

Abstract
Recently, the putative finding of ancient human T cell leukemia virus type 1 (HTLV-1) long terminal repeat (LTR) DNA sequences in association with a 1500-year-old Chilean mummy has stirred vigorous debate. The debate is based partly on the inherent uncertainties associated with phylogenetic reconstruction when only short sequences of closely related genotypes are available. However, a full analysis of what phylogenetic information is present in the mummy data has not previously been published, leaving open the question of what precisely is the range of admissible interpretation. To fulfill this need, were-analyzed the mummy data in a new way. We first performed phylogenetic analysis of 188 published LTR DNA sequences from extant strains belonging to the HTLV-1 Cosmopolitan clade, using the method of statistical parsimony which is designed both to optimize phylogenetic resolution among sequences with little evolutionary divergence, and to permit precise mapping of individual sequence mutations onto branches of a divergence network. We then deduced possible phylogenetic positions for the two main categories of published Chilean mummy sequences, based on their published 157-nucleotide LTR sequences. The possible phylogenetic placements for one of the mummy sequence categories are consistent with a modern origin. However, one of these placements for the other mummy sequence category falls very close to the root of the Cosmopolitan clade, consistent with an ancient origin for both this mummy sequence and the Cosmopolitan clade.
Keywords: Ancient DNA, HTLV-1, Mummy, Phylogenetics, Statistical parsimony, Human migration
1. Introduction
The historical causes underlying the worldwide distribution of human T cell leukemia virus type 1 (HTLV-1) remain incompletely understood (Dekaban et al., 1995). Debate has characterized this area almost as long as the virus has been studied (Gallo et al., 1983; Ishida et al., 1985; Wong-Staal and Gallo, 1985), and new research and divergent interpretation continue to emerge (Miura et al., 1994; Van Dooren et al., 1998). A salient point of discussion concerns whether an “Out-of-Africa” migration in connection with increased global travel over the past several centuries is sufficient to explain the fact that HTLV-1 is found, endemically or sporadically, in most human populations worldwide (Gallo et al., 1983; Wong-Staal and Gallo, 1985; Van Dooren et al., 1998). An alternative view is that the virus was present in Asian and American populations long before the modern era (Ishida et al., 1985; Miura et al., 1994), and has co-descended with its regional human host populations for thousands of years. Recently, the debate has been focused at the empirical level of molecular phylogenetic analysis, especially on how best to explain the phylogenetic origins and structure of a large, globally distributed lineage of HTLV-1 named the “Cosmopolitan” clade into which almost all extant non-African, and a number of African, HTLV-1 strains fall.
In an effort to shed light on this problem, particularly, in relation to the history of HTLV-1 in the Americas, the laboratory of S. Sonoda recently reported several DNA sequences of a 157-bp fragment of the HTLV-1 long terminal repeat (LTR), as well as a 159-bp fragment from the pX gene, recovered after PCR amplification of DNA extracted from bone marrow of a 1500-year-old pre-Columbian mummy from Northern Chile (Li et al., 1999; Sonoda et al., 2000). Although no formal phylogenetic analysis was undertaken, inspection of the pattern of phylogenetically informative sites in the mummy-derived sequences aligned relative to both LTR and pX sequences from several extant HTLV-1 strains was consistent with their having affinities to the Cosmopolitan clade, and more specifically the “transcontinental” subclade that represents the single most widely dispersed lineage of the virus. This in turn led the authors to argue for both (i) an ancient presence of this virus in the Americas and (ii) phylogenetic continuity between, on one hand, HTLV-1 strains present in ancient human populations ancestral to aboriginal peoples of the Americas, and on the other hand, intercontinentally distributed strains of the Cosmopolitan clade as a whole.
These observations and interpretations have been sharply questioned: Gessain et al. (2000) and Vandamme et al. (2000) argued an alternative interpretation, i.e., (i) even the relatively well-studied 157-bp LTR sequence contains insufficient information to definitively establish the phylogenetic affinities of the putatively ancient DNA with conventional, tree-based analyses and (ii) the sequences obtained are most likely of modern origin (i.e., derived from laboratory contaminants). Instead of direct descent between ancient progenitor strains (represented by the mummy sequences) and modern strains, these authors re-emphasized the long-held view that the entire Cosmopolitan clade of HTLV-1 originated in modern times directly from African progenitors.
Interestingly, neither the original nor the commenting research groups explicitly reported any formal phylogenetic analysis of the mummy data, perhaps assuming that insufficient information was available from the 157-bp LTR DNA segment to make such an attempt worthwhile. However, if a high-resolution phylogenetic framework of Cosmopolitan strains estimated with a larger number of informative sites in the HTLV-1 LTR were first constructed, the opportunity exists to (i) narrow the phylogenetic localization of the putatively ancient mummy sequences within the higher resolution framework; (ii) determine how the mummy-derived sequences are related to modern LTR sequences of HTLV-1; (iii) examine the geographic and/or ethnic origins of any corresponding or closely related modern sequences; (iv) ascertain whether the results are more consistent with an ancient or modern origin for the mummy sequences; (v) apply the archaeologically determined age of the mummy remains (Li et al., 1999) as the first available fossil calibration point for phylogenetic dating of HTLV-1. In this way, the full value of the mummy sequence data can be extracted, inherently insufficient as it may be to support a single, unequivocal conclusion on phylogenetic placement.
2. Materials and methods
pX gene sequences derived from geographically diverse populations of HTLV-1 are only sparingly represented in GenBank. The comparative sequence studies have been done for this gene (e.g., Furukawa et al., 2000) have also shown that it is highly conserved and affords little resolution at the phylogenetic level of greatest interest to us, i.e., within the Cosmopolitan clade. Crucially, when we retrieved and aligned the tax– pX sequences that were demonstrated by Furukawa et al. (2000) to provide some resolution at least between Cosmopolitan subclades A and B, we noted that the 159-bp segment analyzed by Li et al. (1999) does not contain any of the phylogenetically informative sites that Furukawa et al. (2000) identified (see Supplementary Fig. 1). Thus, as most research groups focus attention on the LTR for purposes of comparative study, we retrieved from GenBank and aligned, using CLUSTALX (Thompson et al., 1997), a sample of 188 LTR sequences of Cosmopolitan HTLV-1 strains with the criterion that each included at least an LTR segment spanning nucleotides 144–646 of reference strain ATK (GenBank J02029) that spans the 157-bp mummy sequence. The total length of the alignment, available from the authors on request, was 521 nucleotides (including gaps). The widely studied HTLV-1 segment chosen, despite representing less than the full LTR, optimized the breadth of strain sampling in relation to retention of phylogenetically informative sites, and therefore, analytic resolution within the Cosmopolitan clade. In addition to these 168 strains, a total of 24 Central African and Melanesian HTLV-1 strains were included in the alignment to root the Cosmopolitan clade by serving as an outgroup.
For phylogenetic reconstruction, we employed the method of statistical parsimony (Templeton et al., 1992) as implemented in the software package TCS, version 1.13 (program available at http://darwin.uvigo.es) to estimate phylogenetic networks, or cladograms (Clement et al., 2000). This method is based on prior establishment, through population-genetics theory, of a 95% statistical confidence limit for the maximum number of nucleotide sites expected to differ between two given haplotypes without any superimposed substitutions (i.e., the “95% confidence limit of parsimony”) (Templeton et al., 1992). By sequentially joining taxa into progressively larger networks within the parsimony limit, a network of genealogical relationships is reconstructed that is optimized with an explicit, statistically justified criterion of local parsimony. Statistical parsimony has been demonstrated to exhibit its highest resolving power and to significantly outperform traditional phylogenetic approaches when the level of divergence among sequences is low (Crandall, 1994, 1995, 1996; Posada and Crandall, 2001), as it is with strains of HTLV-1 within the Cosmopolitan clade (genetic distances range 0–5% within this clade in our sample). Given such a parsimony-optimized reconstructed network and sufficiently dense taxon sampling, specific mutational changes can be localized discretely on the network and then correlated with nucleotide states of the mummy-derived sequences. There are other advantages to a network-based approach to the study of intraspecies gene genealogy (for further discussion, see the review by Posada and Crandall, 2001). The 157-bp section of the LTR alignment obtained from the Chilean mummy indeed contained too few polymorphic sites to serve as a basis for high-resolution analysis of Cosmopolitan strains of HTLV-1, because the resulting phylogenetic network contained very little structure within the Cosmopolitan clade. Thus, we next undertook to place the mummy sequences within the phylogenetic framework generated by the 521-nucleotide alignment.
The 95% limit of parsimony for the 521-bp LTR alignment was calculated by TCS to be nine mutational steps, and within this connection limit all Cosmopolitan strains represented in the alignment could be linked into a single, statistically parsimonious, main network (Fig. 1). None of the 24 non-Cosmopolitan outgroup strains could be connected to the main network using the 9-step parsimony criterion. A “Pars+1” connection limit of 26 steps (corresponding to 95% cumulative probability that a maximum of one superimposed substitution underlies an observed pairwise distance between genotypes) (Templeton et al., 1992) was therefore invoked in TCS to estimate the position of the root of the Cosmopolitan clade with respect to this outgroup. With a 26-step criterion two equally parsimonious root locations were found, separated from each other by only 2 steps (“Outgroup1” and “Outgroup2” in Fig. 1). Several reticulations (“loops”), representing ambiguities resulting from equally parsimonious points of connection of subnetworks, appear within the cladogram. These ambiguities do not adversely affect interpretation of our specific results or conclusions as discussed below, as none of the possible placements of the mummy sequences depended on prior resolution of a loop. However, the phenomenon does help to explain why traditional methods have often failed to recover useful genealogies when sequence divergence is low, as it often is within species. Indeed, a Bayesian tree (not shown) estimated on the 521-bp alignment using MrBayes 3 (Ronquist and Huelsenbeck, 2003), although highly consistent topologically with the cladogram in Fig. 1, showed only low levels of support for many internal branches. Boundaries of the five Cosmopolitan subclades A–E, previously described as subtype taxa by Gasmi et al. (1994), Miura et al. (1994) and Van Dooren et al. (1998) are readily located on the cladogram (Fig. 1). The presence of five monophyletic Cosmopolitan subclades is, thus, well supported by statistical parsimony.
Fig. 1.
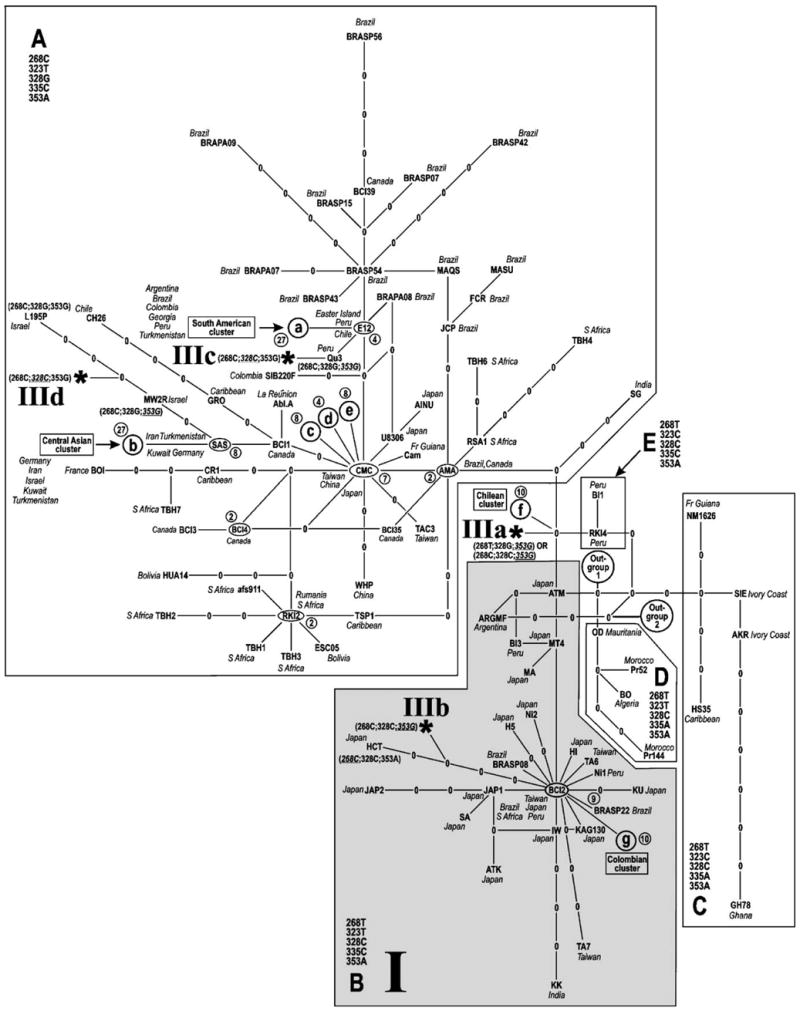
Cladogram of 168 strains belonging to the Cosmopolitan clade of HTLV-1, estimated using statistical parsimony (Templeton et al., 1992) as implemented in the program TCS, version 1.13 (Clement et al., 2000) and a multiple-sequence alignment of 521 bp from the LTR region. Branch tips and internal nodes occupied by extant viral sequences are labelled by the strain name, empty internal nodes by zeroes. Nodes co-occupied by more than one strain with the same genotype are indicated by ellipses, labelled with a single representative strain name from the co-occupying set. Numbers of co-occupant strains at such a node are shown in circles adjacent to each such node. Each line segment, regardless of length as drawn, represents one inferred mutational step as defined by a 95% confidence limit of nine steps. Closed cells in the network result from multiple, equally parsimonious connections between subnetworks at a particular step during execution of the TCS algorithm. For visual clarity locations of five strain clusters are symbolized schematically: (a) South American cluster; (b) Central Asian cluster; (c–e) miscellaneous strains. Two equally parsimonious attachment points of an outgroup composed of 24 non-Cosmopolitan (African and Melanesian) strains to the main network are labelled. Subclades A–E are demarcated in boxes. Predominant nucleotide states found within each subclade at five phylogenetically informative positions (268, 323, 328, 335 and 353) are indicated. Where the genotype of an individual strain differs from the predominant state within a subclade, only the states of divergent sites are given. Occurrence of mummy-associated LTR genotype I (Li et al., 1999) throughout subclade B is indicated by gray shading and the Roman numeral “I”. Possible phylogenetic placements of mummy-associated LTR sequences belonging to genotype III (Li et al., 1999) are marked by asterisks and labelled IIIa–IIId. Note that points of attachment IIIb and IIId of mummy genotype III on the unoccupied branches leading to strains HCT, MW2R and L195 cannot be specified exactly; one possibility was therefore chosen arbitrarily in each case. Nucleotide positions are numbered according to reference strain ATK (GenBank J02029).
3. Results
The LTR clones obtained by Sonoda and co-workers were classified by these authors into three categories, I, II and III (see Fig. 5 of Li et al., 1999). The four clones in category II (SP2-L8, SP2-L9, SP2-L10 and SP2-L11) were identical in sequence to those in category III, aside from the occurrence of one doubleton and two singleton nucleotide substitutions that were seen in no other HTLV-I strain in our LTR alignment. We concluded that no phylogenetic information was present in category II sequences beyond that in category III, and the remaining discussion refers to sequence categories I and III only. In sequence category I (comprising clones SP2-L12 and SP2-L13), the states of nucleotides at phylogenetically informative ATK positions 268, 323, 328, 335 and 353 defined the genotype 268T;323T;328C;335C;353A (here called “genotype I”). This genotype is distributed among observed strains throughout subclade B (with the exception of strain HCT, which appears to have undergone a reverse substitution at nucleotide 268 to confer genotype 268C;323T;328C;335C;353A; see Fig. 1), but nowhere else in the cladogram. Mummy genotype I could also represent one of the unoccupied interior nodes immediately flanking subclade B as it connects with subclades C, D or E. Direct correspondence of mummy genotype I to any of a number of explicitly observed modern HTLV-1 sequences, including some from Japan, is therefore, plausible.
For the second major category of mummy sequences (clones SP1-L1-L11, SP2-L14 and SP2-L15), here called “genotype III”, which differs from genotype I by two nucleotide substitutions at ATK sites 268 and 353, the corresponding 5-site genotype was 268C;323T;328C;335C; 353G. This genotype has neither been reported to occur literally in any extant strain included in our cladogram nor can it be inferred parsimoniously as a necessary missing intermediate. (Note also that a BLASTN search conducted on the NCBI non-redundant nucleotide-sequence database (release 145.0, December 16, 2004; http://www.ncbi.nlm.-nih.gov/BLAST/) on February 7, 2005 did not retrieve any LTR sequences with the genotype 268C;328C;353G from any of a total of 622 significant hits, including all the 188 sequences in our alignment along with 434 other entries.) Nevertheless, by postulating at most one additional nucleotide substitution, mummy genotype III can be inferred to have occurred at a number of disparate positions in the cladogram. Depending on the order in which the 268T →C and 328G →C mutations occurred, one of these positions falls along the internal branch connecting subclade A with subclades B–E, at point IIIa in the cladogram. Depending on details of root location and loop resolution, this node is situated 2–6 steps from the estimated location of the root of the Cosmopolitan clade. A second possibility occurs along the terminal branch carrying strain HCT, at point IIIb in the cladogram. Both placement IIIa and placement IIIb imply the occurrence of one additional substitution, 353A →G, in the terminal branch leading to mummy genotype III.
The only occurrences of 353G in our alignment are in strains Qu3 (Peru), as well as L195P and MW2R (both from Israel and placed within the Central Asian cluster in Fig. 1). A 268C;323T;328C;335G;353G genotype could, therefore, also be placed as a descendant of Qu3 (IIIc in the cladogram), or as a descendant of a missing common ancestor of L195P and MW2R (IIId in the cladogram), in both cases with a single terminal mutation required (328G →C).
Are the placements of mummy genotypes I and III within the Cosmopolitan clade consistent with the phylogenetic positions of HTLV-1 strains that have been recovered from indigenous people in Eastern Asia and Western North America? A total of 14 strains, widely scattered in both subclade A and subclade B in the cladogram in Fig. 1, fall into this category, representing indigenous people from Japan (AINU, U8306); Sakhalin (3S-01, SN-132, SN-94, in strain cluster c); Canada (BCI1, BCI2); Colombia (SIB220F, COL443D), Peru (Qu1, Qu2, Qu3); Chile (CH26); French Guiana (NAR). Intriguingly, one of the possible parsimonious locations for mummy genotype III (location IIIc; Fig. 1) is as an immediate phylogenetic descendant of indigenous strain Qu3 from Peru. If it is allowed that random sampling of genotypes from a common ancestral population could lead to “temporal inversion” of the phylogenetic relationship between physically older and younger strains, this observation would seem consistent with an ancient origin for mummy genotype III. Moreover, the near-ancestral option for placement of mummy genotype III (location IIIa; Fig. 1) is consistent with its representing an ancestor of all 14 indigenous strains.
Although the near-ancestral placement option for mummy genotype III (asterisk IIIa in Fig. 1) potentially attaches it to the network only one step away from subclade E which contains two strains recovered from Peruvians of putative African descent (Van Dooren et al., 1998), this attachment point is also potentially one step removed from strain ATM, which is Japanese in origin. Thus, with near-ancestral placement no unambiguous inference is possible with regard to the geographic provenance of the closest relatives of either mummy genotype.
To summarize our observations, we have shown that, despite the sparseness of the original data, it is indeed possible to localize the mummy-associated sequences reported by Li et al. (1999) and Sonoda et al. (2000) with considerably improved precision in a discrete, character-based phylogenetic framework such as that estimated here by the method of statistical parsimony applied to a 521-bp LTR sequence alignment. Based on its identity with the most common genotype of extant strains found only in the predominantly Japanese Cosmopolitan subclade B, one of the two categories of DNA sequences (genotype I) clearly qualifies as potentially of modern origin, although pending definitive estimation of the age of subclade B this may or may not constitute strong evidence of actual modern origin. In contrast, no exact match to an extant HTLV-1 strain could be identified for mummy-associated genotype III. One of four possible parsimonious placements of genotype III (IIIa) is most consistent with a near-ancestral status in relation to the mostly non-African Cosmopolitan subclades A and B, thus supporting an ancient presence of HTLV-1 in the Americas. Intriguingly, one other possible placement of genotype III (location IIIc in Fig. 1) is as an immediate descendant (one step removed) of a modern strain (Qu3) that was recovered from an indigenous person in the same general geographic region as the mummy location (Van Dooren et al., 1998).
4. Discussion
In our opinion, our analysis highlights the fact that the question of the ancient versus modern status of the putatively mummy-derived HTLV-1 LTR sequences of Li et al. (1999) and Sonoda et al. (2000) remains open, contrary to the opinions expressed by some authors (Gessain et al., 2000; Vandamme et al., 2000). On one hand, the recovery of two phylogenetically distinct HTLV-1 DNA sequences from a single preserved human seems somewhat counterintuitive, and certainly preventing contamination of ancient specimens with modern DNA remains one of the challenging technical problems presented by the study of ancient DNA (Hofreiter et al., 2001). Space does not permit this question to be fully answered here, and its definitive resolution awaits further research. On the other hand, however, the incomplete match between the mummy-associated sequences and known modern genotypes of HTLV-1, as well as the possibility that the unmatched category of sequences (genotype III) may fall in a phylogenetically ancestral position, would seem to argue against an early dismissal of the results (Gessain et al., 2000; Vandamme et al., 2000) as artifactual. If more extensive viral sequence from such preserved ancient human remains could be obtained, yet more precise phylogenetic placement may be possible, and this in turn could contribute to a deeper understanding of the causes and dates of the worldwide dissemination of this important pathogenic retrovirus within the human population.
Acknowledgments
We thank two anonymous reviewers for their insightful comments. DP and KAC gratefully acknowledge support from the US National Institutes of Health and the Brigham Young University Cancer Research Center.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at 10.1016/j.meegid.2005. 02.001.
References
- Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Crandall KA. Intraspecific cladogram estimation: accuracy at higher levels of divergence. Syst Biol. 1994;43:222–235. [Google Scholar]
- Crandall KA. Intraspecific phylogenetics: support for dental transmission of human immunodeficiency virus. J Virol. 1995;69:2351–2356. doi: 10.1128/jvi.69.4.2351-2356.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall KA. Multiple interspecies transmissions of human and simian T-cell leukemia/lymphoma virus type I sequences. Mol Biol Evol. 1996;13:115–131. doi: 10.1093/oxfordjournals.molbev.a025550. [DOI] [PubMed] [Google Scholar]
- Dekaban GA, Digilio L, Franchini G. The natural history and evolution of human and simian T cell leukemia/lymphotropic viruses. Curr Opin Genet Dev. 1995;5:807–813. doi: 10.1016/0959-437x(95)80015-w. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Yamashita M, Usuku K, Izumo S, Nakagawa M, Osame M. Phylogenetic subgroups of human T cell lymphotropic virus (HTLV) type I in the tax gene and their association with different risks for HTLV-I–associated myelopathy/tropical spastic paraparesis. J Inf Dis. 2000;182:1343–1349. doi: 10.1086/315897. [DOI] [PubMed] [Google Scholar]
- Gallo RC, Sliski A, Wong-Staal F. Origin of human T-cell leukaemia-lymphoma virus. Lancet. 1983;2:962–963. doi: 10.1016/s0140-6736(83)90471-3. [DOI] [PubMed] [Google Scholar]
- Gasmi M, Farouqi B, d’Incan M, Desgranges C. Long terminal repeat sequence analysis of HTLV type I molecular variants identified in four north African patients. AIDS Res Hum Retroviruses. 1994;10:1313–1315. doi: 10.1089/aid.1994.10.1313. [DOI] [PubMed] [Google Scholar]
- Gessain A, Pecon-Slattery J, Meertens L, Mahieux R. Origins of HTLV-1 in South America. Nat Med. 2000;6:232. doi: 10.1038/73020. [DOI] [PubMed] [Google Scholar]
- Hofreiter M, Serre D, Poinar HN, Kuch M, Paabo S. Ancient DNA. Nat Rev Genet. 2001;2:353–359. doi: 10.1038/35072071. [DOI] [PubMed] [Google Scholar]
- Ishida T, Yamamoto K, Omoto K, Iwanaga M, Osato T, Hinima Y. Prevalence of a human retrovirus in native Japanese: evidence for a possible ancient origin. J Infect. 1985;11:153–157. doi: 10.1016/s0163-4453(85)92099-7. [DOI] [PubMed] [Google Scholar]
- Li HC, Fujiyoshi T, Lou H, Yashiki S, Sonoda S, Cartier L, Nunez L, Munoz I, Horai S, Tajima K. The presence of ancient human T-cell lymphotropic virus type I provirus DNA in an Andean mummy. Nat Med. 1999;5:1428–1432. doi: 10.1038/71006. [DOI] [PubMed] [Google Scholar]
- Miura T, Fukunaga T, Igarishi T, Yamashita M, Ido E, Funahashi S, Ishida T, Washio K, Ueda S, Hashimoto K, Yoshida M, Osame M, Singhal BS, Zaninovic V, Cartier L, Sonoda S, Tajima K, Ina Y, Gojobori T, Hayami M. Phylogenetic subtypes of human T-lymphotropic virus type I and their relations to the anthropological background. Proc Nat Acad Sci USA. 1994;91:1124–1127. doi: 10.1073/pnas.91.3.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol. 2001;16:37–45. doi: 10.1016/s0169-5347(00)02026-7. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Sonoda S, Li HC, Cartier L, Nunez L, Tajima K. Ancient HTLV type 1 provirus DNA of Andean mummy. AIDS Res Hum Retroviruses. 2000;16:1753–1756. doi: 10.1089/08892220050193263. [DOI] [PubMed] [Google Scholar]
- Templeton AR, Crandall KA, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endo-nuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992;132:619–633. doi: 10.1093/genetics/132.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The clustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandamme AM, Hall WW, Lewis MJ, Goubau P, Salemi M. Origins of HTLV-1 in South America. Nat Med. 2000;6:232–233. doi: 10.1038/73023. [DOI] [PubMed] [Google Scholar]
- Van Dooren S, Gotuzzo E, Salemi M, Watts D, Audenaert E, Duwe S, Ellerbrok H, Grassmann R, Hagelberg E, Desmyter J, Vandamme AM. Evidence for a post-Columbian introduction of human T-cell lymphotropic virus in Latin America. J Gen Virol. 1998;79:2695–2708. doi: 10.1099/0022-1317-79-11-2695. [DOI] [PubMed] [Google Scholar]
- Wong-Staal F, Gallo RC. Human T-lymphotropic retroviruses. Nature. 1985;317:395–403. doi: 10.1038/317395a0. [DOI] [PubMed] [Google Scholar]