

Abstract
Large insert clone libraries have been the primary resource used for the physical mapping of the human genome. Research directions in the genome community now are shifting direction from purely mapping to large-scale sequencing, which in turn, require new standards to be met by physical maps and large insert libraries. Bacterial artificial chromosome libraries offer enormous potential as the chosen substrate for both mapping and sequencing studies. Physical mapping, however, has come under some scrutiny as being “redundant” in the age of large-scale automated sequencing. We report the development and applications of nonelectrophoretic, optical approaches for high-resolution mapping of bacterial artificial chromosome that offer the potential to complement and thereby advance large-scale sequencing projects.
Keywords: genome mapping, fluorescence microscopy, restriction endonuclease, DNA sequencing
Bacterial artificial chromosomes (BACs) (1) have become the preferred large insert cloning system for genomic analysis because such libraries are characteristically stable, show high fidelity, and are compatible with common automated DNA purification procedures (2). Significant problems associated with yeast artificial chromosome libraries, such as insert chimerism and rearrangements, appear to be largely eliminated from BAC resources (3–6). Furthermore, BACs are proven substrates for sequencing when subcloned into plasmids or M13, or used as a primary template for direct end sequencing and internal nucleotide analysis (7). Since the human genome initiative has evolved from primarily mapping chromosomes to sequencing, new strategies are emerging to efficiently sequence BACs covering the entire human genome. Proposals have been made to effectively contig and partially sequence a 30-fold BAC library by clone end-sequencing and mapping using restriction endonucleases (7, 8). According to the authors of this proposal, as many as 600,000 clones would need to be fingerprint-mapped. At the rate of 1,000 fingerprints per day, this effort alone will require close to 2 years for completion. Given these and other considerations, there is a clear need for new approaches to restriction mapping of large insert clones that can be readily automated, work with small amounts of sample, and generate high-resolution ordered restriction maps, instead of merely fingerprints. The information content differences between restriction endonuclease fingerprints and ordered restriction maps are quite large, given a comparable number of restriction endonucleases used for the construction of each type of map (9). This advantage works to facilitate reliable clone contig formation and to confidently verify or align sequence reads.
High-resolution restriction mapping has not been seriously used in massive mammalian sequencing. This lack of use is caused, in part, by the absence of commercially available systems that fully automate the map construction process in a reasonably high-throughput manner. This absence is unfortunate, because restriction maps provide relatively unambiguous markers that are easily interpretable and facilitate sequence read alignment. In fact, a high-resolution map of a BAC clone made by using 5–10 different restriction endonucleases will provide sufficient information to locate the majority of sequence reads derived from ends of plasmid subclones or aligned shotgun data. If the analysis goals are to simply sample-sequence a large insert clone, restriction maps will readily anchor reads, especially from end-sequenced plasmid subclones, and thus provide accurate gap distances between them. These gaps then can be bridged by using primer-based sequencing techniques, or perhaps, the mapped-sequence reads will be used as such to rapidly characterize a given genomic region.
Optical mapping has advanced to become a fully automated high-resolution mapping approach using sophisticated machine vision algorithms and fully integrated statistical approaches for small insert map construction (10). Verification of optical mapping approaches to large insert clones came with the mapping of yeast artificial chromosomes using infrequent cutting restriction enzymes, producing maps with a typical resolution of 80 kb (11). However, no clone contigs were formed and final map construction required using pulsed field gel electrophoresis (PFGE) to size clone inserts. Moreover, the overall map resolution was low. We decided to advance optical mapping of large insert clones by first greatly increasing resolution and later by providing a synergetic foundation for high throughput analysis. We also decided to focus on mapping BACs, for the reasons previously discussed, as well as the fact that the typical BAC clone insert size is ideally suited for optical mapping. Consider that a typical BAC insert is 150 kb in size, or approximately 50 μm in length, and several such molecules are easily imaged within a single field by using a high-power microscope objective. These factors made it possible for us to construct high-resolution, multiple-enzyme restriction maps of several BAC contigs from human chromosomes 11 and 22. Here we describe optically based approaches to large insert clone mapping and verification and also discuss potential applications to large-scale sequencing projects.
MATERIALS AND METHODS
Library Construction and Selection of Clones.
BAC clones were from a human BAC library with 4-fold coverage constructed from a human fibroblast cell line (6, 17). Clones were selected and grouped into contigs as previously described (17).
Sample Preparation.
BAC clones were grown in Luria–Bertani medium (5 ml for each clone) with chloramphenicol (12.5 μg/ml) at 37°C overnight. BAC DNAs were prepared by standard alkaline lysis protocol (22).
BAC Sizing.
The sizes of some BACs were obtained by PFGE analysis of both lambda terminase and NotI linearized BAC DNAs (23, 24). Briefly, BAC DNA was digested to completion with NotI or terminase and fractionated on a 0.8% agarose gel. After electrophoresis, the gel was stained with ethidium bromide and visualized on a UV illuminator. The size of each clone was determined by careful comparison with midrange pulsed-field gel markers (New England Biolabs).
Surface Modification and Calibration.
Coverslips were cleaned as previously described (11). 3-Aminopropyltriethoxysilane (APTES; Sigma) stock (0.10 M) was prepared by dissolving APTES in water and immediately neutralized with 6 M HCl to pH 3.45. Coverslips were treated in 6.3 mM APTES in water diluted from the 0.10 M stock at 50°C for 18 hr. Alternatively, coverslips can be cleaned in concentrated HNO3 overnight and then activated by boiling in 3 M HCl for 3 hr. The coverslips thus treated were incubated in 6.3 mM APTES for 21 hr. Modified surfaces can be preserved in absolute ethanol with 0.1% of 2-mercaptoethanol for more than 5 weeks. The surfaces were assayed by digesting lambda DNA with different enzymes under different conditions optimal for those enzymes to determine the best digestion time. The digestion time ranged from 30 min to 2 hr depending on the specific enzymes and buffer conditions.
Mounting DNA Molecules onto Surfaces.
Triton X-100 was added to the diluted BAC samples (0.03 ng/μl) to the final concentration of 0.1%. Fifteen to 20 microliters of sample was pipetted onto a prewarmed slide on the 45°C heating block. A 22 × 22-mm modified surface was carefully placed on top of the sample drop to spread out the drop. The liquid sandwich was kept on the heating block for 3–5 min until fringes appeared on the coverslip. Appearance of optical fringes indicates that the thickness of the fluid is at submicron level and by experience, the transfer process is complete. Tris-EDTA (TE) buffer (10 mM Tris/1 mM EDTA, pH 8.0) then was added to coverslip edges and drawn in by capillary action. The coverslip was separated from the slide and rinsed in TE buffer. Lambda DNA was mounted by squeezing the sample between a surface and a slide.
Digestion of DNA Molecules on Surfaces.
Five to 10 units of restriction enzymes were diluted in 30 μl of appropriate buffers with 0.02% Triton X-100 and spread onto the DNA mounted coverslips. Digestion was carried out in a humidified closed chamber. After digestion the coverslip was washed with TE twice and stained with 0.1 μM YOYO-1 diluted in 20% 2-mercaptoethanol in TE buffer.
Imaging and Data Analysis.
Images were taken with a cooled charge-coupled device camera (Photometrics) and iplab (Signal Analytics) software (12). The camera setting was adjusted so that the gray level on any part of the molecules was below saturation (4,095 gray levels). This process is done by adjusting the camera collection time with the camera gain fixed at 16×. A 63× oil immersion microscope objective was used for clones larger than 200 kb. The iplab program was used for fluorescence intensity analysis. Molecules usually were divided into about 50-kb segments for intensity analysis. Each segment was separated from the image background through a segmentation function in the program. A threshold value was picked so that the peak on the histogram for an area containing the segment to be measured was the midpoint between the threshold value and the lowest background value. Integrated fluorescence intensity was calculated after segmentation. Local background, which was the average background intensity value of a clean selected area close to the corresponding segment, was subtracted from the sum of segment intensity. The total fluorescence intensity after background subtraction for all the restriction fragments was normalized to the total size of the molecules to translate the size in gray levels into bps.
Alignment of Multiple Enzyme Maps.
Single enzyme maps were constructed by using NotI to linearize samples. By comparing lambda terminase-treated molecules with NotI digestion products (generated in solution), map orientation was possible because terminase-linearized BAC DNA retains the cloning fragment NotI digestion does not. Distinctive patterns of digestion result when comparing terminase vs. NotI-linearized DNA for different enzymes at cloning fragment sites, i.e., BamHI, one extra 6.9-kb fragment; BglII, three extra fragments, 2.7, 2.1, and 1.9 kb; EcoRI, one extra 6.0-kb fragment; NheI, one end longer by 7 kb; SpeI, one 1.2-kb extra fragment and the adjacent end fragment longer by 5.7 kb; XbaI, one extra 4.7-kb fragment and the adjacent end fragment longer by 2 kb; and XboI, one 4.9-kb extra fragment and the adjacent end fragment longer by 2 kb.
RESULTS
Improvements to Optical Mapping.
Our previous work established the feasibility of mapping large insert clones deposited onto derivatized glass surfaces (11). We further advanced our basic optical mapping protocols using BACs, achieving mapping conditions where 30 cuts per clone are routine. In summary, these changes included: development of a simple and reliable procedure to mount large DNA molecules with a usable distribution of molecular extension and minimal breakage; optimization of the surface derivatization, maximizing the range of usable restriction enzymes and retention of small fragments; and development of an open surface digestion format, facilitated access to samples. These developments provided the foundation for automated approaches to mapping large insert clones.
The advancements to optical mapping are manifested by the expansion of the range of restriction fragments sizes that can be measured, as contained within a single BAC clone. We plotted optical mapping vs. electrophoresis-based sizing data to determine the relative error and the precision of optical mapping. Fig. 1 shows these results. For this analysis we chose clone 360E4 (see Fig. 5), and electrophoretically fingerprinted fragments generated using BamHI, BglII, EcoRI, NheI, SpeI, XbaI, and XhoI. These data show optical sizing results ranging from 500 bp to 56 kb with a relative error of 2.9% (comparing optical against pulsed electrophoresis) and a pooled SD = 1.1 kb. Typically, 20–150 BAC molecules are used for construction of a single map. It is important to note that this range of sizing ability is difficult to obtain by using PFGE. We conclude that optical mapping sizing accuracy for restriction fragments generated from BACs is comparable if not superior to conventional and pulsed electrophoresis.
Figure 1.

Comparison of optical mapping restriction fragment sizing vs. PFGE. Maps of BAC 360E4 (see Fig. 5) are plotted vs. PFGE data from digitized images of stained gels. Fragment sizes less than 2 kb were determined by conventional gel electrophoresis. Gel fragments were selected for analysis when unique assignments with optical maps, within experimental error could confidently be made. Some assignments also were confirmed by double digestion. The diagonal line is drawn for reference, and error bars represent the SD on the optical sizing means (each data point represents at least 15 measurements).
Figure 5.

Very high-resolution map of BAC 360E4. Maps were constructed by using seven different restriction enzymes that were overlaid to maintain correct orientation (see Materials and Methods) and verified by double digestion (data not shown). The map is broken into four segments for display. The smallest mapped fragment is 0.50 kb (total insert size: 100 kb).
Our earlier work used PFGE to size yeast artificial chromosome clones inserts before optical mapping (11, 12), because optical mapping results provide only relative fragment sizes instead of absolute values. To improve throughput and increase sizing accuracy we developed optical mapping-based approaches to eliminate any dependence on electrophoretic analysis. We relied on the BAC vector cloning sequence (6.877 kb) cut with NotI to serve as an internal size standard of known fragment mass contained within a cleaved circular molecule. This strategy is similar to one we previously used with linearized bacteriophage clones (13). Two major differences in the analysis of BAC clones included; the target molecule is now circular, and here the cloning fragment-to-insert ratio is much lower (≈1:1 vs. ≈1:25). Fig. 2 shows images of NotI-cleaved relaxed circular BAC molecules, ranging in size from 98 to 195 kb, mounted on optical mapping surfaces. Differentiation of NotI sites defining the cloning fragment from cleavage sites generated within the insert is straightforward given the known size of the cloning fragment, the relative paucity of genomic NotI sites, and the sizing precision of optical mapping. In fact, internal NotI sites within these circular molecules are directly mapped (Fig. 2a A, B, and D). Finally, because broken molecules obviously are excluded from this type of analysis, sizing accuracy was expected to be high. We compared optical mapping sizing errors against PFGE (Fig. 2b), and these results showed a linear mass relationship (65–200 kb) with a pooled SD of 2.5 kb (average of 15 molecules measured per data point) and an average relative error of 1.8%.
Figure 2.

Optical sizing of circular BAC clones. Circular BAC DNA molecules were mounted onto APTES surfaces and digested with NotI. Images of stained, fluorescent molecules were collected and BAC inserts were sized by comparing the fluorescence intensities of inserts (or all fragments generated within the insert) with the BAC cloning vector fragment (6.877 kb). (a) Images of four different circular BAC molecules digested with NotI: A, 150B4 (162.0 kb); B, 280A3 (195.0 kb); C 88H8 (118.0 kb); and D, 999D10 (98.8 kb). (Bar: 5 μm.) (b) BAC insert sizing data compared; optical mapping vs. PFGE. BAC sizes range from 65 kb to 200 kb and error bars represent SD on the means.
Optical Mapping of BAC Clones–Chromosome 11.
Sequence tagged site-content mapping (14), radiation hybrid mapping, clone fingerprinting, and other techniques are used to construct and refine contigs from large insert clones (15). Such contigs contain ordered markers that are frequently ill-defined in terms of the distances between them. Moreover, when few markers are used to define overlap, often the extent of this overlap is difficult to estimate. The point here is that precisely known distances between genomic markers or precisely characterized clone overlaps contribute to formation of a fully competent physical map, readily usable to advance large-scale sequencing or gene-hunting projects. To develop this concept we wanted to construct an optically derived contig from high-resolution maps of BAC clones. We obtained a group (10 clones from 11p13) of BAC clones previously placed into a contig by L.O. and E.R. and screened them with a series of six-cutter restriction endonucleases (EcoRI, SmalI, EagI, NheI, XhoI, and BamHI) to optimize map density to 10–30 cleavage sites per clone.
We found that BamHI digestion suitably created dense maps for simple contig formation without regions containing overly frequent cleavage. Typically, 20–40 molecules of a given clone deposited on a single surface were selected for optical mapping based on judging the completeness of digestion and lack of missing fragments. Fragments smaller than 1.5 kb tended to desorb from the charged optical mapping surface. Fig. 3A shows images of typical molecules selected from each mapped clone arranged and overlapped according to the contig we constructed from overlapping restriction cleavage sites. The maps have an average resolution of 7 kb, with fragment size ranging from 1.5 to 36 kb. Some clones (BAC A) contained as many as 38 fragments. Such maps are nearly impossible to construct by using electrophoresis-based techniques because the large number of similarly sized fragments will generate degenerate bands after full digestion, requiring extensive probing for disambiguation. Furthermore, the generation of Smith-Birnstiel ladders (16), by partial digestion and end labeling, also would be confounded in many instances, because a ladder of labeled products frequently would be too closely spaced for discrimination. Thus, these optically created maps are an uniquely created resource for clone analysis.
Figure 3.

BAC contig map of human chromosome region 11p13 constructed by high-resolution optical mapping. (a) Images of individual clone molecules arranged in a contig. (Bar: 5 μm.) (b) BamHI restriction maps of corresponding clones. The average resolution of these maps is 7 kb; the smallest detectable fragment is about 1.4 kb.
The maps and contig, shown in Fig. 3B, cover approximately 600 kb without gaps. The clones were received with the knowledge that they are overlapping, but unordered. We assembled the contig after optical mapping by simple alignment of restriction sites. Notably, there is a contiguous region, spanning approximately 100 kb containing 7(D–J) of the 10 clones. Optical mapping results also indicate that clones B and C as well as E and F are essentially identical. The redundant maps in this region provide a reliable check for map accuracy in terms of fragment placement and size. The average coefficient of variation for this group (110 fragments), sized 2.5–22.0 kb, is 5.8%, calculating 90% confidence intervals using 4–6 homologous fragments. This variation probably is caused by optical mapping sizing errors, cloning artifacts, restriction length polymorphisms, and small undetectable fragments less than 1 kb in size. Later comparison with Southern blot analysis using cDNA and probes derived from clone ends (data not shown), in addition to electrophoretic fingerprinting, (data not shown) confirmed our results. Here, we assigned each detected gel band to an optical fragment closest in size and determined an average relative error of 4.1%. This result also reflects electrophoresis sizing errors. We conclude that optical mapping data are reproducible and comparable in accuracy to electrophoresis-based measurements.
Optical Mapping of BAC Clones–Chromosome 22.
Another set of previously overlapped clones for mapping was obtained from the California Institute of Technology group covering the chromosome 22 telomeric region (17). Given an already well-established map for this chromosome, our objectives were to prepare high-resolution restriction maps and compare restriction-based contig construction with previous results. Such comparison would clearly define the utility of restriction maps to apply a more universally useful metric to primarily sequence tagged site-based contigs.
For this set of clones, we used several enzymes: XhoI, NotI, and BamHI. The combination of these enzymes yielded maps with a density similar to those constructed for chromosome 11, but multiple enzyme maps are more informative. Fig. 4 shows the optical maps compared with the landmark-based maps, with contigs formed by simple alignment of restriction maps. In total, these maps cover approximately 1.2 Mb, having an average overlap of about three clones. Whereas the landmark-based maps provide clone proximity, the restriction maps accurately align clones with a resolution of about 10 kb, and also approximately place markers on clones, defined by restriction sites. Additional investigation using hybridization or PCR analysis will confidently place markers onto defined restriction fragments. For example, the centromeric end of clone 276D9 and the telomeric end of clone 567H2 bound the marker D22S55 to a 50-kb span. Obviously, the use of additional markers or clones will provide similar results, without resorting to restriction map data, however, these reagents may not be readily available. This point is further illustrated by considering the contig consisting of clones: 57G9, 205F11, 120F5, and 981A9 (Fig. 4f). According to the marker-based map, no overlap is indicated for clone 57G9 and the balance of the mentioned clones. In fact, the optical XhoI map shows significant overlap of this clone with the rest of this group and places markers 132E12 and F1F12 to the far centromeric end. In summary, although the restriction and marker-based maps are in full agreement, the restriction maps bridge gaps and provide high-resolution alignments, even for relatively shallow contigs.
Figure 4.

BAC contig maps of human chromosome 22, telomeric region. Contigs are divided into six groups: a, b, c, d, e and f. Contigs in light blue were constructed by hybridization-based techniques (adapted from ref. 2, not drawn to scale). Optical mapping results for selected clones, indicated in black lines, and formed contigs are shown below. The average resolution of these maps is about 9 kb with a minimum mapped fragment size of 0.80 kb. A putative gap in contig f was closed by optical mapping. Mapping and contig formation results are summarized (g), with red dots indicating clones that were optically mapped.
Because the BAC library was constructed from a human fibroblast cell line, allelic differences and other variations signaled by restriction length polymorphisms should be observed in equal ratio for deep contiguous regions. Several maps (Fig. 4; contigs a and b) indeed show such polymorphisms (Table 1). Here, we scored only polymorphisms arising from deletion or addition of restriction sites; length polymorphisms were ignored. Further restriction mapping with other enzymes probably would reveal additional polymorphic sites.
Table 1.
XhoI polymorphic sites on Chr. 22 BACs detected by high-resolution optical mapping
| Clone | Contig |
XhoI polymorphic coordinate position
|
||||
|---|---|---|---|---|---|---|
| 42 kb | 55 kb | 60 kb | 92 kb | 210 | ||
| 999D10 | a | − | + | |||
| 384D8 | a | − | + | |||
| 88H8 | a | + | − | |||
| 276D9 | b | + | ||||
| 698D7 | b | + | + | |||
| 567H2 | b | + | − | |||
| 34B1 | b | − | + | − | ||
Very High-Resolution Optical Mapping.
High-resolution restriction mapping was used by Kohara and colleagues in 1987 (18) to order a set of small insert clones covering the entire Escherichia coli genome. Although the utility of this map, and the reagents it generated, have proven valuable, enormous effort went into its construction. Unfortunately, their work has remained a unique accomplishment because the lack of appropriate automation has discouraged analogous efforts in other organisms of comparable complexity. Despite this issue, such maps hold promise as scaffolds for large-scale sequencing in addition to serving as a touchstone for analysis of the human genome at a resolution approaching that of actual sequence. This concept becomes more appealing when applied to large insert clones such as BACs. To evaluate this concept we selected a single clone, 360E4, from the previously mapped set of chromosome 22 contigs and mapped further by using six additional enzymes. Multiple restriction enzyme digests obviously increase the number of cleavage sites and yield informationally rich maps, as compared with single digests yields with similar number of cuts. These maps (Fig. 5) then were overlaid with each other, and correct polarity was assured by noting the position of the cloning fragment present on each clone (see Materials and Methods). The average fragment size is 1.2 kb, or roughly 0.07% of the clone sequence is now known. At this resolution, moderately sized deletions, inversions, duplications, and other rearrangements can be noted. For this clone, the distribution of cleavage sites is apparently random with no distinctive cutting patterns showing large barren regions or sites of dense cleavage. Restriction enzymes rich in CG recognition sequences (EagI, SmaI, BssHII, SacII, NarI, SalI, ClaI, and MluI) were tried to detect CpG islands with no apparent cleavage. These data may indicate that this region either contains no detectable genes, or perhaps that the 5′ end of a putative gene lies outside of this particular clone (19).
DISCUSSION
We have demonstrated that optical mapping produces accurate high-resolution maps from BAC molecules, and that these maps can be used to construct accurate contigs, despite incomplete physical landmark data. Given a diploid library, restriction site polymorphisms are useful for phasing clones to chromosomes. Additionally, a very high-resolution map was constructed, and its analysis serves two purposes: to rapidly scan large genomic regions for 5′ ends of genes and to provide a scaffold for sequencing or sequence analysis. Obviously, such analysis also uncovers genomic organizational motifs such as duplications, inversions, and repeats. Hybridization-based techniques (20) also may yield similar results; however, optical mapping does not require previous sequence knowledge and the information density is greater, especially when multiple enzymes are used. Thus, evaluating the ultimate utility of optical mapping to genomic science should center on projected increases to throughput.
We developed a high throughput optical mapping system for the analysis of cosmid and phage clones that uses an effect, called fluid fixation, permitting robotic gridding of many multiple samples onto an optical mapping surface (10). Development of automated microscope imaging systems coupled with sophisticated machine vision approaches and Bayesian statistical approaches to map construction (10, 21) have enabled a fully automated approach to restriction map construction. The task ahead for us, to fully automate BAC mapping, requires straightforward extensions of what we already have accomplished. Preliminary experiments show that the fluid fixation effect also works with BAC molecules and simple enhancements to our map construction algorithms have yielded accurate maps. We thus envision that our laboratory could, in the near future, produce 500 finished maps per day, given 10 optical mapping workstations. Further extensions, with advances in chemistries and imaging techniques, may boost this throughput another 10-fold. Such mapping throughput encourages the consideration of new schemes for map-based sequencing approaches.
The utility of high-resolution maps for large-scale sequencing is hard to dispute if they can be readily constructed at low cost and at a high rate. Although the cost of instrumentation required for optical mapping is high, reagent and disposables costs are very low. High-resolution restriction maps of BACs could be an effective scaffold for the alignment of contigs constructed from shotgun sequencing data. Restriction sites along a BAC would anchor corresponding restriction maps constructed “in silico” from sequence contigs. Increasing the number of restriction enzymes, contig length, and reduction of restriction fragment sizing errors, all play an important role in deciding the optimum scheme for sequence anchoring given considerations of cost, time, and ultimate coverage desired.
To evaluate the experimentally relevant aspects of the anchoring scheme, consider that a sequence contig of length L is to be anchored onto a BAC, of length G, consisting of an ordered restriction map created with m enzymes, where each enzyme is assumed to cut with probability P. Assume that the relative accuracy of the BAC restriction map with respect to any enzyme is β. Consider an arbitrary random location s on the BAC map, and we wish to compute the probability that the sequence contig can be placed there. Let an ordered restriction map be created (in silico) for the sequence contig, corresponding to a particular enzyme, and let this computed map be compared with the BAC map at site s.
It is relevant to estimate the probability of false positive as a function of the number of enzymes (m), length (L), probability that the given enzyme cuts at an arbitrary location (P) and the relative accuracy of the restriction map (β).
First consider the case when m = 1 and the sequence contig is being placed with a fixed orientations (out of two) at site s. The false positive probability for a fixed location is then
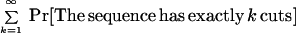 |
1 |
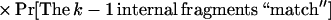 |
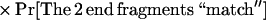 |
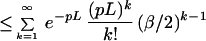 |
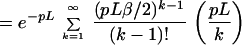 |
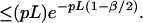 |
Note that the matching rule we have used is fairly simple: given an internal fragment of length x from the sequence contig and a corresponding fragment of length y from the BAC map, we say that they match if
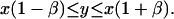 |
2 |
Using m enzymes, and both orientations for the sequence contig, we see that in general this probability generalizes to
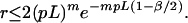 |
3 |
Let us assume that we will consider only the cut sites in the BAC map to anchor the sequence contigs; there are on the average Gp such sites to be considered. Thus the probability that the sequence contig does not get anchored at any of the GP possible false sites then is bounded by e−Gpr and the false positive probability is:
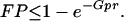 |
4 |
On the other hand, given a sequence contig from a BAC, we will fail to place it at the appropriate location, if it has no cut with respect to any enzyme. The probability with which this event occurs is bounded by FN = e−mpL, the false negative probability. Fig. 6 shows a plot of values obtained from Eq. 4, using a typical BAC clone size of 150 kb. These results show that the number of maps per BAC clone have a dramatic effect on the error rate associated with the anchoring of sequence contigs of varying length. We conclude that for a sufficiently small G, as the number of enzymes m increases or the length of a sequence contig L increases, we almost surely will be able to place these sequence contigs in the correct location.
Figure 6.

Plot of the false positive probability as a function of the number of enzymes and length of sequence contigs. The plot assumes the following parameters: sequence contigs of length ≈5,000–33,000 bp (corresponding to ≈3–4 to 6 genome equivalents), BAC restriction maps constructed using 6-cutter enzymes, and a fragment sizing error of 5%. The plot shows that the anchoring scheme is highly effective for contigs of 5,000 bp, when using seven or more enzymes. However, when contig length is doubled to ≈10,000 bp, only 4–5 enzymes provide similar error.
Thus the overall utility of high-resolution restriction maps may be to enormously facilitate the closure of gaps in several ways: (i) sequence contigs are confidently ordered by alignment to the scaffold maps, and (ii) gap lengths are well characterized, thus enabling closure techniques based on PCR. Additionally, such maps provide a means to verify sequence alignments, especially critical when dealing with large regions of repetitive sequence. A final question remains to be answered: given the complexity of human genome, can the genomics community accurately sequence the entire human genome without high-resolution maps?
Acknowledgments
We thank E. Dimalanta and J. Eddington for experimental assistance. Special thanks go to E. Huff, M. Waterman, M. Urdea, B. Warner, F. Buxton, D. Alexander, and G. Kresbach for helpful discussions. This work was supported by grants from the National Institutes of Health (HG00225-02 and HG00565-03 to E.R.), the National Science Foundation, the W. M. Keck Foundation, and the Lucille P. Markey Charitable Trust.
ABBREVIATIONS
- BAC
bacterial artificial chromosome
- PFGE
pulsed field gel electrophoresis
- APTES
3-aminopropyltriethoxysilane
- TE
Tris-EDTA
References
- 1.Shizuya H, Birren B, Kim U-J, Mancino V, Slepak T, Tachiiri Y, Simon M I. Proc Natl Acad Sci USA. 1992;89:8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmitt H, Kim U-J, Slepak T, Blin N, Simon M I, Shizuya H. Genomics. 1996;33:9–20. doi: 10.1006/geno.1996.0154. [DOI] [PubMed] [Google Scholar]
- 3.Shizuya H, Birren B, Kim U-J, Mancino V, Slepak T, Tachiiri Y, Simon M. Proc Natl Acad Sci USA. 1992;89:8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai L, Taylor J F, Wing R A, Gallagher D S, Woo S-S, Davis S K. Genomics. 1995;29:413–425. doi: 10.1006/geno.1995.9986. [DOI] [PubMed] [Google Scholar]
- 5.Woo S-S, Jiang J, Gill B S, Paterson A H, Wing R A. Nucleic Acids Res. 1994;22:4922–4931. doi: 10.1093/nar/22.23.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim U-J, Birren B W, Spepak T, Mancino V, Boysen C, Kang H, Simon M I, Shizuya H. Genomics. 1996;34:213–218. doi: 10.1006/geno.1996.0268. [DOI] [PubMed] [Google Scholar]
- 7.Venter J C, Smith H O, Hood L. Nature (London) 1996;381:364–366. doi: 10.1038/381364a0. [DOI] [PubMed] [Google Scholar]
- 8.Smith M W, Holmsen A L, Wei Y H, Peterson M, Evans G A. Nat Genet. 1994;7:40–7. doi: 10.1038/ng0594-40. [DOI] [PubMed] [Google Scholar]
- 9.Lander E S, Waterman M S. Genomics. 1988;2:231–239. doi: 10.1016/0888-7543(88)90007-9. [DOI] [PubMed] [Google Scholar]
- 10.Aston C, Hiort C, Schwartz D. Methods in Enzymology. New York: Academic; 1998. , in press. [DOI] [PubMed] [Google Scholar]
- 11.Cai W, Housman D E, Wang Y-K, Schwartz D C. Proc Natl Acad Sci USA. 1995;92:5164–5169. doi: 10.1073/pnas.92.11.5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz D C, Li X, Hernandez L I, Ramnarain S P, Huff E J, Wang Y-K. Science. 1993;262:110–114. doi: 10.1126/science.8211116. [DOI] [PubMed] [Google Scholar]
- 13.Meng X, Benson K, Chada K, Huff E J, Schwartz D C. Nat Genet. 1995;9:432–438. doi: 10.1038/ng0495-432. [DOI] [PubMed] [Google Scholar]
- 14.Olson M, Hood L, Cantor C, Botstein D. Science. 1989;245:1434–1435. doi: 10.1126/science.2781285. [DOI] [PubMed] [Google Scholar]
- 15.Chumakov, I. M., Rigault, P., Le, G., Bellanne-Chantelot, C., Billault, A., Guillou, S., Soularue, G., Poullier, E., Gros, I. & Cohen, D. (1995) Nature (London) 377, Suppl., 175–297. [DOI] [PubMed]
- 16.Smith A M, Birnstiel M L. Nucleic Acids Res. 1976;3:2387–2399. doi: 10.1093/nar/3.9.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim U-J, Shizuya H, Kang H L, Choi S, Garret C L, Smink L J, Birren B W, Korenberg J R, Dunham I, Simon M I. Proc Natl Acad Sci USA. 1996;93:6297–6301. doi: 10.1073/pnas.93.13.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohara Y, Akiyama K, Isono K. Cell. 1987;50:495–508. doi: 10.1016/0092-8674(87)90503-4. [DOI] [PubMed] [Google Scholar]
- 19.Bird A. Nature (London) 1986;321:209–214. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 20.Matera A G, Ward D C. Hum Mol Genet. 1992;1:535–539. doi: 10.1093/hmg/1.7.535. [DOI] [PubMed] [Google Scholar]
- 21.Anantharaman T, Mishra B, Schwartz D C. J Comp Biol. 1997;4:91–118. doi: 10.1089/cmb.1997.4.91. [DOI] [PubMed] [Google Scholar]
- 22.Silhavy Y T J, Berman M L, Enquest L W. Experiments with Gene Fusion. Plainview, NY: Cold Spring Harbor Lab. Press; 1984. [Google Scholar]
- 23.Schwartz D C, Cantor C R. Cell. 1984;37:67–75. doi: 10.1016/0092-8674(84)90301-5. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz D C, Smith L C, Baker M, Hsu M. Nature (London) 1989;342:575–576. [Google Scholar]