

Abstract
Synaptic transmission is regulated by G protein-coupled receptors whose activation releases G protein βγ subunits that modulate presynaptic Ca2+ channels. The sequence motif QXXER has been proposed to be involved in the interaction between G protein βγ subunits and target proteins including adenylyl cyclase 2. This motif is present in the intracellular loop connecting domains I and II (LI-II) of Ca2+ channel α1A subunits, which are modulated by G proteins, but not in α1C subunits, which are not modulated. Peptides containing the QXXER motif from adenylate cyclase 2 or from α1A block G protein modulation but a mutant peptide containing the sequence AXXAA does not, suggesting that the QXXER-containing peptide from α1A can competitively inhibit Gβγ modulation. Conversion of the R in the QQIER sequence of α1A to E as in α1C slows channel inactivation and shifts the voltage dependence of steady-state inactivation to more positive membrane potentials. Conversion of the final E in the QQLEE sequence of α1C to R has opposite effects on voltage-dependent inactivation, although the changes are not as large as those for α1A. Mutation of the QQIER sequence in α1A to QQIEE enhanced G protein modulation, and mutation to QQLEE as in α1C greatly reduced G protein modulation and increased the rate of reversal of G protein effects. These results indicate that the QXXER motif in LI-II is an important determinant of both voltage-dependent inactivation and G protein modulation, and that the amino acid in the third position of this motif has an unexpectedly large influence on modulation by Gβγ. Overlap of this motif with the consensus sequence for binding of Ca2+ channel β subunits suggests that this region of LI-II is important for three different modulatory influences on Ca2+ channel activity.
Neuronal voltage-gated Ca2+ channels are involved in multiple cellular functions including neurotranstransmitter release, Ca2+-mediated regulatory processes, and generation of dendritic action potentials. They consist of complexes of a pore-forming α1 subunit of 190–250 kDa in association with α2δ and β subunits (1, 2). Electrophysiological and pharmacological studies distinguish at least six classes of Ca2+ channel currents designated L-, N-, P-, Q-, R-, and T-type (2, 3). Ca2+ channels containing α1C or α1D subunits are thought to be responsible for L-type currents, α1B for N-type currents, and α1A for both P- and Q-type calcium currents (2, 4). A major goal of current research is to correlate the observed differences among the properties of these channel types with the structures of their α1 subunits.
P/Q-type and N-type Ca2+ channels containing α1A or α1B subunits differ from L-type Ca2+ channels containing α1C or α1D subunits in voltage-dependent inactivation (5–8) and in modulation by G proteins (9). Voltage-dependent inactivation of L-type Ca2+ channels containing cloned α1C is slower than inactivation of P/Q-type Ca2+ channels containing cloned α1A expressed with the same auxiliary subunits in Xenopus oocytes (5, 6). In addition, N-type and P/Q-type Ca2+ channels, but not L-type Ca2+ channels, are modulated by neurotransmitter receptors acting through pertussis toxin-sensitive G proteins via membrane-delimited pathways that cause a positive shift in the voltage dependence of channel activation which is reversed by strong depolarization (9–12). This modulatory effect is mediated by G protein βγ subunits (13, 14), possibly through direct binding to the Ca2+ channel.
A consensus sequence (QXXER) has been proposed to be involved in the interaction of Gβγ subunits with adenylyl cyclase type 2, inwardly rectifying K+ channels, and phospholipase Cβ (15). A related consensus sequence is within or adjacent to the region of the COOH terminus of inwardly rectifying K+ channels which is implicated in binding and regulation by Gβγ subunits (16, 17). It is also found in the intracellular loops connecting domains I and II of α1A and α1B, which are modulated by Gβγ, but not in the corresponding segment of α1C, which is not modulated by this pathway (18–20). Thus, this consensus sequence could be involved in G protein modulation of Ca2+ channels. In these experiments, we probed the role of this consensus sequence in Ca2+ channel function and modulation with synthetic peptides containing this sequence and with mutations of this motif in Ca2+ channel α1 subunits. The results point to an important role for this region of the Ca2+ channel in both voltage-dependent inactivation and G protein modulation.
EXPERIMENTAL PROCEDURES
cDNAs encoding Ca2+ channel subunits α1A [rbA isoform, (19)] and β1b were cloned in pMT2XS (8), α1C [rbC-II isoform (18)] in pZem229, α2δ (21) in pZEM228, and CD8 in EBP-pcD. tsA-201 cells were transfected with the α1, α2δ, and β cDNAs in 1:1:1 molar ratios plus CD8 in either calcium phosphate or lipofectamine (Stratagene) and incubated for at least 48 h. Positively transfected cells were identified by labeling with anti-CD8 antibody tagged with fluorophore and analyzed by whole-cell patch clamp as described (14, 22). Currents were recorded and filtered at 10 kHz (α1A) or 4 kHz (α1C) with an eight-pole Bessel filter. Leak and capacitative currents were measured using hyperpolarizing pulses and subtracted with the p/4 method. Cells were bathed in an external solution containing 100 mM Tris, 4 mM MgCl2, and 10 mM BaCl2 with pH adjusted to 7.3 with methanesulfonic acid. The internal pipette solution consisted of 120 mM aspartic acid, 5 mM CaCl2, 2 mM MgCl2, 10 mM Hepes, 10 mM EGTA, and 2 mM Mg·ATP with pH adjusted to 7.3 with CsOH. When indicated in the figure legends, guanosine 5′-[γ-thio]triphosphate (GTP[γS]) was added to the internal solution at a concentration of 0.6 mM.
The mutants in the α1A subunit were constructed using the method of Ho et al. (23). A XhoI/KpnI restriction fragment containing the 5′ region of the α1A cDNA was subcloned into pBluescript II SK(+) (Stratagene) and used for PCR with Pfu polymerase (Stratagene). Amplified mutant cDNA was subcloned into the BspHI and StuI sites of the XhoI/KpnI construct. This construct was subcloned into full-length α1A in pMT2XS and used for transfection. The mutations in α1C were constructed using a single reaction PCR strategy with Pfu polymerase and the rbC-II cDNA in the expression vector pZem229 as template. The PCR products were subcloned into rbC-II in pZem229 in which two BglII sites had been previously destroyed using NgoMI and BglII. Mutations were verified by cDNA sequencing.
RESULTS
Effects of Peptide Inhibitors on G Protein Modulation.
P/Q-type Ca2+ channels composed of α1A, α2δ, and β1b were expressed in the human embryonic kidney cell line tsA-201, a derivative of HEK293 (22). Ba2+ currents through Ca2+ channels were measured in the whole-cell voltage-clamp configuration. In the presence of GTP[γS] in the intracellular solution, the voltage dependence of activation was shifted toward positive membrane potentials and was less steep than in control intracellular solution (Fig. 1A, compare ▵ and ○), as described (14). This shift reflects modulation of the Ca2+ channel by Gβγ (14). Strong depolarizing prepulses increase Ba2+ currents and shift the activation curve to the left (Fig. 1A) by reversing the voltage-sensitive modulation (14) (Fig. 1A, •). This facilitation of the Ba2+ currents and negative shift of the voltage dependence of activation by depolarizing prepulses provides an index of G protein modulation. This experimental protocol was used to test the ability of synthetic peptides from adenylyl cyclase type 2 (AC2) and from the α1A subunits of Ca2+ channels (rbA) containing the putative QXXER G protein binding sequence (Scheme I) to inhibit G protein modulation competitively following diffusion into the cell from the recording pipette.
 |
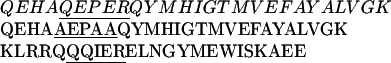 |
 |
Figure 1.

Effects of synthetic peptide inhibitors on G protein modulation of P/Q-type Ca2+ channels. (A) Effect of GTP[γS] on the facilitation of Ba2+ currents of the P/Q-type channel. Tail currents with Ba2+ as current carrier were recorded with (•, ○) or without (▵) GTP[γS] in the intracellular solution. A 4-ms test pulse (test 1) to the indicated test potential was applied from the holding potential of −60 mV. After 1 s, a 10-ms conditioning prepulse to +100 mV was applied, the cell was repolarized to −60 mV for 10 ms, and a second 4-ms test pulse (test 2) to the same potential as test pulse 1 was applied. Tail currents were normalized to the largest tail current in each series of the test pulses, and means ± SEM were plotted against test pulse potential. An increase in tail current after test 2 in comparison to test 1 indicates facilitation. Control intracellular solution, test 1▵; ○, GTP[γS], test 1 •; GTP[γS], test 2. The current traces shown in each panel were recorded during and after test 1 and test 2 at +25 mV. (B–D) Effects of synthetic peptide inhibitors on the facilitation of tail currents of the P/Q-type channel. Peptides (see text for sequence) were applied intracellularly at 100 μM concentration. Tail currents were recorded and analyzed as described in A. (B) AC2 peptide. (C) AC2/AXXAA peptide. (D) rbA peptide.
G protein modulation was blocked by a peptide from adenylyl cyclase type 2 (AC2) which contains the QXXER sequence (Fig. 1B), but not by an identical peptide in which the sequence was changed to AXXAA (Fig. 1C). These results suggest that the AC2 peptide can bind to Gβγ subunits and prevent their interaction with Ca2+ channels. An analogous peptide containing the QXXER sequence from LI-II of α1A also blocks G protein modulation (Fig. 1D). These results suggest a role for the QXXER sequence of α1A in modulation of Ca2+ channel gating by Gβγ.
Comparison of Voltage-Dependent Gating of α1A and α1C.
The Ba2+ currents mediated by α1A inactivate significantly more rapidly than those mediated by α1C (Fig. 2A; τ = 127 ± 14 ms for α1A vs. 915 ± 279 ms for α1C), and the time constant of inactivation for α1C is substantially larger than for α1A across a wide range of test voltages (Fig. 2B, solid symbols). α1A also activates at more negative membrane potentials than α1C (Fig. 2C, solid symbols). The midpoint of the activation curve was 23.6 ± 1.4 mV (n = 15) for α1A compared with 35.4 ± 2.6 mV for α1C, and the voltage dependence was less steep for α1C. Similarly, the voltage dependence of steady-state inactivation at negative membrane potentials where channels inactivate primarily from the closed state is 32 mV more negative for α1A than for α1C (Fig. 1D, solid symbols).
Figure 2.

Effects of mutations in the QXXER motif on voltage-dependent gating of P/Q-type and L-type Ca2+ channels. Control intracellular solutions without GTP[γS] were used in all experiments. (A) Ba2+ currents elicited by a 1000-ms test pulse to +30 mV from a holding potential of −60 mV for the indicated α1 subunits and mutants. (B) Voltage dependence of the time constant for inactivation. Ba2+ currents were recorded during 1000-ms test pulses to the indicated potentials from a holding potential of −60 mV. Currents were fitted with one exponential and the time constants were plotted against the voltage of the test pulse. •, α1A; ○, α1A/QQIEE; ▴, α1C; and ▵, α1C/QQLER. (C) Voltage dependence of activation. Tail currents were recorded at a holding potenial of −60 mV following a 4-ms test pulse to the indicated potential. Tail currents were normalized to the largest tail currents in each series of test pulses, and means ± SEM were plotted as a function of the test voltage. •, α1A; ○, α1A/QQIEE; ▴, α1C; and ▵ α1C/QQLER. (D) Steady-state inactivation. Tail currents were recorded following a 4000-ms prepulse to the indicated potential and a 4-ms test pulse to +30 mV. Tail currents were normalized to the largest tail currents in each series of test pulses, and means ± SEM were plotted as a function of the prepulse voltage. •, α1A; ○, α1A/QQIEE; ▴, α1C; and ▵, α1C/QQLER.
Effects of Mutations in the QXXER Sequence on Voltage-Dependent Gating.
Because Ca2+ channels containing α1A and α1C differ in their gating behavior, we examined whether the amino acid difference in the fifth position of the QXXER motif between α1C (QQLEE) and α1A (QQIER) might alter the gating properties of the channel. The voltage dependence of activation is only slightly shifted to more negative potentials for the mutant channel α1A/QQIEE compared with wild-type (WT) α1A (Fig. 2C). In contrast, large differences were found for the kinetics of inactivation and for the voltage dependence of steady-state inactivation. The time course of inactivation of the mutant is much slower than WT α1A (Fig. 2A) over a broad voltage range (Fig. 2B). The effect is most striking at +15 mV where the mutant is slowed 26-fold (Fig. 2B). The midpoint of the voltage dependence of steady-state inactivation is shifted from −25.4 ± 2.2 mV for α1A to −6.9 ± 1.6 mV for the mutant channel, and the voltage dependence is steeper (Fig. 2D). Thus, the inactivation properties of the mutant α1A containing the QQIEE sequence in LI-II resemble those of α1C.
Complementary effects on inactivation were observed for mutation of the QQLEE sequence in α1C to QQLER. The voltage dependence of activation is shifted slightly to more negative potentials for the mutant channel compared with WT α1C, as observed for the converse mutation in α1A (Fig. 2C). The midpoint for the voltage dependence of steady-state inactivation changed from 7.1 ± 2.3 mV for WT α1C to 1.1 ± 2.0 mV for the mutant (Fig. 2D). The mutant channel inactivated with a time constant of 238 ± 10 ms at +30 mV compared with a time constant of 915 ± 279 ms for WT (Fig. 2A), and more rapid inactivation for the mutant was observed over the whole voltage range tested (Fig. 2B). Thus, conversion of QXXEE to QXXER makes inactivation of Ca2+ channels containing α1C substantially more similar to those containing α1A, although the effects are not as large as the effects of the converse mutation in α1A.
Effects of Mutations in the QXXER Sequence on G Protein Modulation.
Our results with intracellular peptides implicate the QXXER sequence in LI-II in G protein modulation of Ca2+ channels (Fig. 1). As shown in Fig. 3 A and B, activation of α1A is inhibited by GTP[γS] and facilitated by depolarizing prepulses but α1C is not. To test whether mutations in this sequence alter G protein regulation, we analyzed the modulation of Ca2+ channels containing α1A subunits in which the QQIER sequence was mutated to QQIEE and AQIAA. Neither of these mutations in α1A block G protein modulation of the voltage dependence of Ca2+ channel activation (Fig. 3 C and D). In fact, prepulse facilitation is significantly enhanced for these mutations relative to α1A (Fig. 3D). The α1A and α1C subunits also differ in the third position in the QXXER consensus sequence. Conversion of this residue from I in α1A/QQIEE to L as in α1C to yield the double mutant α1A/QQLEE had relatively little effect on inactivation but had a dramatic effect on modulation by Gβγ (Fig. 3E). The voltage dependence of activation in the presence of GTP[γS] was much less shifted to positive membrane potentials, and prepulses to positive voltages caused a correspondingly small facilitation of the Ba2+ current. These results show that the amino acid residue in the third position within the QXXER motif is a crucial determinant of G protein modulation since the subtle change of I to L can nearly completely block facilitation.
Figure 3.

Effects of mutations in the QXXER motif on G protein modulation. Tail currents were recorded following paired test pulses. A 4-ms test pulse (test 1) to the indicated test potential was applied from the holding potential of −60 mV. After 1s, a 10-ms conditioning prepulse to +100 mV was applied, the cell was repolarized to −60 mV for 10 ms, and a second 4-ms test pulse (test 2) to the same potential as test 1 was applied. Tail currents were normalized to the largest tail current in each series of test pulses, and means ± SEM were plotted against test pulse potential. An increase in tail current after test 2 compared with test 1 indicates facilitation. The current traces shown in each panel were recorded during and after test 1 and test 2 at +30 mV. ▵, Control intracellular solution, test 1; ○, GTP[γS], test 1; •, GTP[γS], test 2. (A) α1A. (B) α1C. (C) α1A/AQIAA. (D) α1A/QQIEE. (E) α1A/QQLEE. (F) α1C/QQLER.
We examined whether G protein modulation could be transferred to α1C. Introduction of the QXXER motif into α1C by changing QQLEE to QQLER did not induce facilitation in the presence of GTP[γS] (Fig. 3F). Moreover, further mutation to QQIER as in α1A also did not induce facilitation in the presence of GTP[γS] (data not shown). Thus, the QXXER sequence is not sufficient to confer Ca2+ channel modulation by Gβγ.
To detect quantitative changes in the rates and extents of G protein modulation in these mutants, we investigated the rates of onset and reversal of G protein action in intracellular solution containing GTP[γS]. To measure the rate of onset of G protein action, a test pulse was applied to measure the baseline Ba2+ current, positive prepulses were applied to reverse G protein modulation, an interval of varying duration was interposed to allow G protein modulation to recover, and a second test pulse was applied to measure the extent of recovery of G protein modulation at each time point (Fig. 4A). The facilitation ratio (Itail[test 2]/Itail[test 1]) was largest for α1A/QQIEE, intermediate for WT and α1A/AQIAA, and lowest for α1A/QQLEE. The time constants for the onset of G protein action to reduce IBa under these conditions were similar for α1A with all four sequences (Fig. 4B). To measure the rate of reversal of G protein action, a test pulse was applied to measure the baseline Ba2+ current, a positive prepulse of varying duration was interposed to reverse G protein modulation, and a second test pulse was applied to measure the extent of the G protein effect at each time point (Fig. 4B). The time constant for recovery from G protein modulation was increased from 4.8 ± 0.4 ms (n = 15) for WT α1A to 9.1 ± 1.3 ms (n = 5) for α1A/QQIEE and was reduced to 2.5 ± 0.6 ms (n = 4) for α1A/QQLEE. Because the voltage-dependent reversal of G protein modulation is thought to reflect dissociation of Gβγ from the Ca2+ channel upon depolarization, these results indicate that the change from QQIER to QQIEE stabilizes G protein binding and slows Gβγ dissociation while the further mutation to QQLEE substantially destabilizes G protein binding and accelerates Gβγ dissociation. Evidently, changes in the stability of the bound G protein are largely responsible for the differences in the extent of facilitation for these different α1A subunits.
Figure 4.

Effects of mutations in the QXXER motif of the α1A subunit on the rates of onset and reversal of G protein modulation in the presence of GTP[γS]. (A) Rate of onset. A 4-ms test pulse (test 1) to +30 mV was applied from the holding potential of −60 mV. After 1s, a 10-ms conditioning prepulse to +100 mV was applied to completely relieve G protein inhibition, the cell was repolarized to −60 mV for a period of 1–500 ms, and a second 4-ms test pulse (test 2) to +30 mV was applied. Tail current of the second test pulse was divided by the first test pulse, and means ± SEM were plotted versus the time interval at −60 mV between prepulse and test pulse 2. •, α1A/QQIER; ○, α1A/QQIEE; ▴, α1A/AQIAA; and ▵, α1A/QQLEE. (B) The time courses of G protein action (A) were fitted with one exponential, and the corresponding time constants were plotted. (C) Rate of reversal. A 4-ms test pulse (test 1) to +30 mV was applied from the holding potential of −60 mV. After 1s, a conditioning prepulse to +100 mV for a period of 1 to 18 ms was applied, the cell was repolarized to −60 mV for 10 ms, and a second 4-ms test pulse (test 2) to +30 mV was applied. The tail current of the second test pulse was divided by the first test pulse, and means ± SEM were plotted against time interval at +100 mV. •, α1A/QQIER; ○, α1A/QQIEE; ▴, α1A/AQIAA; and ▵, α1A/QQLEE. (D) Time courses for reversal of G protein action were fitted with one exponential and the corresponding time constants were plotted.
Effects of the Ca2+ Channel β Subunit on Expression of WT and Mutants.
Coexpression of Ca2+ channel β subunits with the α1 subunits increases the current amplitude, shifts the voltage dependence of activation and steady-state inactivation to more negative potentials, and accelerates inactivation (2). The QXXER sequence overlaps the β subunit interaction domain on Ca2+ channel α1 subunits (24). However, as both α1A and α1C bind β subunits, little effect of our mutations on binding of β subunits is expected. The principal effect of coexpression of β subunits with α1A in tsA-201 cells is a substantial increase in current amplitude. For WT α1A, inclusion of the β subunit increased Ba2+ currents from 101 ± 21 pA (n = 11) to 660 ± 622 (n = 28). For α1A/QQIEE, coexpression of β subunits increased Ba2+ currents from 99 ± 26 pA (n = 10) to 1170 ± 250 pA (n = 35) and for α1A/QQLEE the increase was from 120 ± 47 pA (n = 5) to 592 ± 391 pA (n = 9), indicating that the binding and action of the β subunit were retained in these mutants. Similar results were observed with the α1A/AQIAA mutation. Expression of α1C WT and mutants without β subunits did not yield detectable currents, so the α1C mutants that yield normal levels of Ba2+ current must also interact effectively with β subunits.
DISCUSSION
LI-II Is Involved in Binding of Caβ Subunits, Voltage-Dependent Inactivation, and G Protein Modulation.
The intracellular loops that connect the four homologous domains of the Ca2+ channel α1 subunit and the NH2 and COOH termini are the largest domains of the protein and are the most divergent domains among the different isoforms of the α1 subunits. It is likely that these large domains are important in interaction of Ca2+ channels with intracellular proteins that serve as intracellular effectors or modulators. Previous studies have implicated LII-III of the Ca2+ channel α1 subunits in interaction with two different intracellular effectors, the ryanodine-sensitive Ca2+ release channel in the sarcoplasmic reticulum of skeletal muscle (25) and the SNARE (SNAP receptor) proteins involved in docking and exocytosis of neurotransmitter vesicles in neurons (26). These results suggest that LII-III may be specialized as an effector interaction domain. LI-II of the α1 subunit is known to be involved in binding of the β subunits of Ca2+ channels, which have important modulatory effects on the voltage dependence of activation and the kinetics and voltage dependence of inactivation (24). The consensus sequence for binding of Caβ subunits overlaps the QXXER sequence. Our present results implicate LI-II directly in both voltage-dependent inactivation and G protein modulation, suggesting that LI-II may be specialized to serve as a modulatory domain for Ca2+ channel gating. Voltage-dependent conformational changes in LI-II involving the QXXER motif could alter the affinity for Gβγ binding as well as the ease of transition to the inactivated state. Complex regulatory interactions among LI-II, Caβ, and Gβγ may be expected to provide subtle regulation of channel properties. Interestingly, the corresponding intracellular loop of Na+ channels contains a family of phosphorylation sites for cAMP-dependent protein kinase and protein kinase C which also modulate channel function (27), indicating that this loop serves a modulatory function in the structurally related Na+ channel α subunit as well.
Arginine in the QXXER Sequence Is an Important Determinant of Voltage-Dependent Inactivation.
Mutation of R to E in the fifth position of the QXXER sequence has a striking effect on voltage-dependent inactivation. The R/E mutation in α1A slows the rate of inactivation from the open state and also inhibits steady-state inactivation from the closed state by shifting the voltage dependence to more positive membrane potentials. These effects create a channel with inactivation properties very similar to α1C. The converse mutation in α1C accelerates the rate of inactivation and shifts the voltage dependence of steady-state inactivation toward more negative membrane potentials, but its effects are not as pronounced. Our results point to this arginine residue as a key determinant of the rapid, voltage-dependent inactivation and relatively negative steady-state inactivation of Ca2+ channels containing α1A. This single amino acid difference accounts for much of the difference in inactivation properties between these channel types.
In addition to this amino acid residue in LI-II, analysis of chimeras formed from α1A and α1E showed that the membrane-spanning segment S6 in the first domain (IS6) and its flanking regions are also important determinants of the kinetics of voltage-dependent inactivation (28). It seems most likely that the transmembrane segments of the Ca2+ channels contain the main elements required for voltage-dependent inactivation, in analogy with C-type inactivation of K+ channels (29), and that the inactivation process can be further modulated by sequences in LI-II and by interactions with Caβ subunits and Gβγ subunits with this intracellular loop.
Peptides Containing the QXXER Sequence Block G Protein Modulation.
Our results show that synthetic peptides containing the QXXER sequence derived from either adenylyl cyclase type 2 or from α1A can effectively prevent G protein modulation. Similarly the AC2 peptide inhibited regulation of adenylyl cyclase type 2, phospholipase Cβ, and voltage-gated K+ channels (15). It is thought that this peptide binds to Gβγ subunits and prevents their interaction with regulatory targets. This mechanism may also explain the effects of the rbA peptide from LI-II of the α1A subunit that we have used in our experiments. However, the Ca2+ channel peptide may also affect interactions with Caβ subunits that bind to an overlapping segment of α1A (24). Experiments in which the binding of Gβγ is measured directly will be needed to define the mechanism of action of these peptides on Ca2+ channel modulation.
Mutations in the QXXER Sequence Affect G Protein Modulation.
Mutation of QQIER in α1A to QQIEE, AQIAA, or QQLEE substantially alters the extent and the rate of reversal of Gβγ regulation. These results clearly implicate the QXXER sequence in LI-II in G protein modulation of α1A, and indicate that it plays a role in determining the affinity for Gβγ binding and action on α1A. However, mutations in the QXXER sequence do not completely abolish G protein modulation, and introduction of a QXXER sequence into α1C does not transfer G protein modulation. These results are most consistent with the conclusion that this sequence is important for G protein modulation but does not constitute the entire interaction site for Gβγ. Consistent with this idea, Gβγ apparently binds to multiple regions of inwardly rectifying K+ channels, only one of which contains the QXXER sequence in the COOH-terminal domain (17), and x-ray crystallography shows that the interaction between Gβγ and phosducin involves numerous amino acid residues spread through both the NH2 and COOH termini of phosducin (30). Further experiments will be necessary to define all of the requirements for binding and modulation of Ca2+ channels by Gβγ.
Acknowledgments
We thank Drs. Bertil Hille and Neil Nathanson for critical comments on the manuscript. This research was supported by National Institutes of Health Research Grant NS22625 to W.A.C., a postdoctoral research fellowship from the Deutsche Forschungsgemeinschaft to S.H., and a postdoctoral research fellowship from the National Institutes of Health to G.H.H.
Footnotes
Abbreviations: GTP[γS], guanosine 5′-[γ-thio]triphosphate; WT, wild type.
References
- 1.Catterall W A. Annu Rev Biochem. 1995;65:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann F, Biel M, Flockerzi V. Annu Rev Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J-F, Randall A D, Ellinor P T, Horne W A, Sather W A, Tanabe T, Schwarz T L, Tsien R W. Neuropharmacology. 1993;32:1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]
- 4.Snutch T P, Reiner P B. Curr Opin Neurobiol. 1992;2:247–253. doi: 10.1016/0959-4388(92)90111-w. [DOI] [PubMed] [Google Scholar]
- 5.Tomlinson W J, Stea A, Bourinet E, Charnet P, Nargeot J, Snutch T P. Neuropharmacology. 1993;32:1117–1126. doi: 10.1016/0028-3908(93)90006-o. [DOI] [PubMed] [Google Scholar]
- 6.Stea A, Tomlinson W J, Soong T W, Bourinet E, Dubel S J, Vincent S R, Snutch T P. Proc Natl Acad Sci USA. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sather W A, Tanabe T, Zhang J-F, Mori Y, Adams M E, Tsien R W. Neuron. 1993;11:291–303. doi: 10.1016/0896-6273(93)90185-t. [DOI] [PubMed] [Google Scholar]
- 8.Stea A, Dubel S J, Pragnell M, Leonard J P, Campbell K P, Snutch T P. Neuropharmacology. 1993;32:1103–1116. doi: 10.1016/0028-3908(93)90005-n. [DOI] [PubMed] [Google Scholar]
- 9.Hille B. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 10.Marchetti C, Carbone E, Lux H D. Pflügers Arch. 1986;406:104–111. doi: 10.1007/BF00586670. [DOI] [PubMed] [Google Scholar]
- 11.Bean B P. Nature (London) 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 12.Dolphin A C. Exp Physiol. 1995;80:1–36. doi: 10.1113/expphysiol.1995.sp003825. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda S R. Nature (London) 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 14.Herlitze S, Garcia D E, Mackie K, Hille B, Scheuer T, Catterall W A. Nature (London) 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, DeVivo M, Dingus J, Harry A, Li J, Sui J, Carty D J, Blank J L, Exton J H, Stoffel R H, Inglese J, Lefkowitz R J, Logothetis D E, Hildebrandt J D, Iyengar R. Science. 1995;268:1166–1169. doi: 10.1126/science.7761832. [DOI] [PubMed] [Google Scholar]
- 16.Kunkel M T, Peralta E G. Cell. 1995;83:443–449. doi: 10.1016/0092-8674(95)90122-1. [DOI] [PubMed] [Google Scholar]
- 17.Slesinger P A, Reuveny E, Jan Y N, Jan L Y. Neuron. 1995;15:1145–1156. doi: 10.1016/0896-6273(95)90102-7. [DOI] [PubMed] [Google Scholar]
- 18.Snutch T P, Tomlinson W J, Leonard J P, Gilbert M M. Neuron. 1991;7:45–57. doi: 10.1016/0896-6273(91)90073-9. [DOI] [PubMed] [Google Scholar]
- 19.Starr T V B, Prystay W, Snutch T P. Proc Natl Acad Sci USA. 1991;88:5621–5625. doi: 10.1073/pnas.88.13.5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubel S J, Starr T V B, Hell J, Ahlijanian M K, Enyeart J J, Catterall W A, Snutch T P. Proc Natl Acad Sci USA. 1992;89:5058–5062. doi: 10.1073/pnas.89.11.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellis S B, Williams M E, Ways N R, Brenner R, Sharp A H, Leung A T, Campbell K P, McKenna E, Koch W J, Hui A, Schwartz A, Harpold M M. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 22.Margolskee R F, McHendry-Rinde B, Horn R. BioTechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- 23.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 24.Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch T P, Campbell K P. Nature (London) 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- 25.Tanabe T, Beam K G, Adams B A, Niidome T, Numa S. Nature (London) 1990;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- 26.Sheng Z-H, Rettig J, Takahashi M, Catterall W A. Neuron. 1994;13:1303–1313. doi: 10.1016/0896-6273(94)90417-0. [DOI] [PubMed] [Google Scholar]
- 27.Li M, West J W, Numann R, Murphy B J, Scheuer T, Catterall W A. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J-F, Ellinor P T, Aldrich R W, Tsien R W. Nature (London) 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]
- 29.Hoshi T, Zagotta W N, Aldrich R W. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 30.Gaudet R, Bohm A, Sigler P B. Cell. 1996;87:577–588. doi: 10.1016/s0092-8674(00)81376-8. [DOI] [PubMed] [Google Scholar]