce inhibition, even at high concentrations. Similar results were obtained with monoclonal IgG SAU (not shown). To gain further insight into the complexity of the binding site, we performed crosscompetition experiments with the three positive peptides used in the different possible combinations. Individually, when tested at a 5 μM concentration, peptides SP93855 and SP89261 produced 80% and 75% inhibition, respectively. When both peptides were simultaneously used in an equimolar mixture, they had no synergistic or additive effect on Ig SAg binding to gp120. A similar trend was seen in inhibition experiments with other combinations of the peptides. These results imply that the binding site probably is centered on the V4 variable domain (peptides SP93855 and SP89261) of gp120 with contribution from the C2 constant domain (peptide SP89045), and that it is clearly not sensitive to the presence of N-linked glycosylation sites in these areas of the molecule.
Figure 2.

Inhibition of SAg binding by synthetic peptides from distant sites of gp120. Dilutions of the synthetic peptides were mixed with an equal volume of IgM LAN at a concentration that achieved 50% of maximal binding. After incubation, the mixtures were transferred to gp120MN-coated wells, and residual binding was revealed as indicated in the text. The results are from one representative experiment.
Delineation of Critical Amino Acid Positions by Mutagenesis.
To further characterize the SAg-binding site, we determined whether rgp120 binding was affected by amino acid substitutions. First, the fact that peptide SP89045 (residues 252-272) was inhibitory of SAg activity, and peptide 1960 (residues 241-260) was inactive, suggests that amino acids 252-260 are not engaged in gp120–Ig interaction. We therefore synthesized M1 mutant peptide, where the polar amino acids Gln263 and Asn267 were replaced by the apolar amino acid Ala. Second, for peptide SP93855, which maps to the V4 domain, sequence comparison of the gp120 from the various isolates tested revealed that, despite the sequence variability of this domain, some positions are conserved (Fig. 3). We have therefore used M2a mutant peptide, where amino acids Leu395, Phe396, and Asn397 were replaced by Ala. Finally, for SP89261 we designed two mutant peptides, M3a and M3b, where the conserved amino acids Ile419 and Leu421, and Ile425, His426, and Gln427 were replaced by Ala, respectively. Competition experiments showed that only the amino acid substitutions in M2a and M3b mutant peptides significantly impaired or abolished SAg binding (Fig. 4), implying that Leu395–Asn397 and Ile425–Gln427 amino acid positions are critical for gp120 SAg binding.
Figure 3.

Sequence comparison of the gp120 molecules from different isolates. Shown are the locations of the variable regions. The sequences corresponding to the peptides that inhibit gp120 SAg binding are boxed. Gaps are introduced to maximize homology and dots indicate homology. Numbering is according to Ratner et al. (10).
Figure 4.

Mutagenesis analysis of the sequence motifs involved in SAg binding. Dilutions of the wild-type and mutant peptides (M1, M2a, M3a, and M3b) were mixed with an equal volume of IgM LAN at a concentration that achieved 50% of maximal binding. After incubation, the mixtures were transferred to gp120MN-coated wells, and residual binding was revealed as indicated in the text. Represented is the inhibition achieved by the peptides used at a 5 μM concentration. The results are from one representative experiment.
DISCUSSION
A definitive assignment of Ig interaction sites with foreign ligands requires information on the three-dimensional structure derived, for example, from crystallographic data for Ig–ligand complexes. In most cases, however, epitopes can be identified by indirect means such as reactivity with phylogenetically related ligands, or by protein modification using either chemical procedures or site-directed mutagenesis (12). For infectious agents exhibiting antigenic variation and evolving at a high rate, such as HIV-1, study of proteins from various isolates provides a convenient means for identifying critical residues necessary for immune recognition. In this report, we have applied this approach to map the SAg-binding site of gp120 for normal human Ig. This was possible because HIV-1 isolates from different infected individuals vary in primary sequences. In addition, there is a deeper level of variation between isolates from different phylogenetically defined clades, with nine clades (A through I) identified as a result of coordinated studies of HIV-1 diversity (13). Using gp120s from different clades, we have shown that the SAg-binding activity is well conserved, a feature reminiscent of the conservation of the CD4-binding site of gp120 between different viral clades (14). That gp120s from different clades exhibit the SAg property is not inherently surprising. HIV-1 clades are defined on the basis of primary sequence, whereas SAg binding is a much more complex process that depends on interactions with motifs that are likely to be influenced by the tertiary and quaternary structures of the protein (see below). It will be important to see whether human monoclonal antibodies elicited in response to HIV infection interfere with the SAg-binding activity.
Our studies with gp120 synthetic peptides indicate that the SAg-binding region of gp120 is composed of two discrete binding sites, which allow for the simultaneous binding of VH3+ Ig molecules. We have shown that residues at the base of the V4 domain and the amino-terminal region flanking the C4 domain of gp120 (positions 392-434) are critical for binding. It is remarkable that this latter domain is also the target of other molecular interactions. Discontinuous epitopes overlapping the C4 region are the target of a large proportion of anti-HIV antibodies from most seropositive subjects, and neutralizing antibodies bind linear or discontinuous C4 epitopes (2). This domain also participates, with the C2 and C3 domains of gp120, in forming the CD4-binding domain to the CD4 coreceptor (2), implying that the C4 domain is accessible for molecular interactions. It is of interest that the C4 epitope associated with the SAg activity also contributes to the CD4 binding site and the mutations that alter SAg activity within this site also may affect CD4 binding to gp120 (2). In addition, our data suggest that SAg binding requires contributions of residues from the C2 domain (positions 252-272).
On the basis of their ability to be presented in the form of short peptides, we have categorized the SAg-binding site as being composed of two widely separated linear segments. Our data show that the core motif is a discontinuous epitope spanning the V4 domain and the amino-terminal region flanking the C4 domain, with a contribution from a segment of the distant C2 domain. The finding that two motifs located on different domains of the gp120 molecule are involved in SAg binding would suggest that they are located in close proximity in the native structure. There is, however, strong evidence demonstrating physical interactions between distant domains of the gp120 molecule, including the V2 and V3 domains (15), the V2 and C4 domains (16), the V3 and C4 domains (17), and all three domains (15). These interdomain interactions suggest that other regions of the molecule distant from the linear sequences we have identified might be indirectly implicated in optimal expression of the SAg-binding motif.
Although the secondary structure of gp120 has not been elucidated, it has been proposed that it may contain as many as six α-helices and a number of β-strands (1, 2). To provide a theoretical model of the secondary structure of the SAg-binding site and its adjacent areas, we analyzed the gp120 segments between residues 252-272 and 392-434 with regard to the distribution of polar and nonpolar residues, and α-helices and β-strands. Amino acid analysis of the V4 loop domain in the seven isolates derived from distinct geographic origins shows that the amino acids adjacent to the amino-terminal Cys are quite conserved (Fig. 3). In addition, a previous study predicted that residues 394-412 in this domain comprise a β-turn region, with amino acids that are hydrophilic and flexible and, hence, have a higher probability of being exposed at the surface of the gp120 molecule (1). These features may account for our finding that mutations at the V4 epitope (residues 392-414) significantly affect SAg binding. On the other hand, the V4/C4 epitope (residues 414-434) contains a potentially surface-exposed α-helix (2). Changing three amino acids of this helix dramatically impairs SAg binding (Fig. 4), a finding in agreement with another study (18). Thus, the results presented herein support the notion that the core SAg binding is mediated by two discrete, but separate, sites that map to amino acid positions 392-434. The most critical residues at the amino terminus appear to be Leu395–Asp397. At the carboxyl terminus Ile425, His426, and Gln427 are also critical. In addition, residues from the C2 domain (positions 252-272) seem to play an accessory role in gp120 SAg binding to normal human Igs.
These findings are potentially important in the design of an efficient gp120-based vaccine against HIV-1. The SAg-binding site must endow gp120 with the ability to crosslink membrane IgM on normal human B cells to induce their activation in a VH3 gene-family-specific manner (7). Because this VH family is the largest in the human repertoire (19, 20), this property is thought to have deleterious consequences for the immune system, including a progressive decline of B cells with progression of the disease (9, 21) and formation of complement-activating immune complexes with subsequent activation of the complement cascade (22, 23). These potentially deleterious SAg effects might be offset by the use of a gp120 vaccine devoid of its SAg-binding site.
Acknowledgments
We are indebted to Drs. J. P. Bouvet (Institut Pasteur, Paris) and P. Lambin (Institut National de Transfusion Sanguine, Paris) for the monoclonal antibodies, the Agence Nationale de Recherche sur le SIDA (Paris) for the HIV synthetic peptides, and Dr. M.-P. Kieny (Transgène, Strasbourg, France) for HIV recombinant proteins. We thank Dr. G. Bentley (Institut Pasteur, Paris) for helpful discussions. This work was supported by grants from the Institut Pasteur and the Fondation pour la Recherche Médicale. S.K. is an Investigator of the Centre National de la Recherche Scientifique, and M.Z. is an Investigator of the Institut National de la Santé et de la Recherche Médicale.
Footnotes
Abbreviation: SAg, superantigen.
References
- 1.Modrow S, Hahn B H, Shaw G M, Gallo R C, Wong-Staal F, Wolf H. J Virol. 1987;61:570–578. doi: 10.1128/jvi.61.2.570-578.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poignard P, Klasse P J, Sattentau Q J. Immunol Today. 1996;17:239–246. doi: 10.1016/0167-5699(96)10007-4. [DOI] [PubMed] [Google Scholar]
- 3.Levy J A. Microbiol Rev. 1993;57:183–289. doi: 10.1128/mr.57.1.183-289.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziegler J L, Stites D P. Clin Immunol Immunopathol. 1986;41:305–313. doi: 10.1016/0090-1229(86)90001-2. [DOI] [PubMed] [Google Scholar]
- 5.Robinson J, Edward W, Mitchell W M, Chambers W H, Schuffman S S, Montefiori D C, Oeltman T N. Hum Pathol. 1988;19:535–540. doi: 10.1016/s0046-8177(88)80200-4. [DOI] [PubMed] [Google Scholar]
- 6.Robinson W E J, Montefiori D C, Mitchell W M. Lancet. 1988;i:790–794. doi: 10.1016/s0140-6736(88)91657-1. [DOI] [PubMed] [Google Scholar]
- 7.Berberian L, Goodglick L, Kipps T J, Braun J. Science. 1993;261:1588–1591. doi: 10.1126/science.7690497. [DOI] [PubMed] [Google Scholar]
- 8.Berberian L, Shukla J, Jefferis R, Braun J. J Acquired Immune Defic Syndr. 1994;7:641–646. [PubMed] [Google Scholar]
- 9.Zouali M. Immunol Today. 1995;16:399–405. doi: 10.1016/0167-5699(95)80009-3. [DOI] [PubMed] [Google Scholar]
- 10.Ratner L, Haseltine W, Patarca R, Livak K J, Starcich B, Josephs S F, Doran E R, Rafalski J A, Whitehorn E A, Baumeister K, Ivanoff L, Petteway S R, Jr, Pearson M L, Lautenberger J A, Papas T K, Ghrayeb J, Chang N N, Gallo R C, Wong-Stall F. Nature (London) 1985;313:277–284. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- 11.Silverman G J, Pirès R, Bouvet J P. J Immunol. 1996;157:4416–4502. [PubMed] [Google Scholar]
- 12.Benjamin D C, Berzofsky J A, East I J, Gurd F R N, Hannum C, Leach S J, Margoliash E, Michael J G, Miller A, Prager E M, Reichlin M, Sercarz E E, Smit-Gill S J, Todd P E, Wilson A C. Annu Rev Immunol. 1984;2:67–101. doi: 10.1146/annurev.iy.02.040184.000435. [DOI] [PubMed] [Google Scholar]
- 13.Gao F, Morrison S G, Robertson D L, Thornton C L, Craig S, Karlsson G, Sodroski J, Morgado M, Galvao-Castro B, von Briesen H, Beddows S, Weber J, Sharp P M, Shaw G M, Hahn B H. J Virol. 1996;70:1651–1657. doi: 10.1128/jvi.70.3.1651-1667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore J P, McCutchan F E, Poon S W, Mascola J, Liu J, Cao Y, Ho D D. J Virol. 1994;68:8350–8364. doi: 10.1128/jvi.68.12.8350-8364.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choe H R, Sodroski J. J Virol. 1995;69:2801–2810. doi: 10.1128/jvi.69.5.2801-2810.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore J P, Sattentau Q J, Yoshiyama H, Thali M, Charles M, Sullivan N, Poon S W, Fung M S, Traincard F, Pinkus M, Robey G, Robinson J E, Ho D D, Sodroski J. J Virol. 1993;67:6136–6151. doi: 10.1128/jvi.67.10.6136-6151.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore J P, Thali M, Jameson B A, Vignaux F, Lewis G K, Poon S W, Charles M, Fung M S, Sun B, Durda P J, Akerblom L, Wahren B, Ho D D, Sattentau Q J, Sodroski J. J Virol. 1993;67:4785–4796. doi: 10.1128/jvi.67.8.4785-4796.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodglick L, Zevit N, Neshat M S, Braun J. J Immunol. 1995;155:5151–5159. [PubMed] [Google Scholar]
- 19.Zouali M. Nat Genet. 1994;7:118–120. doi: 10.1038/ng0694-118. [DOI] [PubMed] [Google Scholar]
- 20.Cook G P, Tomlinson I M. Immunol Today. 1995;16:237–242. doi: 10.1016/0167-5699(95)80166-9. [DOI] [PubMed] [Google Scholar]
- 21.Goodglick L, Braun J. Am J Pathol. 1994;144:623–636. [PMC free article] [PubMed] [Google Scholar]
- 22.Goodglick, L., Townsley-Fuchs, J., Fairhurst, R. M., Neshat, M., Marcelo, D., Mosier, D. E. & Braun, J. in Human B-Cell Superantigens, ed. Zouali, M. (Landes, Austin, TX), pp. 145–160.
- 23.Karray S, Juompan L, Zouali M. In: Human B-Cell Superantigens. Zouali M, editor. Austin, TX: Landes; 1996. pp. 161–177. [Google Scholar]