

Abstract
Metallothionein (MT), despite its high metal binding constant (KZn = 3.2 × 1013 M−1 at pH 7.4), can transfer zinc to the apoforms of zinc enzymes that have inherently lower stability constants. To gain insight into this paradox, we have studied zinc transfer between zinc enzymes and MT. Zinc can be transferred in both directions—i.e., from the enzymes to thionein (the apoform of MT) and from MT to the apoenzymes. Agents that mediate or enhance zinc transfer have been identified that provide kinetic pathways in either direction. MT does not transfer all of its seven zinc atoms to an apoenzyme, but apparently contains at least one that is more prone to transfer than the others. Modification of thiol ligands in MT zinc clusters increases the total number of zinc ions released and, hence, the extent of transfer. Aside from disulfide reagents, we show that selenium compounds are potential cellular enhancers of zinc transfer from MT to apoenzymes. Zinc transfer from zinc enzymes to thionein, on the other hand, is mediated by zinc-chelating agents such as Tris buffer, citrate, or glutathione. Redox agents are asymmetrically involved in both directions of zinc transfer. For example, reduced glutathione mediates zinc transfer from enzymes to thionein, whereas glutathione disulfide oxidizes MT with enhanced release of zinc and transfer of zinc to apoenzymes. Therefore, the cellular redox state as well as the concentration of other biological chelating agents might well determine the direction of zinc transfer and ultimately affect zinc distribution.
Metallothionein (MT) is a protein that has long been in search of a function. Its composition and structure (1–3) have clearly identified it as a biological metal-chelating agent, yet it does not resemble conventional proteins and its two distinct zinc–sulfur clusters are unlike any known inorganic zinc(II) complexes. Two properties of MT might reveal aspects of its cellular function(s). Its binding of zinc is exceptionally strong owing to the exclusive coordination of the metal with cysteine sulfur ligands [stability constant of Zn7MT-2 = 3.2 × 1013 M−1 at pH 7.4 (4)]. Thionein (T), the apoform, is a potent zinc acceptor. On the other hand, the sulfur ligands are highly reactive and determine not only the binding of zinc to T but also its release from MT (5, 6), which then becomes a zinc donor. This raises questions as to the circumstances under which T removes zinc from proteins and/or MT donates it to apoproteins. Zinc would be expected to be transferred from the protein with the lower stability constant to that with the higher one (reaction 1), which in most cases would determine unidirectional zinc flow from the zinc protein to T.
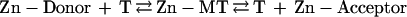 |
1 |
Indeed, T has been shown to block the action of zinc-dependent transcription factors—e.g., TFIIIA and Sp1—presumably by means of metal chelation (7, 8). MT, on the other hand, transfers at least some of its complexed zinc ions to a number of apoproteins (9). The process by which the latter is accomplished remains enigmatic, because the zinc binding constants of most of the enzymes studied are at least 1,000 times lower than that of MT. What conditions and factors allow T to remove zinc from proteins and for MT to donate zinc to apoproteins? The dual capacity of this system to function as both a zinc donor and acceptor (reaction 1) clearly calls for critical examination.
To our knowledge, attempts have not been made to study bidirectional zinc transfer between a specific protein and T/MT to examine the acceptor/donor properties of the T/MT system. We have now employed two different zinc enzymes for this purpose, Escherichia coli alkaline phosphatase (AP) and bovine carboxypeptidase A (CPA). They were chosen because their catalytic mechanisms and structures are well characterized, their apoforms can be prepared and reconstituted with Zn(II) salts (9–11), and they differ in their capacity to bind zinc. Moreover, AP is a dimer containing one catalytic and one cocatalytic zinc atom, as well as one magnesium atom per monomer (12). On the other hand, CPA requires only one zinc atom per molecule to achieve full activity. The distinct differences between the two enzymes are important to rule out specific interactions between MT and either one of them.
The present studies of zinc transfer focus on additional factors that determine its direction. The removal of zinc from zinc enzymes does not occur simply by an interaction with T but requires the participation of other agents. Moreover, we have identified groups of agents that drive the zinc acceptor/zinc donor equilibrium in the direction of zinc release, in particular thiol-centered reactions of MT (6), leading to the postulate that the redox behavior of the sulfur ligands controls the availability of the redox-inert zinc (13).
MATERIALS AND METHODS
Materials.
Human and rabbit liver MT-2 were prepared as described (14). Rabbit liver Cd5Zn2MT-2 was kindly provided by J. H. R. Kägi (University of Zürich). Bovine CPA, E. coli AP, and chemicals were purchased from Sigma. D. S. Auld (Harvard Medical School) provided the CPA substrate dansylglycylglycyltryptophan (Dns-G-G-W). Deionized water (resistivity of ≥15 MΩ⋅cm) and metal-free pipette tips (Fisher) were used throughout. Adventitious metals were removed from all buffer stock solutions by treatment with 5% (vol/wt) Chelex (Bio-Rad) for 2 hr at room temperature and subsequent filtration through a Millex-GS microfilter (Millipore).
Preparation of T and Zn7MT-2.
T and zinc-substituted Zn7MT-2 were prepared as described (6). T prepared this way can be stored at −180°C under nitrogen for more than 3 months without loss of thiol groups. Stock solutions of Zn7MT-2 were prepared prior to use, and excess zinc or Tris was removed by four to six cycles of dilution and concentration with appropriate buffer in a 2-ml Centricon concentrator, molecular weight cutoff = 3,000 (Amicon).
Characterization of T.
The reaction of T with 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) in the absence and presence of different aliquots of zinc sulfate was followed by spectrophotometry at 412 nm in 20 mM Hepes, pH 7.5 at 25°C.
Preparation of Apo-AP.
An aliquot of 4 mg of AP suspended in 2.5 M ammonium sulfate was transferred to a 1.5-ml Eppendorf microcentrifuge tube. The enzyme was collected by centrifugation and the supernatant was discarded. AP was dissolved in 1.5 ml of 10 mM 8-hydroxyquinoline-5-sulfonic acid (HQSA), pH 8.0, and incubated at 4°C overnight. The sample was transferred to a 2-ml Centricon concentrator (molecular weight cutoff = 30,000) that had been treated with 10 mM HQSA, pH 8.0, concentrated to a final volume of ≈250 μl, diluted to a volume of 2 ml with chelating agent, and reconcentrated, all at 4°C. This procedure was repeated four times. HQSA was then replaced with 10 mM Tris, pH 8.0, and the procedure was repeated another 14 times. The enzyme was stored at −20°C. AP and its apoform were >95% pure as judged by SDS/PAGE (stacking gel 5%, resolving gel 12% acrylamide). Protein concentrations were determined by spectrophotometry (ɛ278 = 6.77 × 104 M−1⋅cm−1). The apoenzyme preparation contained <3% of the original zinc and magnesium.
Zinc Transfer from Zn7MT-2 to Apo-AP.
Stock solutions of AP and apo-AP in 10 mM Tris⋅HCl, pH 8.0, were diluted with the same buffer to make 0.5 μM working solutions. A solution of 100 mM p-nitrophenyl phosphate in water was diluted with 10 mM Tris⋅HCl, pH 8.0, to obtain a 1 mM substrate working solution. Enzyme (50 μl) and substrate were rapidly mixed in an acid-washed quartz cuvette and the formation of p-nitrophenolate was monitored at 400 nm for 2 min (9). A linear best fit was used to calculate the turnover rates between 50 and 150 s for slow and between 20 and 60 s for fast reactions. Unless otherwise stated, activity is expressed as percent of that measured for native AP.
Zinc transfer experiments were conducted by preincubation of T and AP, or apo-AP with Zn7MT-2 or Cd5Zn2MT-2 and potential agents that enhance transfer in 10 mM Tris⋅HCl, pH 8.0, for 2 hr at 20°C and subsequent activity analysis.
Preparation of Apo-CPA.
CPA was purified on a CABS-Sepharose column (15) and stored as a crystal suspension in 10 mM Tris⋅HCl, pH 7.5, at 4°C. Apo-CPA was prepared from CPA crystals (16) and stored as a suspension at 4°C. The purity of CPA and its apoform was at least 95% as determined by SDS/PAGE (see above). The zinc-to-protein ratio for CPA was 1.1 and for the apoenzyme it was <0.001. Protein concentrations were determined by spectrophotometry (ɛ280 = 6.42 × 104 M−1⋅cm−1).
Zinc Transfer from Zn7MT-2 to Apo-CPA.
CPA activity was measured by changes in the fluorescence of the substrate Dns-G-G-W (17) with a Biosequential SX-18MV stopped-flow reaction analyzer (Applied Photophysics, Leatherhead, U.K.). Stock solutions of CPA and apo-CPA, in 20 mM Hepes/1 M NaCl, pH 7.5, were diluted to 2 μM with the same buffer. A stock solution of 20 mM Dns-G-G-W in acetonitrile was prepared and diluted with 20 mM Hepes/1 M NaCl, pH 7.5, to 20 μM.
Activity measurements were carried out at an excitation wavelength of 285 nm (slit width 0.3 nm), a detector sensitivity of ≈400 V, and time intervals of 10 ms at 25°C. The emission increase of tryptophan fluorescence was monitored at 340 nm for 50 s. A single-exponential best fit was used to calculate the catalytic rates. These were converted into percentages of the rate of native CPA measured under the same experimental conditions.
Zinc transfer experiments were conducted by preincubation of apo-CPA with Zn7MT-2 and potential agents that enhance transfer for defined periods of time at 20°C and subsequent activity analysis. Zinc transfer from CPA to T was measured by incubating the enzyme and T in 1 M Tris⋅HCl, pH 8.0, for 90 min at 20°C and subsequently determining the CPA activity.
Zinc Transfer from Zn7MT-2 to 4-(2-Pyridylazo)resorcinol (PAR).
MT was incubated with PAR (100 μM) in 20 mM nitrogen-saturated 20 mM Hepes, pH 7.5, and the release of zinc in the absence and presence of sodium selenite and selenocystamine was measured by spectrophotometry [ɛ500 of Zn(PAR)2 = 65,000 M−1⋅cm−1 (18)].
RESULTS
Reactivity of MT
Zinc Transfer from MT-2 to Apo-AP.
The activity of a reconstituted zinc enzyme is a measure of the extent of metal transfer from MT-2 to the apoenzyme. The phosphotransferase activity of the apo-AP preparation used in these experiments was less than 10% of that of the native enzyme. A 4-fold molar excess of zinc sulfate fully reactivates apo-AP, corresponding to a stoichiometric amount of zinc (four zinc ions, two catalytic and two cocatalytic, per enzyme dimer are required). Zn7MT-2 also reactivates the apoenzyme, but somewhat slower than zinc sulfate (>15 min vs. 5 min). However, maximal activation requires more than a 15-fold molar excess of Zn7MT-2 (Fig. 1). Thus, on average, less than one zinc atom is transferred from Zn7MT-2 to the apoenzyme.
Figure 1.

Concentration dependence of reconstitution of apo-AP with Zn7MT-2 (⧫) and Cd5Zn2MT-2 (▪). Apo-AP, 0.5 μM, was incubated with various concentrations of Zn7MT-2 and Cd5Zn2MT-2 in 10 mM Tris⋅HCl, pH 8.0. Aliquots were taken after 2 hr and assayed spectrophotometrically for enzymatic activity.
The extent of reactivation depends on the type of buffer used and its concentration. High concentrations of Tris (above 100 mM as used conventionally for assay of phosphotransferase activity (9, 19, 20) and millimolar concentrations of citrate affect the extent of reactivation with MT-2 and zinc sulfate markedly. Increasing the concentration of Tris from 10 mM to 1 M decreases the final phosphatase activity by more than 3-fold, demonstrating competition between Tris and the apoenzyme for zinc. These effects clearly account for the observation that only 40% of apo-AP could be reactivated with MT in 1 M Tris⋅HCl, pH 8.0, in previous experiments (9).
Since only about one zinc atom is transferred from MT to the apoenzyme, the question arises from which cluster and from which particular location in the cluster this zinc atom originates. Therefore, Cd5Zn2MT-2 was used as metal donor, because both zinc atoms are located at crystallographically defined positions in the β-domain (2). Reactivation of apo-AP by this MT-2 species, if it occurs, would have to be due to zinc transfer from the β-domain, because cadmium AP, if formed, has only a residual activity of <1% (21). Cd5Zn2MT-2 reactivates apo-AP in a pattern virtually identical to that of Zn7MT-2 (Fig. 1), confirming that zinc is transferred and that it originates in the β-domain, and further indicating that only 1 or 2 zincs are transferred from MT-2. Detailed structural studies of these transfer reactions will be required to determine the precise origins and destinations of the zinc ions involved.
Enhancement of Zinc Transfer in the Presence of Disulfide Reagents.
Disulfides interact with MT-2 and release zinc ions (6). These reagents also would be expected to enhance zinc transfer from MT-2 to apoenzymes, hence reactivation of apo-AP in the presence of two such reagents, DTNB and glutathione disulfide (GSSG), was investigated. DTNB is highly reactive, and consequently it reacts at a much lower excess and orders of magnitude faster than GSSG (6, 22, 23). Micromolar concentrations of DTNB more than triple the reactivation efficiency of Zn7MT-2, whereas millimolar concentrations of GSSG are required to achieve the same effect (Fig. 2).
Figure 2.

Reconstitution of apo-AP with Zn7MT-2 in the presence of oxidizing agents. Apo-AP, 0.5 μM, was incubated with Zn7MT-2, 0.29 μM, and various concentrations of DTNB (⧫) or GSSG (▪) in 10 mM Tris⋅HCl, pH 8.0. Aliquots were taken after 2 hr and assayed spectrophotometrically for enzymatic activity. AP activity of 100% corresponds to reactivated apo-AP in the absence of oxidizing agents under otherwise identical conditions.
Zinc Transfer from MT-2 to Other Systems.
To investigate whether these observations apply more generally to the properties of MT in zinc transfer, two other systems were investigated, another apoenzyme and a system that is based not on measuring zinc transfer by enzymatic assays of zinc enzymes but on detecting transferred zinc directly. For this purpose we selected the chromophoric zinc-chelating agent PAR.
Both zinc sulfate and Zn7MT-2 reconstitute apo-CPA. With the former, activity is completely restored in a few seconds [kf = 7 × 105 M−1⋅s−1 (24)], whereas with the latter it is slower, requiring more than 30 min to reach completion. Activity increases as a function of the amount of MT-2 added and is restored fully by an equimolar amount of Zn7MT-2 (Fig. 3). Since maximum activity is reached at a stoichiometry of approximately 1:1 (apoenzyme:MT-2), on average only one zinc atom of MT-2 is transferred. When the transfer reaction is carried out in the presence of 0.1 mM zincon (2-carboxy-2′-hydroxy-5′-sulfoformazylbenzene), the absorbance remains unchanged (ΔA620 ≤ 0.005), indicating that no “free” Zn(II) is released into the medium and that there is no competition between zincon and the apoenzyme.
Figure 3.

Concentration dependence of reconstitution of apo-CPA with Zn7MT-2. Apo-CPA, 2 μM, was incubated with various concentrations of Zn7MT-2 in 20 mM Hepes/100 mM NaCl, pH 7.5. Measurements were taken after 30 min.
Zinc may also be transferred from MT to metal chelating agents and the transfer is influenced by a great variety of oxidizing agents and cellular agents that can oxidize MT (13). Among these, the effect of selenium compounds on zinc transfer from MT to PAR is particularly significant. Micromolar concentrations of either selenite or selenocystamine release about four zinc atoms per MT molecule in 1 hr (Fig. 4). The rate and extent of this reaction are comparable to the reaction of MT with DTNB.
Figure 4.

Reaction of MT-2 with PAR in the presence of selenite or selenocystamine. MT-2, 0.5 μM, was incubated with PAR, 100 μM, in the absence (•) and presence of 50 μM selenocystamine (▪) or 50 μM sodium selenite (⧫).
Reactivity of T
Zinc Transfer from AP to T.
We have also begun to study the chemical properties of T, the apoform of MT. There have been almost no studies of the apoprotein reported aside from its capacity to bind zinc, owing to its presumed instability. As an important component of the MT system, the reactivity of its cysteine thiols is a critical parameter for our understanding of the reactivity of the zinc–sulfur bonds in MT. We have now found that T is stable if stored at −180°C in liquid nitrogen and that it can be employed for several hours in biochemical studies without significant oxidation of its thiols. This has allowed us to investigate T in reactions in which it acts as a metal acceptor toward a zinc enzyme, and in redox reactions with disulfides.
The activity of AP depends critically on the concentration of buffer ions. In the presence of Tris, citrate, or reduced glutathione (GSH), the catalytic activity of AP decreases over time (Table 1). These agents also influence zinc transfer from AP to T. Addition of T to AP in 10 mM Tris⋅HCl, pH 8.0, only marginally affects the activity of the enzyme, but when the identical experiment is performed in 1 M Tris⋅HCl, pH 8.0, AP activity decreases to below 10% of its original value within approximately 30 min (Fig. 5). The concentration of T at which 50% of the original activity of AP is inhibited (IC50) is about 0.25 μM in 1 M Tris⋅HCl, pH 8.0. This dramatic decrease in enzymatic activity is completely reversible: addition of a 200-fold excess of zinc sulfate fully restores its activity. The loss of enzyme activity is likely due to removal of Zn(II) from the active site. Thus, buffer ions such as Tris, or cellular agents such as citrate and GSH, clearly mediate zinc transfer. In contrast to Tris, the concentrations at which citrate and GSH are effective are below 1 mM—i.e., in the physiological range.
Table 1.
Effect of buffers and chelating agents on the inactivation of AP by T
| Agent added* | [Agent], mM | AP activity, %
|
|
|---|---|---|---|
| In the absence of T | In the presence of T | ||
| None | 0 | 100 | >80 |
| Tris | 500 | 90.8 | 11.5 |
| Citrate | 0.5 | 94 | 40.4 |
| GSH | 0.5 | 94.8 | 77.2 |
AP, 0.5 μM, was incubated with T, 1 μM, in 10 mM Tris·HCl, pH 8.0, in the presence of Tris, sodium citrate, or GSH. Aliquots were taken after 2 hr and assayed spectrophotometrically for enzymatic activity at 20°C.
Figure 5.

Time dependence of the inactivation of AP by T. AP, 0.5 μM, was incubated with T, 1 μM, in 1 M Tris⋅HCl, pH 8.0. Aliquots were taken at defined time intervals and assayed spectrophotometrically for enzymatic activity.
The effects of T on CPA activity are comparable to those on AP activity: First, addition of T to CPA in 1 M Tris⋅HCl, pH 8.0, progressively decreases the enzyme’s activity (data not shown). Second, at substoichiometric amounts of T, the inactivation reaction reaches equilibrium within 30 min (IC50 of 0.4 μM). Third, loss of enzyme activity is also most likely due to removal of Zn(II) from the active site, because addition of a 100-fold excess of zinc sulfate restores 65% of the enzyme’s original activity.
Effect of Zinc on the T/Disulfide Interactions.
MT reacts with disulfides at neutral pH. So does T, which reacts about 30 times faster than MT with DTNB (23). Given this difference, it is unclear how the zinc in MT influences the reactivity of thiols in this reaction. Therefore, we have determined the reactivity of T toward DTNB as a function of increasing concentration of zinc. Remarkably, there is a linear decrease of the extent of DTNB reduction to 10% of its original value until four equivalents of zinc are added to T (Fig. 6). The remaining reactivity upon adding more zinc corresponds to that of MT at this particular time interval.
Figure 6.

Effect of Zn(II) on the reaction of T with DTNB. T, 0.5 μM, was incubated with various concentrations of zinc sulfate in 20 mM Hepes, pH 7.5, for 30 s, and DTNB was added to a final concentration of 50 μM. Relative reactivity of thiols was determined from measurements taken after 30 s.
DISCUSSION
MT as Zinc Donor.
Several important new aspects of zinc transfer emerge from these studies of the reconstitution of the apoforms of zinc enzymes by Zn7MT-2. First of all, it should be emphasized that these reactions are thermodynamically unfavorable. The zinc binding constant of T [stability constant of Zn7MT-2 = 3.2 × 1013 M−1 at pH 7.4 (4)] and those of the apoproteins studied [stability constant of AP = 107 to 108 for both pairs of Zn(II) at pH 6.5 (25) and of CPA = 2.1 × 108 M−1 at pH 8.0 (26)] predict that zinc transfer in this direction should not be significant (reaction 2).
 |
2 |
where P is any apoprotein. However, reaction 2 is based on two assumptions. One is that MT has a metal transfer potential of seven based on cooperative binding of seven zinc ions all with similar binding constants. The other is that the products do not undergo further reactions and thereby shift the equilibrium to the right.
To date, most reports have accepted that zinc binds cooperatively and that only T and Zn7MT-2 exist, without stable intermediates (27). That being so, zinc transfer from MT-2 to apoenzymes would proceed with the dissociation of all zinc atoms from MT-2 (reaction 3).
 |
3 |
Neither of these reactions seems to occur in the manner indicated. Thus, the stability constant of Zn7MT-2 is too high to account for zinc transfer from a thermodynamically relatively stable system (MT-2) to others that are orders of magnitude more labile (CPA, AP) under stoichiometric concentrations of the reactants. If the binding constants were the same for all seven zinc atoms in MT-2, only a negligible fraction of zinc should be transferred. Indeed, without a driving force, the occurrence of zinc transfer would be a surprising result. The observed metal transfer potential of MT-2 is approximately one in the case of CPA and perhaps less than that in the case of AP. Because addition of zincon to this reaction did not reveal any increase in the pool of free zinc, transfer does not induce further dissociation of the zinc as postulated by reaction 3. Therefore formation of T during the transfer reaction can be ruled out. Yet, six of the seven zinc ions in the system have to be accounted for. Quite possibly, they initially remain bound as a Zn6MT-2 species. At present, the precise structure of this species is unknown. Rearrangement of Zn6MT-2, perhaps with the formation of two Zn3 clusters, might occur because the coordination of MT is quite flexible as demonstrated for copper MT, where an MT species containing one Cu6S9 and one Cu6S11 unit has been reported (28). MT exhibits high structural flexibility, and cadmium exchange inside the β-cluster and between the β-clusters of two MT molecules is fast (29). A second possibility is that the β-cluster is stabilized through extensive delocalization of zinc, as is known for cadmium in a Cd6MT species obtained after treating Cd7MT-2 with EDTA (30). In the absence of any enhancers (see below), such types of rearrangements indeed might be not only the rate-determining step in the zinc transfer reaction—the simple uptake of zinc ions by both apoenzymes studied is faster than the transfer reaction—but also the driving force that brings about this thermodynamically unfavorable process. Removing Zn6MT-2 from the equilibrium with Zn7MT-2 would indeed explain why zinc can be released from the latter at all (reaction 4). Thiol oxidation might be yet another mechanism facilitating this process because this would decrease ligand strength. In particular, we have found that the uncoordinated thiols in T react much faster with oxidizing agents such as DTNB than do the metal-bound thiols in MT-2 and that this reactivity is modulated by zinc (Fig. 6).
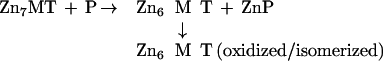 |
4 |
The transfer of a single zinc ion from Zn7MT-2 raises the question of why MT-2 complexes seven zinc ions but transfers only one. Since Zn(II) binding to apo-CPA (24) or apo-AP (9) is either comparable to or faster than zinc transfer from MT-2 to these apoenzymes, processes involved in the release of ionic zinc from MT-2 seem to contain the rate-determining step in the transfer reaction. On a physiological time scale nonenhanced transfer appears rather slow, hence it seems likely that the rate and extent of zinc release from MT-2 should be enhanced in vivo. The unique structure of MT offers several possibilities of how such enhancement could be achieved in vitro.
One way to release zinc ions from Zn7MT-2 would be by displacing them with different metal ions that can form tighter complexes with MT-2. Interestingly, zinc is orders of magnitude less tightly bound to T than, for example, Cd(II) or Cu(I) (31). Cd(II) or Cu(I) replaces zinc in MT-2 within seconds. Therefore, metal replacement reactions could constitute a means by which more than one zinc atom from Zn7MT-2 is made available to activate apoproteins. Owing to the unusually high binding constant of Cd7MT-2 in coordination sites that contain thiolate sulfur [KCd = 3.2 × 1017 M−1 at pH 7.4 (29)] Cd(II) will preferentially replace Zn(II) in MT-2 instead of binding to nitrogen and oxygen ligands in enzyme sites. The release of one zinc ion by metal exchange requires one cadmium or cuprous ion. Because in most organisms both cadmium and copper are far less abundant than zinc the likely physiological importance of this metal exchange observed in vitro remains obscure.
Thiol modification reactions are particularly attractive as a means to release zinc because they affect the zinc ligands of MT (5, 6) and are biochemically feasible. Such reactions, possibly involving cellular GSSG, would not only facilitate release but also increase the number of zinc ions transferred per MT molecule (with the thiolation reaction as the rate-determining step) in vivo. From a range of possible transfer-enhancing reagents two thiol/disulfide-exchange reagents, DTNB and GSSG, were investigated here as examples of disulfides with greatly differing reactivities. DTNB is a highly reactive thiolation agent. It rapidly thiolates the sulfhydryl groups of MT-2, with concomitant release of ionic zinc. Moreover, it significantly increases the number of zinc ions transferred per MT-2 molecule. The reactivation of apo-CPA or -AP in the presence of micromolar concentrations of DTNB is more than 3 times as great as in its absence, indicating that only three to four, but not all seven, zinc ions become available for transfer. GSSG has similar effects, although it requires millimolar concentrations to achieve the same enhancement. Obviously the lower reactivity of GSSG toward MT-2 (6) as compared with DTNB (22) affects the rate of zinc release and consequently the rate of zinc uptake of the apoenzyme on the time scale of the experiment. In the case of CPA, DTNB also increases the extent of zinc transfer, whereas no clear effect was seen with GSSG up to 1 mM. In addition, we have now identified selenium compounds as possible agents that increase the rate of zinc transfer from MT to an acceptor at micromolar concentrations of reagents. Selenium is an essential trace element that is a biologically active component of several proteins. Our experiments indicate that zinc transfer reactions may well be a possible target for the molecular action of selenium.
T as Zinc Acceptor.
In the absence of a driving force for the forward or backward reaction, the equilibrium of removal of zinc from a zinc protein by T (reaction 5) should be controlled thermodynamically. Because for most proteins the binding constant for zinc is at least 1,000 fold lower than that for T, zinc transfer from zinc enzymes to T should be preferred thermodynamically.
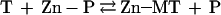 |
5 |
In the case of AP, however, despite expectations based on the relative stability constants of MT and AP, T does not appear to sequester significant amounts of zinc from the active site of the enzyme. Thus, T does not necessarily act as a strong chelating agent toward enzymes. If this were so, then the induction of T in the cell might interfere with many zinc-dependent enzymatic processes. However, T does inactivate AP in the presence of buffer ions such as Tris or citrate. One possible explanation is that the buffer ions help deplete the enzyme of its zinc, and thereby act to shuttle zinc between the enzyme and T. The fact that T is ineffective in 10 mM Tris strongly suggests that there is no direct interaction of T and the zinc enzyme. Nor is a specific inhibitory effect of T very plausible, because T acts in substoichiometric amounts and also inactivates CPA, mitigating against a specific AP–T interaction. In effect, Tris and citrate lower the stability constant of AP. Tris is not only a buffer ion but also a cosubstrate for AP (20, 21) and a chelating agent. Zinc-chelating agents such as citrate and GSH occur in the cytosol in millimolar concentrations (32–34) and could serve such a purpose (reaction 6). These agents do not release zinc from MT, presumably because they cannot gain access to the zinc ions in the clusters. They are important determinants for the direction of zinc transfer. Under the conditions investigated here they favor the unidirectional transfer of zinc from AP to T.
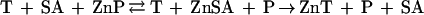 |
6 |
where SA is shuttle agent.
The concentrations of both citrate and GSH can vary over a relatively wide range in the cell (32–34). It is, therefore, conceivable that such changes of concentrations affect the direction of zinc transfer in vivo and could render T a specific chelating agent under yet to be defined circumstances. Moreover, T is a very strong reductant. It reacts significantly faster with disulfides than does MT. The reactivity of T toward disulfides and other oxidizing agents is efficiently quenched in the presence of zinc (Fig. 6). Whether such reducing power of T is needed in the cell and whether such reducing power—or as a matter of fact the reductive functions of any cellular thiols—is controlled by binding of zinc are currently unknown.
Analytical methods for the direct determination of T in biological material do not exist. Therefore, neither the concentrations nor the turnover of this species is known. However, it has been proposed that T exists in a large number of tumors (35).
Implications.
Our results suggest that zinc transfer between zinc enzymes and MT should not be considered a simple two-component system as earlier interpretations suggested (9). We have already found two types of agents that modulate the release of zinc from its thermodynamically tight binding sites in the clusters of MT (oxidizing agents) and that carry zinc between MT and a participating protein (zinc-chelating agents). We have shown that GSSG releases zinc, but we also find that certain buffers, such as Tris and citrate, chelate metals, including zinc; under certain circumstances they form complexes that significantly affect the state of zinc and have a marked influence on metal transfer. In fact, they might become the cellular factors that form intermediates that could enhance or control transfer. As such they could participate in the conveyance of zinc from MT to other proteins and in the mechanism of transfer. Zinc is always associated with another cellular ligand and is not transferred as the free zinc ion but in the form of a complex with any one or several of these agents that together constitute a distribution system. Clearly, the number and identities of such agents are unknown at this point, as are the details and extent to which they would be sensitive to the oxidative and reductive state of the environment for properties of MT as a zinc donor or zinc acceptor. The nature and identity of the shuttle and distribution system are currently under investigation.
Acknowledgments
We thank Drs. J. F. Riordan and G.-F. Hu for helpful discussions. This work was supported by the Endowment for Research in Human Biology, Inc. C.J. was supported in part by a fellowship from the Alexander von Humboldt-Stiftung.
ABBREVIATIONS
- T
thionein
- MT
metallothionein
- MT-2
metallothionein isoform 2
- CPA
carboxypeptidase A
- AP
alkaline phosphatase
- HQSA
8-hydroxyquinoline-5-sulfonic acid
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- GSH
reduced glutathione
- GSSG
glutathione disulfide
- PAR
4-(2-pyridylazo)resorcinol
- EDTA
ethylenediaminetetraacetic acid
- zincon
2-carboxy-2′-hydroxy-5′-sulfoformazylbenzene
- Tris
2-amino-2-hydroxymethyl-1,3-propanediol
- Hepes
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
Footnotes
A commentary on this article begins on page 3333.
References
- 1.Margoshes M, Vallee B L. J Am Chem Soc. 1957;79:4813. [Google Scholar]
- 2.Robbins A H, McRee D E, Williamson M, Collett S A, Xuong N H, Furey W F, Wang B C, Stout C D. J Mol Biol. 1991;221:1269–1293. [PubMed] [Google Scholar]
- 3.Arseniev A, Schultze P, Wörgötter E, Braun W, Wagner G, Vašák M, Kägi J H R, Wüthrich K. J Mol Biol. 1988;201:637–657. doi: 10.1016/0022-2836(88)90644-4. [DOI] [PubMed] [Google Scholar]
- 4.Otvos J D, Petering D H, Shaw C F. Comments Inorg Chem. 1989;1:1–35. [Google Scholar]
- 5.Fliss H, Ménard M. Arch Biochem Biophys. 1992;293:195–199. doi: 10.1016/0003-9861(92)90384-9. [DOI] [PubMed] [Google Scholar]
- 6.Maret W. Proc Natl Acad Sci USA. 1994;91:237–241. doi: 10.1073/pnas.91.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng J, Heuchel R, Schaffner W, Kägi J H R. FEBS Lett. 1991;279:310–312. doi: 10.1016/0014-5793(91)80175-3. [DOI] [PubMed] [Google Scholar]
- 8.Zeng J, Vallee B L, Kägi J H R. Proc Natl Acad Sci USA. 1991;88:9984–9988. doi: 10.1073/pnas.88.22.9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Udom A O, Brady F O. Biochem J. 1980;187:329–335. doi: 10.1042/bj1870329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson R T, Vallee B L. Biochemistry. 1968;7:4343–4349. doi: 10.1021/bi00852a029. [DOI] [PubMed] [Google Scholar]
- 11.Coleman J E, Vallee B L. J Biol Chem. 1961;236:2244–2249. [PubMed] [Google Scholar]
- 12.Bosron W F, Kennedy F S, Vallee B L. Biochemistry. 1975;14:2275–2282. doi: 10.1021/bi00681a036. [DOI] [PubMed] [Google Scholar]
- 13.Maret W, Vallee B L. Proc Natl Acad Sci USA. 1998;95:3478–3482. doi: 10.1073/pnas.95.7.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vašák M. Methods Enzymol. 1991;205:41–44. doi: 10.1016/0076-6879(91)05082-7. [DOI] [PubMed] [Google Scholar]
- 15.Cueni L B, Bazzone T J, Riordan J F, Vallee B L. Anal Biochem. 1980;107:341–349. doi: 10.1016/0003-2697(80)90394-2. [DOI] [PubMed] [Google Scholar]
- 16.Auld D S. Methods Enzymol. 1988;158:71–79. doi: 10.1016/0076-6879(88)58048-5. [DOI] [PubMed] [Google Scholar]
- 17.Ng M, Auld D S. Anal Biochem. 1989;183:50–56. doi: 10.1016/0003-2697(89)90170-x. [DOI] [PubMed] [Google Scholar]
- 18.Shaw C F, III, Laib E, Savas M M, Petering D H. Inorg Chem. 1990;29:403–408. [Google Scholar]
- 19.Simpson R T, Vallee B L, Tait G H. Biochemistry. 1968;7:4336–4342. doi: 10.1021/bi00852a028. [DOI] [PubMed] [Google Scholar]
- 20.Sone M, Kishigami S, Yoshihisa T, Ito K. J Biol Chem. 1997;272:6174–6178. doi: 10.1074/jbc.272.10.6174. [DOI] [PubMed] [Google Scholar]
- 21.Coleman J E, Gettins P. Adv Enzymol Relat Areas Mol Biol. 1983;55:381–480. doi: 10.1002/9780470123010.ch5. [DOI] [PubMed] [Google Scholar]
- 22.Li T-Y, Minkel D T, Shaw C F, III, Petering D H. Biochem J. 1981;193:441–446. doi: 10.1042/bj1930441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savas M M, Shaw C F, III, Petering D H. J Inorg Biochem. 1993;52:235–249. doi: 10.1016/0162-0134(93)80028-8. [DOI] [PubMed] [Google Scholar]
- 24.Billo E J, Brito K K, Wilkins R G. Bioinorg Chem. 1978;8:461–475. doi: 10.1016/0006-3061(78)80001-5. [DOI] [PubMed] [Google Scholar]
- 25.Coleman J E, Nakamura K, Chlebowski J F. J Biol Chem. 1983;258:386–395. [PubMed] [Google Scholar]
- 26.Coleman J E, Vallee B L. J Biol Chem. 1960;235:390–395. [PubMed] [Google Scholar]
- 27.Nielson K B, Winge D R. J Biol Chem. 1983;258:13063–13069. [PubMed] [Google Scholar]
- 28.Presta A, Green A R, Zelazowski A, Stillman M J. Eur J Biochem. 1995;227:226–240. doi: 10.1111/j.1432-1033.1995.tb20380.x. [DOI] [PubMed] [Google Scholar]
- 29.Otvos J D, Liu X, Li H, Shen G, Basti M. In: Metallothionein III. Suzuki K T, Imura N, Kimura M, editors. Basel: Birkhäuser; 1993. pp. 57–74. [Google Scholar]
- 30.Vazquez F, Vašák M. Biochem J. 1988;253:611–614. doi: 10.1042/bj2530611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kägi J H R. In: Metallothionein III. Suzuki K T, Imura N, Kimura M, editors. Basel: Birkhäuser; 1993. pp. 29–55. [Google Scholar]
- 32.Meister A. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 33.Westergaard N, Banke T, Wahl P, Sonnewald U, Schousboe A. Proc Natl Acad Sci USA. 1995;92:3367–3370. doi: 10.1073/pnas.92.8.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dulhunty A F. Biophys J. 1988;53:609–616. doi: 10.1016/S0006-3495(88)83139-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pattanaik A, Shaw C F, III, Petering D H, Garvey J, Kraker A J. J Inorg Biochem. 1994;54:91–105. doi: 10.1016/0162-0134(94)80023-5. [DOI] [PubMed] [Google Scholar]