

Abstract
The mechanisms that initiate liver regeneration after resection of liver tissue are not known. To determine whether cytokines are involved in the initiation of liver growth, we studied the regeneration of the liver after partial hepatectomy (PH) in mice lacking type I tumor necrosis factor receptor (TNFR-I). DNA synthesis after PH was severely impaired in these animals, and the expected increases in the binding of the NF-κB and STAT3 transcription factors shortly after PH failed to occur. Binding of AP-1 after PH was decreased in TNFR-I knockout mice compared with animals with the intact receptor whereas C/EBP binding was not modified. Injection of interleukin 6 in TNFR-I-deficient animals 30 min before PH corrected the defect in DNA synthesis and restored STAT3 and AP-1 binding to normal levels but had no effect on NF-κB binding in the regenerating liver. The results indicate that TNF, signaling through the TNFR-I, can initiate liver regeneration and acts by activating an interleukin 6-dependent pathway that involves the STAT3 transcription factor.
The liver has the unique capacity to regenerate after removal of part of its mass. This growth response is particularly remarkable because hepatocytes that constitute ≈65% of the cells of the mammalian liver have low proliferative activity and long life spans. Nevertheless, hepatocytes readily proliferate after partial hepatectomy (PH) and undergo one or more rounds of semisynchronous replication before returning to quiescence (1, 2). In young rats and mice, >95% of hepatocytes replicate after PH, and the hepatic mass is restored in 7–10 days. The growth process is tightly regulated and terminates when it reaches a set point, defined as the optimal ratio between hepatic functional mass and body mass. The same principles that govern liver regeneration after PH in rats and mice apply to the growth response of human livers transplanted to a new host. In this situation, a small transplant grows, but a large transplanted liver decreases in size, so in each case, the optimal liver mass/body mass set point for the individual host is attained (1).
During the last few years, much new information has become available on the events that may initiate liver regeneration (3–6). Many growth factors can stimulate DNA replication of hepatocytes in primary culture, and at least two of these factors, transforming growth factor α and hepatocyte growth factor/scatter factor, participate in the growth response after PH in vivo. However, the precise point at which these factors might act has not been identified. Furthermore, although transforming growth factor α and hepatocyte growth factor/scatter factor greatly stimulate DNA synthesis of cultured hepatocytes, infusion of these factors directly into the liver in vivo during a 24-h period causes only a minor increase in hepatocyte DNA synthesis (7). However, hepatocytes in the intact liver become capable of responding to these growth factors if they receive stimuli that “prime” quiescent hepatocytes to undergo replication (8–10). These and other experiments indicate that the initiation of liver regeneration requires an initial stage in which quiescent hepatocytes acquire proliferative competence (1, 3, 7). During this stage, which roughly corresponds to the first 4 h after PH, binding of the transcription factors NF-κB, AP-1, and STAT3 increases. Activation of NF-κB occurs minutes after PH and is transient (11, 12). AP-1 and STAT3 binding increase more slowly after the operation, and STAT3 binding remains high for 6 h or more (13–16). Tumor necrosis factor (TNF) activates NF-κB in many cell systems and causes strong NF-κB binding in rat liver within 30 min after i.p. injection (11). A potential role for TNF in liver regeneration is indicated by the work of Akerman et al. (17), who showed that TNF antibodies can delay and diminish DNA synthesis in regenerating rat liver and inhibit the increase in circulating levels of interleukin 6 (IL-6) after PH. We have proposed that liver regeneration may be initiated by one or more cytokines and that growth factors act later on hepatocytes that have become competent to proliferate (1, 3). To directly test the role of TNF in the initiation of liver regeneration after PH, we analyzed the liver growth response after PH in mice lacking TNF receptor 1 (TNFR-I). These animals develop and grow normally but are resistant to TNF-mediated toxicity and are highly susceptible to infection by Listeria monocytogenes and Mycobacterium tuberculosis (18–20). We report that liver regeneration is severely impaired in TNFR-I-deficient mice and that the defect in DNA synthesis can be corrected by IL-6 injection.
MATERIALS AND METHODS
Animals.
TNFR-I knockout mice (p55−/−) of the C57BL/6 strain were used in these experiments (21, 22). Wild-type C57BL/6 mice originally purchased from The Jackson Laboratory served as controls. All experiments were performed with male mice weighing 25–30 g kept in a temperature-controlled room with alternating 12-h dark/light cycles. PH consisting of the removal of the anterior and left lateral hepatic lobes was performed by the procedure of Higgins and Anderson (23) as described (24). The experiments were conducted in accordance with the institutional guidelines of the University of Washington School of Medicine.
Nuclear Extracts.
Mice were killed at various times after PH as indicated for each experiment. All solutions used for the preparation of nuclear extracts contained protease inhibitors as described (11). Tissue was homogenized and nuclear extracts prepared as described (11). Nuclear extracts were frozen and stored at −80°C until use. Protein concentrations were measured by the Bradford method.
Electrophoretic Mobility-Shift Assays (EMSA). Probes.
The following double-stranded probes were used: NF-κB binding sequence from the class 1 major histocompatibility enhancer element (H2K) as described (11); AP-1, consensus oligonucleotide probe (Santa Cruz Biotechnology); STAT3, oligonucleotide corresponding to the binding site for the Sis inducible factor (Santa Cruz Biotechnology); and C/EBP, oligonucleotide corresponding to nucleotides −112 to −86 of the rat albumin gene promoter (25). The NF-κB and C/EBP probes were prepared by annealing the complementary oligonucleotides in a thermal cycler in 50 mM Tris (pH 8.0) and 1 mM EDTA. Annealed oligonucleotides were purified by PAGE. NF-κB, AP-1, and STAT3 oligonucleotide probes were prepared by end labeling with γ32P-adenosine 5′-triphosphate using T4 polynucleotide kinase. The C/EBP was end-labeled with α32P-deoxycytidine 5′-triphosphate using Klenow DNA polymerase.
Assays.
Nuclear protein (10 μg) was used in each assay and was incubated with 0.2 ng of 32P-end-labeled, double-stranded oligonucleotide probe as described (11). The mixture was incubated for 30 min at room temperature and electrophoresed through 5% polyacrylamide Tris–glycine–EDTA gels. For antibody supershift assays, 1 μl of the antibody (1 μg/μl; all from Santa Cruz Biotechnology) was added to the respective samples after 30 min of incubation with the labeled probe. Samples were incubated at room temperature for an additional 30 min before electrophoresis. The following antibodies were used: anti-p50- and anti-p65-specific polyclonal antibodies for the NF-κB components; c-Jun/AP-1 polyclonal antibody specific for c-Jun; c-Jun/AP-1 polyclonal antibody reactive with c-Jun, Jun B, and Jun D; Jun B-specific polyclonal antibody; c-Fos-specific polyclonal antibody; STAT3-specific polyclonal antibody; C/EBP polyclonal antibody for C/EBPα and C/EBP polyclonal antibody specific for C/EBPβ. Gels were dried and exposed to Kodak X-AR film from 2 h to 2 days.
RNA Preparation and Northern Blot Hybridization.
RNA was isolated by homogenization of the livers in 4 M guanidine thiocyanate followed by ultracentrifugation through cesium chloride as described (7). Samples (20 μg/lane) were separated by electrophoresis through 1.1% formaldehyde–agarose gels and transferred to a nylon membrane (MagnaGraph, Micron Separations, Westboro, MA). After 2–4 h of prehybridization at 42°C, the filters were hybridized with the following probes labeled with α32P-deoxycytidine 5′-triphosphate by random priming: TNFR-I, 240-bp SpeI–BglII fragment from murine TNFR-I cDNA; TNFR-II, 450-bp XbaI–SalI fragment from murine TNFR-II cDNA; c-fos, 915-bp EcoRI–SphI fragment from human c-fos cDNA; c-jun, 1800-bp EcoRI fragment from murine c-jun cDNA; jun B, 1700-bp EcoRI fragment from jun B cDNA; and β2-microglobulin (loading control), 350-bp PstI fragment from mouse cDNA. After hybridization at 42°C for 12–24 h, the filters were washed and exposed to Kodak XAR film with intensifying screens at −80°C. After each hybridization, probes were removed by washing with 50% formamide/6× SSC at 65°C for 30 min.
Determination of IL-6 mRNA by Reverse Transcriptase–PCR Assay.
Liver IL-6 mRNA was determined by coupled, reverse transcription–PCR. cDNA was prepared from 1 μg of total RNA from each liver sample using a Gene Amp RNA PCR kit (Perkin–Elmer), in a buffer containing 2.5 units of murine leukemia virus reverse transcriptase and 2.5 μM of oligo(dT) primer. Samples were incubated at 42°C for 15 min, 99°C for 5 min, and 5°C for 5 min. An aliquot of cDNA representing 50 ng of input RNA was amplified using the same kit. The PCR contained the same buffer as the reverse transcriptase reaction, 0.4 μM IL-6 primers (CLONTECH), and 2.5 units of AmpliTaq DNA polymerase (Perkin–Elmer). The reaction was performed at 94°C for 1 min, 60°C for 1 min, and 74°C 1.5 min for 30 cycles. The optimal number of cycles needed to obtain detectable product (638-bp product size) under nonsaturating conditions was determined by performing the PCR for 10–40 cycles. No product was detectable after 10 or 20 cycles; saturation of the reaction occurred at 40 cycles. Amplified products obtained with 30 cycles were electrophoresed in 2% agarose gels and stained with ethidium bromide. Quantitation of IL-6 mRNA was done by competitive PCR using the PCR Mimic Protocol (CLONTECH). IL-6 competitor primer (2 μl) yielding a product size of 435 bp was used per reaction in concentrations of 10–10−6 attomoles.
Histology.
Livers from BrdUrd-injected mice (30 μg/g; 2 h before killing) were fixed in methyl Carnoy’s fluid for 4–6 h, prepared for histological analysis, and stained using the Amersham cell proliferation kit.
RESULTS
Transcription Factor Binding.
C57BL/6 wild-type and TNFR-I knockout mice were partially hepatectomized, and liver nuclear extracts were prepared from animals killed 30 min to 5 h after the operation. We determined the binding of the transcription factors NF-κB, AP-1, STAT3 and C/EBP (Fig. 1) by EMSA. In nuclear extracts from wild-type mice, there was a large increase in NF-κB (p50/p65 heterodimer) binding as well as that of p50 homodimers 30 min to 4 h after PH, as described (11, 12). In marked contrast, practically no increase in the binding of NF-κB and p50 homodimers was detected in extracts of knockout mice. The faster migrating band appearing below the p50 homodimers in Fig. 1 corresponds to a factor referred to as “PH factor,” which was originally thought to displace NF-κB and prevent its binding after PH (26), an observation not supported by subsequent work (11, 12). PH factor binding was detectable in the nuclear extracts of some TNFR-I knockout mice (Fig. 1, lanes 3 and 4). The AP-1 transcription factor is comprised of heterodimers of c-Fos and Jun family proteins or of Jun homodimers (27). AP-1 binding was increased in nuclear extracts from regenerating livers of wild-type mice 2–4 h after PH (at 2 h, only one of the extracts showed increased binding), as reported (13, 14). However, this induction was reduced in the knockout mice (Fig. 1). STAT3 is a member of the family of transcription factors known as the signal transduction and activation of transcription proteins (28). Binding of STAT3 in the liver is increased during liver regeneration (15, 16), during the acute phase response, and after injections of epidermal growth factor and IL-6 (29, 30). We confirmed published observations that STAT3 increases after PH in wild-type mice (31) but did not detect STAT3 binding after PH in mice lacking TNFR-I (Fig. 1). We also examined the binding of C/EBP in wild-type and knockout mice. This transcription factor is highly abundant in hepatocytes and is an important regulator of many liver-specific genes (5, 32–34). The mobility-shift analysis shown in Fig. 1 reveals that C/EBP binding after PH was not impaired in TNFR-I knockout mice and may even have been higher than that in wild-type mice. We used specific antibodies against various protein components of the transcription factors to analyze the nature of the binding complexes (Fig. 2). Mobility-shift assays for NF-κB performed in presence of p65 and p50 antibodies confirmed that the bands present in the nuclear extracts obtained from livers of wild-type mice indeed corresponded to NF-κB (p50/p65 heterodimer) and p50 homodimers. Neither of these bands was detectable in extracts obtained from livers of TNFR-I knockouts. Similarly, the STAT3 band in nuclear extracts from wild-type mice was shifted entirely by STAT3 antibodies, but the band was not detectable in liver extracts from knockout mice. Analysis of AP-1 complexes showed that c-Fos, c-Jun, Jun B, and other Jun family proteins are components of the AP-1 complex in nuclear extracts from regenerating livers of wild-type mice. Bands corresponding to c-Jun and other Jun family proteins were also present in the extracts of knockout mice, but no bands were shifted with c-Fos antibodies (Fig. 2). Analysis of C/EBP complexes using C/EBPα and C/EBPβ antibodies in extracts from wild-type as well as TNFR-I knockout mice revealed an increase in overall C/EBP binding as well as of each isoform between 1 and 3 h after PH (data not shown). In sum, NF-κB and STAT3 activation was drastically inhibited, if not abolished, overall AP-1 binding was diminished, and the AP-1 c-Fos component was not detectable in liver nuclear extracts of TNFR-I knockout mice after PH.
Figure 1.

Transcription factor binding after PH in wild-type (WT) and TNFR-I knockout mice (TNFR-I KO). Mice were partially hepatectomized and killed 0.5 and 5 h after the operation, as indicated at the top of the figure (two mice per time point). Nuclear extracts were prepared, and EMSAs were performed using 10 μg of nuclear protein in each lane. Reticulocyte lysate was used as a marker to determine the position of the p50/p65 NF-κB heterodimer (11). p50 homodimer position is indicated as (p50)2. There was variability in AP-1 binding in wild-type mice killed 2 h after PH. PHF, PH factor
Figure 2.

Mobility-shift analysis of transcription factor binding using specific antibodies (Ab). Wild-type (WT) and TNFR-I knockout mice (TNFR-I KO) were partially hepatectomized and killed 4 h after the operation for NF-κB, STAT3, and AP-1 analysis. Nuclear extracts were incubated for 30 min with 32P-labeled probes followed by addition of 1 μl of the antibody (1 μg/μl) to 10 μg of extract. After incubation for 30 min, nuclear extracts (10 μg per lane) were analyzed by EMSA. The antibody used in each assay is indicated at the top of the figure. For each transcription factor, the first lane shows EMSA of the nuclear extract without antibody preincubation.
mRNA Expression.
The cellular effects of TNF can be initiated by the ligand-dependent activation of two major receptors, TNFR-I and TNFR-II, which do not form heterodimers. The knockout mice used in our experiments lack only TNFR-I and should have intact TNFR-II. In livers of wild-type mice, TNFR-II mRNA increased >10-fold 4 h after PH, and a relatively minor change was detected for TNFR-I (Fig. 3). Thus, TNFR-I mRNA is constitutively and highly expressed in mouse liver, but TNFR-II mRNA is inducible after PH (35). As expected, no TNFR-I mRNA was detectable in the liver of the knockout mice before or after PH. On the other hand, TNFR-II mRNA increased after PH in these animals at an apparently faster rate than in wild-type mice, indicating that TNFR-II mRNA synthesis is not impaired in TNFR-I knockout mice. Consistent with previous results from this and other laboratories (1, 4, 5, 36), c-fos mRNA increased ≈10-fold in the liver of wild-type mice after PH with maximal expression at 0.5–1 h. This early elevation of c-fos mRNA expression was absent in livers of knockout mice. Expression of c-jun and jun B mRNAs after PH was induced to the same extent in wild-type and knockout mice.
Figure 3.

Expression of TNFR-I, TNFR-II, c-jun, c-fos, and Jun B mRNAs in wild-type (WT) and TNFR-I knockout mice (TNFR-I KO). Animals were partially hepatectomized and killed 0.5–5 h after operation. Samples (20 μg/lane) were separated by electrophoresis and hybridized with α32P-deoxycytidine 5′-triphosphate probes. β2-Microglobulin (β2M) mRNA (bottom row) was used as a loading control.
DNA Synthesis and Liver Mass After PH.
After PH in mice, DNA synthesis remains very low and unchanged until ≈30–32 h after the operation (24), when it starts to increase, and it reaches a maximum at ≈40 h. To determine whether the lack of TNFR-I would alter DNA synthesis in the regenerating liver, TNFR-I knockout and wild-type mice were partially hepatectomized and killed 24–68 h after the operation (Fig. 4). All mice were injected with BrdUrd 2 h before killing. In wild-type mice, BrdUrd incorporation in hepatocytes reached a peak at 40 h after PH, decreased by almost 75% during the next 10 h, but was still elevated above control levels at 68 h. In contrast, BrdUrd incorporation in hepatocytes of TNFR-I knockout mice after PH was severely impaired. Compared with wild-type animals, hepatocyte replication in knockout animals was reduced by >60% at 40 h and was even lower between 44 and 68 h after PH. During the period of time that precedes the major peak of DNA synthesis during liver regeneration, hepatocytes often accumulate a small amount of lipid droplets in the cytoplasm. These droplets were detected in hepatocytes of wild-type mice used in these experiments at 30 h after the operation. However, hepatocytes of TNFR-I knockout animals had massive fat infiltration (which was very prominent during the second day after PH), consisting of very large cytoplasmic lipid vacuoles that distorted the cell architecture (data not shown).
Figure 4.

Hepatocyte DNA synthesis after PH in wild-type and TNFR-I knockout mice. Wild-type and TNFR-I knockout mice were partially hepatectomized and killed 24–68 h after the operation (three animals per time point) as indicated. All animals received an i.p. injection of 30 μg/g BrdUrd (BrdU) 2 h before killing. The ordinate shows the percentage of hepatocytes labeled by BrdUrd at the various time points. The bars indicate the SD of the means. ▪, Wild-type C57BL/6 mice; ○, TNFR-I knockout mice.
Because hepatocyte proliferation was severely inhibited 40–68 h after PH in knockout animals, it became important to determine whether partially hepatectomized TNFR-I knockout mice would survive and eventually regenerate their livers. Mortality of TNFR-I knockouts after PH was negligible during the first 2 days after PH but was 50% or more between days 3 and 5. Compared with wild-type animals, surviving knockout mice had a deficit of regeneration at 7 and 14 days after the operation, as estimated by determination of liver-to-body weight ratios. In wild-type mice, the liver weight-to-body weight ratios at 7 and 14 days after PH corresponded, respectively, to 78% and 95% of the ratio for nonoperated, intact animals. Ratios for TNFR-I knockouts at 7 and 14 days after PH were, respectively, 55% and 78% of the liver-to-body weight ratio of nonoperated knockout mice (statistically significant at P < 0.005). Liver weight gain in the knockout animals 1 week after PH was, at least in part, a consequence of hepatocyte replication because hepatocyte labeling by BrdUrd at this time was higher in knockout than in wild-type mice (data not shown). In sum, DNA replication was severely impaired in TNFR-I knockout mice for at least 4 days after PH, and ≈50% of these animals died. Knockout mice that survive can regenerate their livers although regeneration is slower than normal and a deficit in the restoration of the mass persists for at least 2 weeks.
Detection of IL-6 mRNA in the Liver After PH.
IL-6 is a major regulator of the hepatic acute phase response in vivo and a STAT3 inducer (31, 37–39). Its transcription is stimulated by TNF through NF-κB activation (40, 41). We initially attempted to detect IL-6 mRNA in the liver of nonoperated and partially hepatectomized mice by Northern blot analysis but were not successful, as already reported (42). We then developed a reverse transcriptase PCR assay to compare the expression of IL-6 mRNA after PH in wild-type and TNFR-I knockout mice (Fig. 5). The amplification reaction was carried out under nonsaturating conditions using 30 amplification cycles (see Materials and Methods). IL-6 mRNA was increased in the livers of wild-type and knockout mice after PH, reaching a peak 4 h after the operation. To quantitate the mRNA present in these extracts, competitive PCR assays were performed with 1 μg of RNA obtained from livers of wild-type and knockout mice 4 h after PH. RNA from wild-type mice showed complete competition with 10−1–10−2 attomoles/μl competition primer. Competition of IL-6 mRNA from knockout mice was obtained with a competitor concentration of 10−3 attomoles/μl, demonstrating that IL-6 mRNA is decreased by >10-fold in the liver of TNFR-I knockout mice after PH.
Figure 5.

IL-6 mRNA expression after PH. (A) Wild-type (WT) and TNFR-I knockout (TNFR-I KO) mice were partially hepatectomized and killed 0.5–5 h after the operation, as indicated at the top of A. M, molecular weight marker lane; Pos, positive control for IL-6 mRNA (CLONTECH); Neg, mRNA not added for reverse transcription step. (B) Quantitation of IL-6 mRNA by competitive PCR in livers of wild-type and knockout mice 4 h after PH. Competitor concentrations are indicated at the top of B. The product sizes are 638 bp for IL-6 mRNA and 435 bp for the competitive fragment.
Hepatocyte Replication and STAT3 Binding Defects in TNFR-I Knockout Mice Are Corrected by IL-6.
Based on the results of the measurements of IL-6 mRNA described above, we attempted to determine whether administration of IL-6 would reverse the DNA synthesis and transcription factor binding deficiencies present in TNFR-I knockout mice. We injected mice with IL-6 30 min before PH and killed the animals 40 h after the operation, 2 h after receiving BrdUrd. Fig. 6 shows that IL-6 injection into TNFR-I knockout mice completely corrected the DNA replication deficiency present in these animals. IL-6-injected knockout mice had a similar mortality rate as that of wild-type mice and did not show the heavy accumulation of large fat vacuoles that developed in the livers of TNFR-I knockout animals after PH. BrdUrd incorporation in hepatocytes of IL-6-injected TNFR-I knockouts at 40 h after PH did not differ from that of wild-type mice and was 3-fold higher than in noninjected TNFR-I knockout mice. In marked contrast, injection of the same dose of IL-6 into wild-type mice shortly before PH caused inhibition of DNA synthesis. Gel mobility-shift analysis of NF-κB, AP-1, and STAT3 binding 3 h after PH in IL-6-injected TNFR-I knockout mice revealed that injection of the cytokine completely corrected the deficiency in STAT3 binding. However, IL-6 did not cause an increase in NF-κB binding (Fig. 7). Although IL-6 injection in TNFR-I knockout mice augmented AP-1 binding, the AP-1 complexes formed in the liver of these animals remained deficient in the c-Fos component (data not shown).
Figure 6.

Effect of IL-6 on hepatocyte DNA synthesis in TNFR-I knockout mice (TNFR-I KO). Wild-type (WT) and TNFR-I mice were partially hepatectomized and killed 40 h after the operation. Three TNFR-I knockout animals and three wild-type mice received an s.c. injection of recombinant human IL-6 (1 mg/kg) 30 min before PH. All mice were injected with BrdUrd (BrdU) 2 h before killing (see Fig. 4). The ordinate shows the percentage of hepatocytes labeled by BrdUrd.
Figure 7.

Effect of IL-6 on transcription factor binding in TNFR-I knockout mice. Wild-type and TNFR-I mice were partially hepatectomized and killed 3 h after PH. A group of knockout mice received an s.c. injection of IL-6 (see Fig. 5). Nuclear extracts (prepared from two animals in each group) were analyzed by EMSA as described in Fig. 1 (10 μg/lane). Lanes: 1, wild-type mice; 2, TNFR-I knockout mice; and 3, TNFR-I knockout mice injected with IL-6. The arrow on the right indicates the position of the STAT3 band.
DISCUSSION
This work establishes that TNF signaling through TNFR-I can initiate liver regeneration after PH and that IL-6 is a key target of TNF gene activation in the regenerating liver. Knockout mice that lack TNFR-I showed a severe defect in hepatocyte replication during the first 4 days after PH. Surviving animals (<50% of the total) eventually regenerated their livers, but restoration of the normal liver weight-to-body weight ratio was still not completed 2 weeks after PH, a time at which the regenerative process was already terminated in wild-type mice. IL-6 reversed the deficiency in hepatocyte replication imposed by the lack of TNFR-I, corrected the defects in STAT3 and AP-1 binding, but did not reverse the almost complete inhibition of NF-κB binding after PH. These experiments solve a long-standing riddle in the understanding of liver regeneration by identifying TNF as an initiator of the process and by showing that signaling through one of its receptors can initiate the cascade of events that lead to DNA synthesis many hours later. However, these experiments do not address the important question of whether TNF is the sole initiator of liver regeneration or, more likely, if it is one among multiple factors required but not sufficient by themselves to elicit a full regenerative response. We also did not examine whether IL-6 injection lead to normalization of other indices of liver growth besides DNA replication. After this paper was submitted for publication, Cressman et al. (31) demonstrated that liver regeneration is severely impaired in IL-6-deficient mice. After PH, no STAT3 binding could be detected in the liver of IL-6 knockouts, and the expression of AP-1, myc, and cyclin D1 was depressed. IL-6 injection prevented liver damage, restored STAT3 binding, and corrected the impaired hepatocyte DNA synthesis in these animals (31). These results complement our own observations and, taken together, are consistent with the following sequence of events for the initiation of liver regeneration:
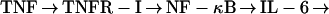 |
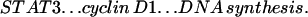 |
TNF is known to be required for hepatocyte proliferation induced by direct liver mitogens such as lead nitrate (43). Our work and data obtained using TNF antiserum (17) establish that TNF can also initiate hepatocyte proliferation in response to tissue loss. Although TNF is the major inducer of NF-κB after PH, high levels of hepatocyte replication in the regenerating liver can occur in the absence of NF-κB binding, as long as IL-6 is present in appropriate concentrations. This conclusion is based on the observation that IL-6-injected TNFR-I knockout mice have normal levels of hepatocyte replication after PH but little if any NF-κB binding.
In both TNFR-I and IL-6 knockout mice, correction of the deficit in DNA replication by IL-6 injection was associated with STAT3 activation. This transcription factor also transactivates genes involved in the hepatic acute phase response initiated by TNF, IL-6, and other cytokines (15, 16, 28, 30). Although further studies are required, the results suggest that STAT3 and cytokines capable of leading to STAT3 activation play a key role in proliferative and inflammatory responses in the liver. How similar initiating mechanisms can lead to different biological responses is an unresolved problem. IL-6 inhibits growth factor-induced DNA replication in cultured hepatocytes (44) but may increase hepatocyte DNA synthesis in vivo in intact, nonpartially hepatectomized animals (45). In our experiments, IL-6 injection of wild-type mice had an inhibitory effect on DNA synthesis after PH whereas it stimulated DNA replication in TNFR-I knockout mice that are deficient in hepatic IL-6 mRNA. Thus, the stimulatory effect of IL-6 on hepatocyte DNA replication may occur only at a relatively narrow range of intrahepatic IL-6 concentrations.
Injection of anti-TNF serum has been shown to inhibit C/EBPβ binding and c-Jun synthesis after PH (13). Neither of these alterations were present in TNFR-I knockout mice. A possible explanation for these discrepancies is that the activation of these pathways may involve TNFR-II, which is intact in TNFR-I knockout mice. On the other hand, the knockout mice were deficient in the c-Fos component of AP-1, an alteration that was not observed in the antibody blockage experiments (13). In mouse fibroblasts, NF-κB activation and proliferation in response to TNF are exclusively mediated by TNFR-I whereas AP-1 and the protein kinase pathways can be activated through both TNFR-I and TNFR-II (46). Preliminary experiments suggest that NF-κB activation in hepatocytes is also exclusively mediated by TNFR-I, but experiments with mice lacking TNFR-II as well as with double receptor knockouts are necessary to precisely establish the role of each receptor in TNF signaling associated with hepatocyte proliferation.
It has been suggested that lipopolysaccharide released from the gut into the portal circulation induces hepatic TNF production after PH (47), but it has not been established whether its concentration in portal blood increases after PH. TNF is not detectable in hepatic vein blood (that is, blood that has passed through the liver) in normal rats, but it is elevated between 1.5 and 4 h after PH (16). This increase may be the result of an alteration in LPS processing and TNF synthesis by nonparenchymal cells (48). Thus, nonparenchymal cells may be required for processing hepatocyte mitogenic signals at the start of liver regeneration. We have proposed that hepatocyte replication after PH requires the sequential action of cytokines and growth factors (13). We suggest that activation of the TNFR-I pathway involving IL-6 and STAT3 is a mechanism by which hepatocytes acquire proliferative competence in the regenerating liver.
Acknowledgments
We thank J. Bruix and R. Pierce for their comments and T. Carlson for the preparation of the manuscript. This work was supported by Grants CA 23226 and 35249.
Footnotes
Abbreviations: PH, partial hepatectomy; TNF, tumor necrosis factor; IL, interleukin; TNFR-I, TNF receptor 1; EMSA, electrophoretic mobility-shift analysis.
References
- 1.Fausto N, Webber E M. In: The Liver Biology and Pathobiology. Arias I, Boyer J, Fausto N, Jakoby W, Schachter D, Shafritz D, editors. New York: Raven; 1994. pp. 1059–1084. [Google Scholar]
- 2.Bucher N L R. In: Liver Regeneration and Carcinogenesis: Molecular and Cellular Mechanisms. Jirtle R L, editor. San Diego: Academic; 1995. pp. 1–25. [Google Scholar]
- 3.Fausto N, Laird A D, Webber E M. FASEB J. 1995;9:1527–1536. doi: 10.1096/fasebj.9.15.8529831. [DOI] [PubMed] [Google Scholar]
- 4.Diehl A M, Rai R M. FASEB J. 1996;10:215–227. doi: 10.1096/fasebj.10.2.8641555. [DOI] [PubMed] [Google Scholar]
- 5.Taub R. FASEB J. 1996;10:413–427. [PubMed] [Google Scholar]
- 6.Michalopoulos G K. FASEB J. 1990;4:176–187. [PubMed] [Google Scholar]
- 7.Webber E M, Godowski P J, Fausto N. Hepatology. 1994;19:489–497. [PubMed] [Google Scholar]
- 8.Mead J E, Braun L, Martin D A, Fausto N. Cancer Res. 1990;50:7023–7030. [PubMed] [Google Scholar]
- 9.Liu M-L, Mars W M, Zarnegar R, Michalopoulos G K. Hepatology. 1994;19:1521–1527. [PubMed] [Google Scholar]
- 10.Moolten F L, Oakman N J, Bucher N L R. Cancer Res. 1970;30:2353–2357. [PubMed] [Google Scholar]
- 11.FitzGerald M, Webber E, Donovan J, Fausto N. Cell Growth Differ. 1995;6:417–427. [PubMed] [Google Scholar]
- 12.Cressman D E, Greenbaum L E, Haber B A, Taub R. J Biol Chem. 1994;269:30429–30435. [PubMed] [Google Scholar]
- 13.Diehl A M, Yin M, Fleckenstein J, Yang S Q, Lin H Z, Brenner D A, Westwick J, Bagby G, Nelson S. Am J Physiol. 1994;267:G552–G561. doi: 10.1152/ajpgi.1994.267.4.G552. [DOI] [PubMed] [Google Scholar]
- 14.Hsu J-C, Bravo R, Taub R. Mol Cell Biol. 1992;12:4654–4665. doi: 10.1128/mcb.12.10.4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cressman D E, Diamond R H, Taub R. Hepatology. 1995;21:1443–1449. [PubMed] [Google Scholar]
- 16.Trautwein C, Rakemann T, Niehof M, Rose-John S, Manns M P. Gastroenterology. 1996;110:1854–1862. doi: 10.1053/gast.1996.v110.pm8964411. [DOI] [PubMed] [Google Scholar]
- 17.Akerman P, Cote P, Yang S Q, McClain C, Nelson S, Bagby G J, Diehl A M. Am J Physiol. 1992;263:G579–G585. doi: 10.1152/ajpgi.1992.263.4.G579. [DOI] [PubMed] [Google Scholar]
- 18.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Nature (London) 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 19.Pfeffer K, Matsuyama T, Kundig T, Wakeman A. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 20.Flynn J L, Goldstein M M, Chan J, Triebold K J, Pfeffer K, Lowenstein C J, Schreiber R, Mak T W, Bloom B R. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto M, Mariathasan S, Nahm M H, Baranyay F, Peschon J J, Chaplin D D. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 22.Zheng L, Fisher G, Miller R E, Peschon J, Lynch D H, Lenardo M J. Nature (London) 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 23.Higgins G M, Anderson R M. Arch Pathol. 1931;12:186–202. [Google Scholar]
- 24.Webber E M, Wu J C, Wang L, Merlino G, Fausto N. Am J Pathol. 1994;145:398–408. [PMC free article] [PubMed] [Google Scholar]
- 25.Cereghini S, Blumenfeld M, Yaniv M. Genes Dev. 1988;2:957–974. doi: 10.1101/gad.2.8.957. [DOI] [PubMed] [Google Scholar]
- 26.Tewari M, Dobrzanski P, Mohn K L, Cressman D E, Hsu J-C, Bravo R, Taub R. Mol Cell Biol. 1992;12:2898–2908. doi: 10.1128/mcb.12.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karin M. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 28.Ihle J N. Cell. 1996;84:331–334. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 29.Ruff-Jamison S, Zhong Z, Wen Z, Chen K, Darnell J E, Cohen S. J Biol Chem. 1994;269:21933–21935. [PubMed] [Google Scholar]
- 30.Horn F, Lutticken C, Wegenka U, Yuan J, Heinrich P C. In: Cytokines and The Liver. Gerok W, Decker K, Andus T, Gross V, editors. Boston: Kluwer; 1995. pp. 14–28. [Google Scholar]
- 31.Cressman D E, Greenbaum L E, DeAngelis R A, Ciliberto G, Furth E E, Poli V, Taub R. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 32.Wang N D, Finegold M J, Bradley A, Ou C N, Abdelsayed S V, Wilde M D, Taylor L R, Wilson D R, Darlington G J. Science. 1995;269:1108–1112. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- 33.Rana B, Xie Y, Mischoulon D, Bucher N L, Farmer S R. J Biol Chem. 1995;270:18123–18132. doi: 10.1074/jbc.270.30.18123. [DOI] [PubMed] [Google Scholar]
- 34.Diehl A M, Yang S Q, Yin M, Lin H Z, Nelson S, Bagby G. Hepatology. 1995;22:252–261. doi: 10.1016/0270-9139(95)90379-8. [DOI] [PubMed] [Google Scholar]
- 35.Decker K F, Obolenskaya M Y. J Gastroenterol Hepatol. 1995;10:S12–S17. doi: 10.1111/j.1440-1746.1995.tb01789.x. [DOI] [PubMed] [Google Scholar]
- 36.Fausto N, Mead J E. Lab Invest. 1989;60:4–13. [PubMed] [Google Scholar]
- 37.Baumann H, Gauldie J. Immunol Today. 1994;15:74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 38.Geisterfer M, Richards C D, Baumann M, Fey G, Gwynne D, Gauldie J. Cytokine. 1993;5:1–7. doi: 10.1016/1043-4666(93)90017-y. [DOI] [PubMed] [Google Scholar]
- 39.Xing Z, Richards C D, Braciak T, Thibault V, Gauldie J. In: Cytokines and The Liver. Gerok W, Decker K, Andus T, Gross V, editors. Boston: Kluwer; 1995. pp. 164–171. [Google Scholar]
- 40.Libermann T A, Baltimore D. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimizu H, Mitomo K, Watanabe T, Okamoto S, Yamamoto K. Mol Cell Biol. 1990;10:561–568. doi: 10.1128/mcb.10.2.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higashitsuji H, Arii S, Furutani M, Mise M, Monden K, Fujita S, Ishiguro S, Kitao T, Nakamura T, Nakayama H, Fujita J, Imamura M. J Surg Res. 1995;58:267–274. doi: 10.1006/jsre.1995.1042. [DOI] [PubMed] [Google Scholar]
- 43.Columbano A, Shinozuka H. FASEB J. 1996;10:1118–1128. doi: 10.1096/fasebj.10.10.8751714. [DOI] [PubMed] [Google Scholar]
- 44.Satoh M, Yamazaki M. J Cell Physiol. 1992;150:134–139. doi: 10.1002/jcp.1041500118. [DOI] [PubMed] [Google Scholar]
- 45.Koga M, Ogasawara H. Life Sci. 1991;49:1263–1270. doi: 10.1016/0024-3205(91)90139-3. [DOI] [PubMed] [Google Scholar]
- 46.Kalb A, Bluethmann H, Moore M W, Lesslauer W. J Biol Chem. 1996;271:28097–28104. doi: 10.1074/jbc.271.45.28097. [DOI] [PubMed] [Google Scholar]
- 47.Cornell R P, Liljequist B L, Bartizal K F. Hepatology. 1990;11:916–922. doi: 10.1002/hep.1840110603. [DOI] [PubMed] [Google Scholar]
- 48.Tran-Thi T-A, Decker K, Baeuerle P A. Hepatology. 1995;22:613–619. doi: 10.1002/hep.1840220235. [DOI] [PubMed] [Google Scholar]