

Abstract
Alpha-synuclein (αSyn) misfolding is associated with several devastating neurodegenerative disorders, including Parkinson’s disease (PD). In yeast cells and in neurons αSyn accumulation is cytotoxic, but little is known about its normal function or pathobiology. The earliest defect following αSyn expression in yeast was a block in endoplasmic reticulum (ER)–to–Golgi vesicular trafficking. In a genomewide screen, the largest class of toxicity modifiers were proteins functioning at this same step, including the Rab guanosine triphosphatase Ypt1p, which associated with cytoplasmic αSyn inclusions. Elevated expression of Rab1, the mammalian YPT1 homolog, protected against αSyn-induced dopaminergic neuron loss in animal models of PD. Thus, synucleinopathies may result from disruptions in basic cellular functions that interface with the unique biology of particular neurons to make them especially vulnerable.
Parkinson's disease (PD) is the second most common neurodegenerative disorder (1,2). Accruing evidence points to a causative role for the presynaptic protein alpha-synuclein (αSyn) in PD pathogenesis. αSyn is a major constituent of Lewy Bodies—cellular inclusions that are the hallmark pathological feature of PD and other neurodegenerative disorders collectively referred to as synucleinopathies (3). Moreover, missense mutations in the αSyn gene (A53T, A30P, E46K) (4–6) and duplication or triplication of the locus cause PD (7–9). In mouse, rat, fly, and nematode models of PD, increased levels of αSyn lead to neurodegeneration (10–13). Elucidating the mechanisms underlying the cytotoxic effects of αSyn will be essential for the development of treatments to ameliorate or prevent the synucleinopathies.
Despite extensive study, little is known about αSyn's normal function or how αSyn contributes to disease. Many cellular defects have been implicated in the etiology of synucleinopathies, including impairment of the ubiquitin-proteasome system, mitochondrial dysfunction, accumulation of lipid droplets, production of reactive oxygen species (ROS), and stress within the ER (14). A yeast PD model, with dosage sensitivity for αSyn expression, recapitulates many of these defects (15). But which are cause and which effect remain unclear. Here, two independent approaches, genetic and cell biological, converged to identify inhibition of ER-Golgi trafficking as a major component of synuclein-dependent toxicity.
αSyn accumulation causes ER stress
An increase in αSyn gene dosage in yeast from one copy (no growth defect) to two copies results in growth arrest and cell death (15) (Fig. 1A). To investigate the earliest defects caused by αSyn, we took advantage of the ability to rapidly and synchronously induce its expression from a galactose-inducible promoter. A slight decline in viability was observed after 4 hours of induction, and 60% of cells lost colony-forming ability by 8 hours (Fig. 1, A and B). ER stress, measured by a reporter for the unfolded protein response, appeared earlier. Expression of wild-type αSyn (αSyn-WT) or disease-associated αSyn (αSyn-A53T) caused a fourfold increase in ER stress relative to control cells after 6 hours (Fig. 1C).
Fig. 1.
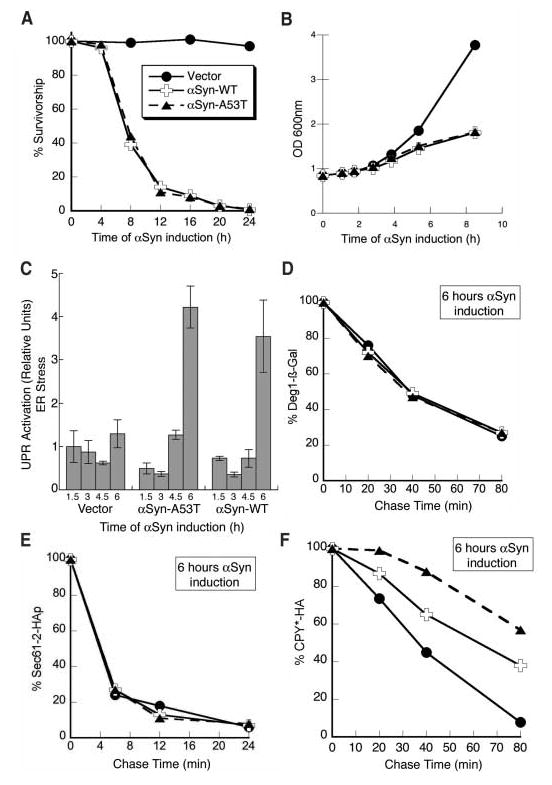
Expression of αSyn causes cell death and ER stress and impairs ERAD. (A) Survivorship curve during αSyn induction. After induction of αSyn-WT, αSyn-A53T expression, or control cells (Vector), cells with an optical density at 600 nm (OD600nm) of 1 were harvested and treated as described (24). Colony-forming units were determined and converted to relative percentages. (B) Growth curve during αSyn induction. After induction, the OD600nm for each sample was measured at the indicated times. (C) Cells induced for expression of αSyn-WT, αSyn-A53T, or control cells (Vector) were harvested at the times indicated; the level of UPR activation was then determined and plotted as relative units of ER stress. The degradation rate of Deg1-βGal (D), Sec61-2p (E), and CPY* (F), after 6 hours of either αSyn-WT or αSyn-A53T expression, was determined by pulse-chase immunoprecipitation as described (24) and compared to that of control cells (Vector).
αSyn accumulation impairs degradation of selective ERAD substrates
ER stress typically results from the accumulation of misfolded proteins within the ER. Such malformed proteins are retrotranslocated from the ER to the cytoplasm for degradation by the proteasome through a process termed ERAD (endoplasmic reticulum associated degradation) (16). Misfolded cytosolic αSyn might impair the proteasome’s capacity for protein degradation and so cause an accumulation of misfolded proteins in the ER and associated ER stress. To investigate this possibility, we examined the degradation rate of a well-characterized cytosolic proteasome substrate, Deg1–β-Gal (17). Its rate of degradation was identical to that of control cells after 6 hours of αSyn expression (Fig. 1D). Thus, general proteasome activity was unaffected at a time when the level of an ER stress reporter was elevated fourfold (Fig. 1C) and a significant percentage of cells had lost viability (Fig. 1A).
A second cause of ER stress might be impairment of ERAD at a step before protea-some degradation. We therefore examined the degradation of two different misfolded proteins within the ER: (i) CPY*, a soluble misfolded substrate (18); and (ii) Sec61-2p, a misfolded membrane-spanning substrate (19). The degradation of Sec61-2p was unaffected (Fig. 1E), yet the turnover of CPY* was impaired in cells expressing αSyn-WT, and more so in cells expressing the disease-associated αSyn-A53T (Fig. 1F). Thus, paradoxically, toxic levels of αSyn inhibited the degradation of one ERAD substrate (CPY*) without perturbing the turnover of another (Sec61-2p).
αSyn accumulation inhibits ER-Golgi trafficking
A distinction between CPY* and Sec61-2p is that CPY* degradation requires trafficking from the ER to the Golgi (20, 21). We investigated if αSyn affects vesicular trafficking between the ER and Golgi, by following two wild-type proteins that traffic through this pathway, correctly folded CPY and alkaline phosphatase (ALP). Their subcellular location is easily determined by compartment-specific glycosylations and proteolytic cleavages that alter the molecular mass of each protein in a well-characterized manner (22, 23). In cells expressing either αSyn-WT or αSyn-A53T, a pronounced defect in CPY (Fig. 2, A and B) and ALP (Fig. 2, C and D) trafficking was observed 3 hours after αSyn induction; by 4 hours their transport from the ER to the Golgi was almost completely blocked. For both CPY and ALP, expression of mutant αSyn-A53T (which causes early-onset PD in humans) caused a more rapid onset of the trafficking block than did αSyn-WT at equivalent levels of expression (Fig. 2, B and D). Notably, the earliest detectable impairment of growth (Fig. 1B) corresponded to the earliest detectable impairment in vesicular transport (Fig. 2, A and B) and preceded the onset of ER stress (Fig. 1C).
Fig. 2.
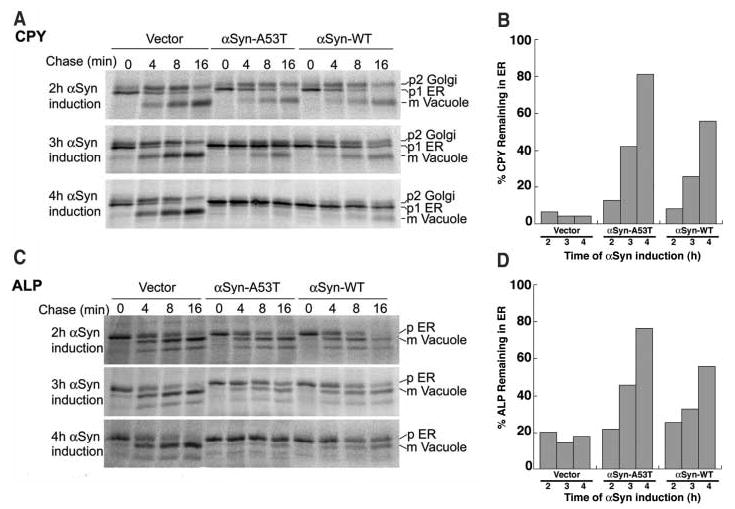
αSyn accumulation causes a severe block in vesicular trafficking in the early secretory pathway. The trafficking of CPY (A and B) and ALP (C and D) was monitored in cells expressing αSyn-WT or αSyn-A53T at the times indicated by pulse-chase immunoprecipitation and compared to that of control cells (Vector). (B) Graphic representation of the amount of CPY remaining in the ER [p1/(p1 + p2 + mCPY)]. (D) Graphic representation of the amount of ALP remaining in the ER [p1/(p1 + p2 + mALP)]. For CPY, p1 is the ER form, p2 is the Golgi form, and m is the mature vacuolar form. For ALP, p is the ER form and m is the mature vacuolar form.
Genomewide overexpression screen identifies modifiers of αSyn toxicity
A genetic approach was employed to advance from determining the timing of affected cellular processes to identifying critical lethal lesions. We used an overexpression library in which individual yeast open reading frames (ORFs) were fully sequenced and placed, without protein tags, under the control of a galactose-inducible promoter. The 3000 randomly selected genes in this library, representing all functional classes, were individually transformed into a strain expressing αSyn-WT (Fig. 3). We used a strain with an intermediate level of αSyn expression (24), and thus an intermediate level of toxicity, enabling us to screen simultaneously for enhancers and suppressors. We identified 34 genes that suppressed and 20 genes that enhanced αSyn toxicity when overexpressed (table S1). One functional class enriched in our screen provided proof-of-principle for the effectiveness of the screen. This class included genes involved in carbohydrate metabolism or galactose-regulated gene expression specifically. Not surprisingly, these modifiers were not specific for αSyn toxicity; most were also recovered in a screen for suppressors of a galactose-regulated toxic huntingtin protein.
Fig. 3.
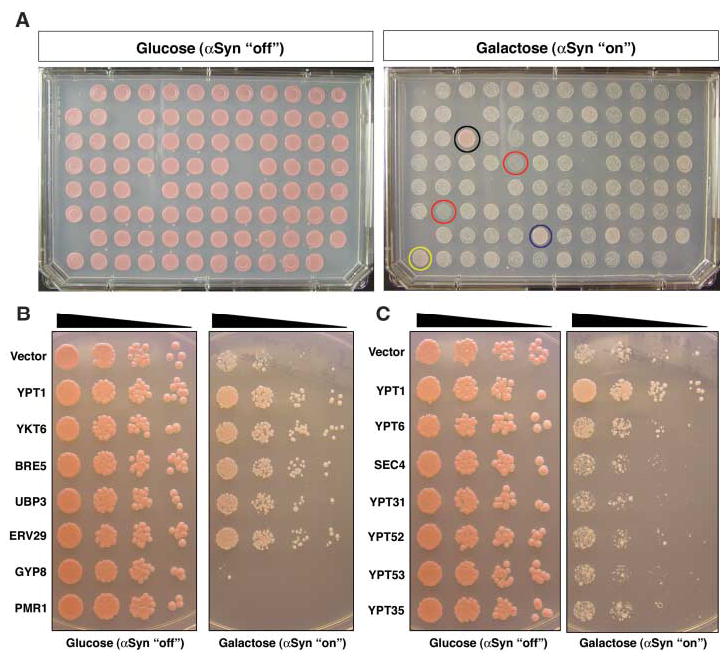
Plasmid overexpression screen identifies ER-Golgi trafficking genes as modifiers of αSyn toxicity. (A) Representative plates from αSyn modifier screen (24). αSyn-expressing cells were transformed individually with each of 3000 random ORFs under the control of a galactose-inducible promoter. Transformants were grown on synthetic media containing either glucose (control, αSyn ‘‘off’’) or galactose (to induce expression of αSyn and candidate ORFs, αSyn ‘‘on’’). Examples of strong- and moderate-toxicity suppressors are shown as black and blue circles, respectively. Examples of enhancers of αSyn-induced toxicity are shown as red circles, and a false-positive that did not reproduce upon further analysis is shown as a yellow circle. (B) Spotting assay shows that overexpression of ER-Golgi trafficking genes YPT1, YKT6, BRE5, UBP3, and ERV29 suppress αSyn-induced toxicity, whereas GYP8 and PMR1 overexpression enhances toxicity. (C) Suppression of toxicity is specific to the transport step facilitated by YPT1, because overexpression of other Rab GTPases has no effect on growth.
ER-Golgi vesicle trafficking genes modify αSyn toxicity
The largest and most effective class of suppressors, all highly specific for αSyn toxicity (fig. S1), were involved in vesicle-mediated membrane trafficking. Notably, all act at the same step of ER-to-Golgi trafficking or are known suppressors of defects in this step: the Rab guanosine triphosphatase (GTPase), Ypt1p; SNARE [soluble NSF (N-ethylmaleimide–sensitive factor) attachment protein receptor] protein, Ykt6p; Ubp3p and Bre5p, a ubiquitin protease and its cofactor required for deubiquitination of coat protein complex II (COPII) component Sec23p; and Erv29p, an ER-exit cargo receptor (table S1 and Fig. 3B). We also recovered Gyp8p as an enhancer of toxicity. GYP8 encodes a Rab GTPase activating protein whose preferred substrate is Ypt1p. Thus, overexpression of genes promoting forward ER-Golgi transport suppresses αSyn toxicity, and those negatively regulating this step enhance toxicity.
A Ypt1-regulated step is particularly important
There are many Rab GTPases, which function at different points of the secretory pathway. Genes that encode other Rab proteins were present in the library but were not identified as αSyn suppressors. Because false-negatives are common in high-throughput screens, these Rabs were carefully and quantitatively retested. Whereas overexpression of Ypt1p rescued toxicity, six other Rab GTPases functioning at more distal points in vesicular trafficking (Ypt6p, Sec4p, Ypt31p, Ypt52p, Ypt53p, or Ypt35p) did not (Fig. 3C).
If inhibition of ER-Golgi trafficking is indeed a critical aspect of αSyn-induced toxicity, then ameliorating toxicity by Ypt1p overexpression should increase forward trafficking. Indeed, overexpression of Ypt1p markedly enhanced forward transport of CPY (Fig. 4, A and B). Overexpression of GYP8, a negative regulator of Ypt1p, exacerbated the trafficking defect (Fig. 4, A and B). A dominant-negative version of Ypt1p, a protein fusion that obviates the function of Ypt1p’s C-terminal geranylgeranyl membrane anchor signal, enhanced αSyn toxicity. Defects in Ypt1p can be suppressed by SLY1-20, which encodes a dominant form of the ER-to-Golgi target (t)-SNARE associated protein Sly1p (25). In a corresponding manner, SLY1-20 strongly suppressed both the αSyn-induced growth defect (fig. S2) and the CPY trafficking defect (Fig. 4, C and D). The ability of these specific suppressors and enhancer alleles to rescue or exacerbate trafficking defects, as well as to rescue or exacerbate αSyn toxicity, confirms that forward ER-to-Golgi vesicular transport is particularly sensitive to αSyn accumulation. Ypt1p frequently localized to αSyn cytoplasmic inclusions (Fig. 4E), suggesting that the cytotoxic form of αSyn may associate with transport vesicles as αSyn normally does with synaptic vesicles (26, 27).
Fig. 4.
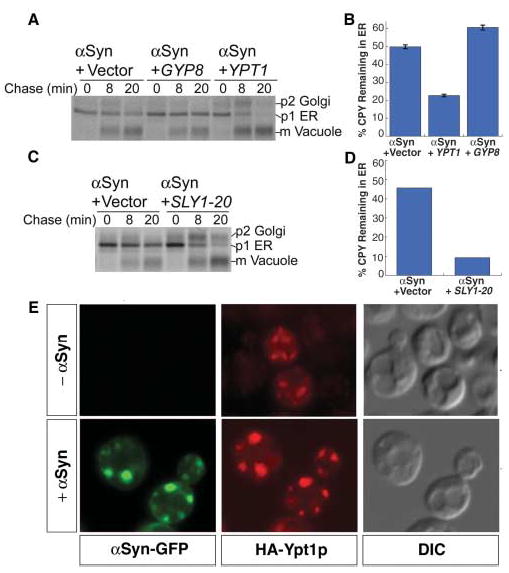
αSyn-induced cyto-toxicity and vesicular trafficking defects are modified by ER-Golgi trafficking components. The trafficking of CPY in cells expressing αSyn-WTand harboring either galactose-inducible GYP8 (A and B), galactose-inducible YPT1 (A and B), or SLY1-20 (C and D) was monitored by radio-labeling after 7 hours (C and D) or 8 hours (A and B) of induction and compared to trafficking in control cells (Vector). (B and D) Graphic representation of the amount of CPY remaining in the ER [p1/(p1 + p2 + mCPY)]. (E) Cells expressing αSyn-WT–GFP (green fluorescence protein) and HA (hemagglutin)–Ypt1p were examined by fluorescence and indirect immunofluorescence microscopy after 6 hours of αSyn induction.
Rescue of αSyn-induced dopaminergic neuron loss by Ypt1p/Rab1
Next we tested the ability of our strongest yeast suppressor to rescue αSyn-associated dopaminergic (DA) neuron loss in animal models of PD (10, 13). In Drosophila, the ability of Rab1 (the murine YPT1 ortholog) to mitigate toxicity of αSyn-WT as well as of the disease-associated αSyn-A53T was determined. Adult flies expressing αSyn, in the presence or absence of added Rab1, were aged, and DA neuron numbers were assessed in the dorsomedial (DM) cluster after immunostaining for tyrosine hydroxylase (TH), which specifically identifies DA neurons. Consistent with previous studies (10), flies expressing αSyn-WT or αSyn-A53T alone exhibited DA neuron loss (Fig. 5, A to C). Coexpression of Rab1 was sufficient to fully rescue this loss (Fig. 5, A to C; two independent transgenic lines). Rescue was specific to Rab1 because directed expression of the control protein β-galactosidase (β-Gal) has no effect on αSyn toxicity (10). Suppression of αSyn toxicity by Rab1 was at least as strong as that of the strongest suppressor previously identified in this system, the chaperone protein Hsp70 (10).
Fig. 5.
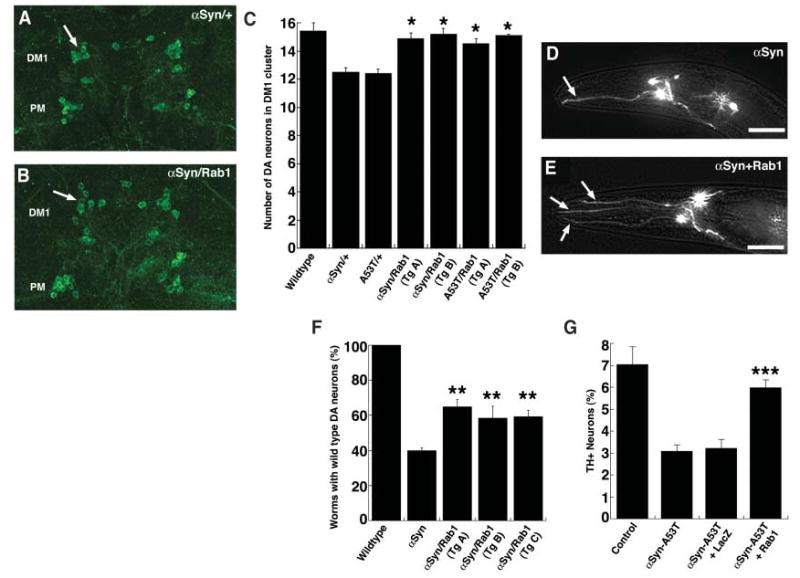
Expression of Rab1 rescues DA neuron loss in animal models of PD. (A to C) Studies in Drosophila. (A and B) Aged fly brains immuno-stained for TH to highlight DA neurons; selective loss of TH immunostaining in the DM1 cluster is fully prevented by Rab1. Genotypes TH-GAL4 UAS-αSyn in trans to + or UAS-Rab1. (C) Quantitation of TH-positive neurons in DM1 cluster. Wild-type (αSyn) or mutant (A53T) αSyn causes loss of TH that is prevented by Rab1. Genotypes: TH-GAL4/+ (wildtype), TH-GAL4 UAS-αSyn or TH-GAL4 UAS-A53T in trans to + or UAS-Rab1 (two independent lines). (*) Values significantly different from αSyn or A53T, P < 0.00001, Student’s t test. (D) DA toxicity in C. elegans, reflected by degeneration of cephalic sensilla (CEP) neuronal processes in Pdat-1::GFP + Pdat-1::αSyn at the 7-day stage. The arrow highlights the single CEP process remaining. (E) Intact DA neurons in Pdat-1::GFP + Pdat-1::α Syn + Pdat-1::Rab1 at the 7-day stage. (F) Quantitation of Rab1 rescue of αSyn-induced neuron loss in three independent transgenic lines. (**) Values significantly different from αSyn, P < 0.05, Student’s t test. Scale bars: 20 μm. (G) Studies in rat midbrain primary neurons. Rab1 protects primary DA neurons from toxicity induced by αSyn-A53T. Primary midbrain cultures were transduced with lentivirus encoding αSyn-A53T alone, or αSyn-A53T plus β-Gal (LacZ), or αSyn-A53T plus Rab1. Control cells were cultured in the absence of lentivirus. Selective DA cell death was evaluated immunocytochemically. Viability is expressed as the percentage of MAP2-positive neurons that were also TH-positive (three independent experiments, at least 100 cells counted per experiment for each condition). The data are presented as the mean ± SD, n = 3 experiments; ***P < 0.01, analysis of variance with Newman-Keuls post-test.
We also tested Rab1 in a Caenorhabditis elegans model (13). The dopamine transporter (DAT-1) gene promoter was used to direct expression of Rab1 along with αSyn in DA neurons. Expression of αSyn alone resulted in 60% of animals with reduced numbers of DA neurons at the 7-day stage compared to controls. Coexpression of Rab1 significantly rescued neurodegeneration in all three of the independent transgenic lines established (Fig. 5, D to F). Suppression by Rab1 was as strong as that seen with the strongest suppressor yet identified in this system, torsinA, an ER-associated protein with chaperone activity (13).
Finally, we tested the ability of Ypt1p/Rab1 to protect against αSyn toxicity in mammalian DA neurons. We produced lentiviruses expressing αSyn-A53T, Rab1, and a control protein, β-Gal. Primary cultures of rat midbrain neurons were transduced with viruses encoding αSyn-A53T, αSyn-A53T plus Rab1, or αSyn-A53T plus β-Gal (Fig. 5, G to I). The viability of DA neurons was assessed relative to the number of total neurons by staining with antibodies specific for TH and the neuronal marker microtubule-associated protein 2 (MAP2). Cultures transduced with αSyn-A53T–encoding lentivirus had decreased numbers of DA neurons (~50% loss) relative to cultures infected with control virus. The selective toxicity of αSyn-A53T to the DA neurons was robustly attenuated by coexpression of Rab1. Thus, the ability of Ypt1p/Rab1 to protect from αSyn toxicity is conserved from yeast cells to DA neurons in animal models of PD.
Discussion
Inhibition of ER-Golgi trafficking by αSyn is a critical cellular lesion contributing to toxicity and cell loss. Moreover, increased Rab1 production is sufficient to protect against αSyn-associated DA neuron loss in animal models of PD.
Our current understanding of Rab function involves Ypt1p/Rab1 playing an essential role in the tethering and docking of the transport vesicle with the Golgi. αSyn likely inhibits this stage of ER-Golgi transport rather than vesicle generation at the ER: αSyn was not observed associated with the ER, and the trafficking-related modifiers act at this stage. The detrimental relation between αSyn and Rab1 is supported by the Golgi fragmentation that is caused by either a dominant mutant Rab1 or forced expression of αSyn (28, 29). A reduction in ER-Golgi transport caused by αSyn would result in an accumulation of proteins in the ER and produce ER stress, potentially accounting for the ER stress observed in PD disease models (30) and in yeast. A trafficking block associated with the Golgi would also explain the endocytosis defect we previously reported (15), because we observe a similar block in FM4-64 dye internalization in the temperature-sensitive ypt1-3 strain in which a defect in endocytosis occurs secondarily to an ER-Golgi trafficking block.
Genes whose overproduction increases forward transport between ER and Golgi would allow cells to overcome the αSyn-induced transport block. Conversely, genes whose overproduction negatively regulates ER-Golgi trafficking exacerbate the transport block caused by αSyn. The results of our genetic screen in yeast are consistent with this scenario: Over-expression of Ypt1p and Ykt6p both likely increase forward transport by increasing the likelihood of membrane vesicles from the ER tethering to Golgi target membranes. Likewise, overexpression of the negative regulator of Ypt1p, Gyp8p, would inhibit this process. Increasing exit of vesicles from the ER would also improve forward transport. Accordingly, overexpression of a ubiquitin protease (Ubp3p) and its cofactor (Bre5p), which together function to deubiquitinate the COPII coat protein Sec23p, would promote vesicle exit from the ER (31).
Recent experiments demonstrated a previously unappreciated, normal function for αSyn (32). Increased expression of αSyn is sufficient to rescue a lethal neurodegenerative phenotype in mice lacking cysteine-string protein α (CSPα). CSPα may thus act as a chaperone to assemble or maintain synaptic SNARE components in functional states over the many repeated SNARE assembly/disassembly cycles expected in neurons (32). Although αSyn does not appear to simply substitute for the lost CSPα chaperone role, it might act downstream or in a parallel pathway involving SNARE complex assembly. This might well include interactions with Rabs, tethering factors, or SNARE proteins, an intriguing aspect because our yeast screen identified both Ypt1p/Rab1 and Ykt6p, a vesicle (v)-SNARE that has been shown to interact genetically with Ypt1p, as potent suppressors. Our findings, that inappropriate αSyn accumulation is toxic owing to specific cellular defects involving an ensemble of proteins that function with SNAREs to mediate vesicle trafficking—coupled with the ability of Rab1 to protect against neurodegeneration in animal models of PD—suggest that toxic activities of αSyn may be related to its normal function.
αSyn is expressed throughout the brain, yet DA neurons are particularly sensitive in PD. Our work suggests that αSyn accumulation is likely to impede the early secretory pathway in many cell types, potentially helping to explain the nonDA lesions resulting from αSyn duplication or triplication (7–9). What, then, might render DA neurons particularly sensitive to an ER-Golgi transport block? Dopamine is inherently unstable and can oxidize to generate ROS, with enzymatic metabolism by monoamine oxidase producing H2O2 (33). Dopamine is synthesized in the cytosol and rapidly pumped by the vesicular monoamine transporter 2 (VMAT2) transporter into synaptic vesicles, where the low vesicular pH and the absence of monoamine oxidase limits dopamine breakdown. Defects in the early secretory pathway could cause a shortage of synaptic vesicles and reduce delivery of VMAT2 to the synapse. Both would impede dopamine loading and produce a rise in cytosolic dopamine concentration. The inhibition of vesicular trafficking by αSyn may affect dopamine-producing neurons more particularly, because neurotransmitters produced by other neurons are less toxic.
The ability of Rab1 to protect against αSyn-induced neuron loss in three independent animal models is strong evidence for a specific link between αSyn and ER-Golgi trafficking. Neurons express additional Rab GTPases not present in yeast, and some of these might be affected by αSyn in a similar manner. Notably, our yeast screen identified additional modifiers of αSyn toxicity, involved in cell stress responses, signaling, and metalion transport, suggesting that there may be further links between the pathobiology of αSyn in yeast and neuronal cells. Our work cross-validates several different model systems for the study of PD and establishes that simple model systems can be useful in the investigation of even complex neurodegenerative diseases.
Footnotes
Supporting Online Material
References
- 1.Vila M, Przedborski S. Nat Med. 2004;10(Suppl):S58. doi: 10.1038/nm1068. [DOI] [PubMed] [Google Scholar]
- 2.Forman MS, Trojanowski JQ, Lee VM. Nat Med. 2004;10:1055. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Proc Natl Acad Sci USA. 1998;95:6469. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, et al. Science. 1997;276:2045. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Kruger R, et al. Nat Genet. 1998;18:106. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 6.Zarranz JJ, et al. Ann Neurol. 2004;55:164. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 7.Singleton AB, et al. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 8.Chartier-Harlin MC, et al. Lancet. 2004;364:1167. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 9.Ibanez P, et al. Lancet. 2004;364:1169. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 10.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Science. 2002;295:865. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 11.Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. Proc Natl Acad Sci USA. 2002;99:10813. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masliah E, et al. Science. 2000;287:1265. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 13.Cao S, Gelwix CC, Caldwell KA, Caldwell GA. J Neurosci. 2005;25:3801. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dauer W, Przedborski S. Neuron. 2003;39:889. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 15.Outeiro TF, Lindquist S. Science. 2003;302:1772. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCracken AA, Brodsky JL. Bioessays. 2003;25:868. doi: 10.1002/bies.10320. [DOI] [PubMed] [Google Scholar]
- 17.Hochstrasser M, Varshavsky A. Cell. 1990;61:697. doi: 10.1016/0092-8674(90)90481-s. [DOI] [PubMed] [Google Scholar]
- 18.Knop M, Hauser N, Wolf DH. Yeast. 1996;12:1229. doi: 10.1002/(sici)1097-0061(19960930)12:12<1229::aid-yea15>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 19.Biederer T, Volkwein C, Sommer T. EMBO J. 1996;15:2069. [PMC free article] [PubMed] [Google Scholar]
- 20.Caldwell SR, Hill KJ, Cooper AA. J Biol Chem. 2001;276:23296. doi: 10.1074/jbc.M102962200. [DOI] [PubMed] [Google Scholar]
- 21.Spear ED, Ng DT. Mol Biol Cell. 2003;14:2756. doi: 10.1091/mbc.E02-11-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klionsky DJ, Emr SD. EMBO J. 1989;8:2241. doi: 10.1002/j.1460-2075.1989.tb08348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevens T, Esmon B, Schekman R. Cell. 1982;30:439. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- 24.Materials and methods are available as supporting material on Science Online.
- 25.Ossig R, Dascher C, Trepte HH, Schmitt HD, Gallwitz D. Mol Cell Biol. 1991;11:2980. doi: 10.1128/mcb.11.6.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fortin DL, et al. J Neurosci. 2004;24:6715. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rochet JC, et al. J Mol Neurosci. 2004;23:23. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- 28.Wilson BS, et al. J Cell Biol. 1994;125:557. doi: 10.1083/jcb.125.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ. J Biol Chem. 2002;277:48984. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- 30.Ryu EJ, et al. J Neurosci. 2002;22:10690. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen M, Stutz F, Belgareh N, Haguenauer-Tsapis R, Dargemont C. Nat Cell Biol. 2003;5:661. doi: 10.1038/ncb1003. [DOI] [PubMed] [Google Scholar]
- 32.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Cell. 2005;123:383. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 33.Lotharius J, et al. J Biol Chem. 2002;277:38884. doi: 10.1074/jbc.M205518200. [DOI] [PubMed] [Google Scholar]
- 34.We thank D. Eide, M. Hochstrasser, J. Warner, and R. Collins for plasmids. We are grateful to members of the Lindquist lab for helpful suggestions and comments on the manuscript, especially J. Shorter and B. Bevis for advice and encouragement and T. Outeiro for making the screening strain. A.D.G. is a Lilly Fellow of the Life Sciences Research Foundation. The YEASTFlex collection was supported by National Human Genome Research Institute grant R01-HG002923. This work was supported by NIH (A.A.C.; Udall Center, S.L.; J.-C.R.; G.A.C.), Missouri Alzheimer’s Grant (A.A.C.), National Institute on Aging (N.M.B.), and the McKnight Fund for Neuroscience (S.L.). S.L. and N.M.B. are investigators of the Howard Hughes Medical Institute. S.L. is a founder and former member of the Board of Directors of, and has received consulting fees from, FoldRx Pharmaceuticals, a company that investigates drugs to treat protein-folding diseases. She holds a patent on modulators of α-synuclein toxicity; genes described in the patent are licensed to FoldRx Pharmaceuticals. She is also a member of the Board of Directors of Johnson & Johnson. A.A.C. and J.-C.R. have received consulting fees from FoldRx Pharmaceuticals, and J.-C. R. has received payment from FoldRx for testing drugs in his laboratory.