

Abstract
A convenient two-step, one-pot procedure was developed for the conversion of primary alcohols to carboxylic acids. The alcohol was first treated with NaOCl and TEMPO under phase-transfer conditions, followed by NaClO2 oxidation in one pot. This reaction is applicable to a wide range of alcohols and the mild reaction conditions are compatible with many sensitive functional groups, including electron-rich aromatic rings, acid-labile isopropylidene ketal and glycosidic linkages, and oxidation-prone thioacetal, p-methoxybenzyl, and allyl moieties. Several glycosaminoglycans such as heparin, chondroitin, and hyaluronic acid oligosaccharides have been synthesized in high yields by using this new oxidation protocol.
Keywords: carbohydrates, glycosaminoglycan, oxidation, synthetic methods
Introduction
Oxidation of primary alcohols to carboxylic acids is a fundamental transformation in organic synthesis, albeit with relatively few good general methods available.[1] This is particular the case for assembly of complex oligosaccharides such as glycosaminoglycans (GAGs). The existence of a large number of protective groups for selective GAG functionalization in addition to the acid lability of glycosidic linkages, severely limits available methodologies due to cross-reactivity. Furthermore, in GAG synthesis, the need to simultaneously convert multiple primary alcohols to carboxylic acids demands a robust and high-yielding method. Herein, we report a new convenient two-step, one-pot protocol for oxidizing primary alcohols to carboxylic acids and its application.
GAGs are a family of highly functionalized, linear and negatively charged oligosaccharides, consisting of repeating disaccharide units of a 2-deoxy-2-amino hexose linked to a pyranosyl uronic acid.[2] Depending upon the identity of the amino sugars and uronic acids, the GAG family can be divided to hyaluronic acid, heparin/heparan sulfate, chondroitin/chondroitin sulfate, and dermatan. GAGs play diverse and critical roles in many important biological processes such as lymphocyte trafficking, inflammatory response, wound healing, and tumor metastasis.[3] Biomedical applications of GAGs in areas such as antiviral, anti-angiogenesis, and anticoagulation are enormous, exemplified by the development of Arixtra, a fully synthetic heparin pentasaccharide drug, for the treatment of deep vein thrombosis.[4]
With the recognition of their biological importance, chemical syntheses of GAGs are undergoing extensive studies;[2,5] such syntheses are typically carried out following two general approaches. In the first method, protected pyranosyl uronic acids are directly utilized. However, with the strongly electron-withdrawing 6-carboxyl moiety, these building blocks often have low reactivities both as donors and as acceptors, resulting in low glycosylation yields. Furthermore, base sensitivity of these compounds conferred by the carboxyl group complicates protective group manipulations.[6,7] These limitations also preclude the direct usage of uronic acids in solid-phase oligosaccharide syntheses.[8] An attractive alternative is to use pyranosides as building blocks, which can give high glycosylation yields. With this strategy, one of the key challenges is the need to convert primary hydroxyl groups at C-6 positions into carboxylic acids post glyco-assembly. The traditional pyridinium dichromate (PDC) mediated oxidation,[9–11] was found to be inefficient; it often required large excess of PDC subsequently resulting in separation difficulties and low yields.[6,9] Two-step protocols, such as Swern oxidation followed by treatment of NaClO2 or PDC,[7,12] not only are inconvenient, but also can produce elimination side products due to the strongly basic condition utilized.[6,7] Besides the undesirable usage of toxic chromium(VI) agents, Jones oxidation[13] and the combination of chromium trioxide and periodic acid[6] are less useful in oligosaccharide synthesis with the employment of strong acids. Recently, TEMPO (2,2,6,6-tetramethylpiperidinyl-1-oxy)-catalyzed oxidations with a co-oxidant (e.g. NaOCl[14–16] and bis(acetoxy)iodobenzene (BAIB) [17,18]) have become a popular choice. However, substantial side reactions with thioacetal,[13] allyl[9] and electron-rich aromatic rings such as methoxybenzyl[19] moieties have been reported. Moreover, reduced oxidation efficiencies were observed with large oligosaccharides.[15]
Results and Discussion
During our study of GAG synthesis, we were faced with the task of oxidation state adjustment of hexasaccharide 1. Despite prolonged reaction time and repeated trials, the reaction of 1 with TEMPO/NaOCl[14–16] led to multiple partially oxidized products with a trace amount of the desired tricarboxylic acid 2. Attempts with TEMPO/BAIB[18] or NaClO2 catalyzed by NaOCl/TEMPO[19] met with similar fate. A two-step process of Dess–Martin oxidation followed by NaClO2[20] gave inconsistent results. Finally, we discovered that a convenient two-step, one-pot protocol with TEMPO/NaOCl followed by treatment of NaClO2 afforded the desired carboxylic acid in high yield and good purity; this result led us to further explore the scope of this method.
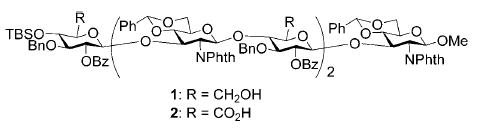
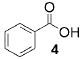
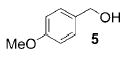
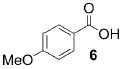
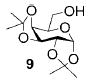
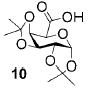
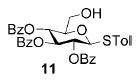
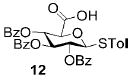
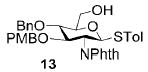
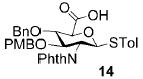
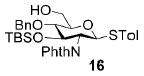
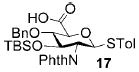
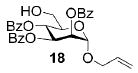
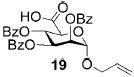
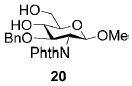
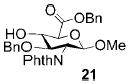
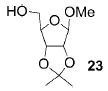
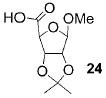
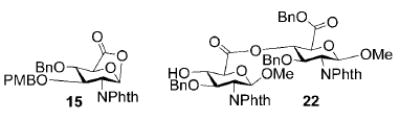
A panel of primary alcohols and monosaccharides (Table 1) were examined first to test the functional group compatibility. Simple benzylic alcohols (Table 1, entries 1,2) including the electron-rich p-methoxy benzyl alcohol (5; Table 1, entry 2) can be oxidized in high yields without chlorinating the aromatic rings. Aliphatic alcohol 1-decanol (7; Table 1, entry 3) was converted in 90% yield to decanoic acid (8). Pyranosyl uronic acids such as glucoronic acid, galactonic acid, and mannosinic acid often exist in oligosaccharides and selectively protected uronic acids are useful for carbohydrate synthesis.[11,18] Conversion of the free primary hydroxyl groups in galactoside 9, glucoside 11,[21] 2-deoxy-2-amino-glucoside derivatives 13 and 16, and mannoside 18[22] to the corresponding uronic acids proceeded smoothly in 82–92% yields (Table 1, entries 4–8). Acid-sensitive isopropylidene and p-methoxybenzyl (PMB) groups were stable under reaction conditions (Table 1, entries 4 and 6). Thioglycosides have been extensively used in oligosaccharide assembly.[18,23] The presence of thioacetal in thioglycosides precludes the usage of noble-metal oxidation conditions, such as PtO2 and O2.[13] Previous attempts of TEMPO/NaOCl oxidation of thioglycosides generated a mixture of sulfoxides and sulfones in preference to alcohol oxidation.[13] By using our reaction protocol, both disarmed (compound 11) and armed (compounds 13 and 16) thioglycosides were oxidized in 82, 85, and 86% yields respectively (Table 1, entries 5–7). No glycosyl sulfoxides or sulfones were identified from these reactions, with trace amount (~6%) of lactone 15 isolated from oxidation of thioglycoside 13. The allyl group, which is a popular linker for the bioconjugation of carbohydrates with proteins,[24] remained intact following oxidation of mannosyl pyranoside 18[22] (Table 1, entry 8). A primary alcohol can be selectively converted to a carboxylic acid in the presence of a free secondary hydroxyl group as 21 was obtained in 75% yield following oxidation of diol 20[25] and benzyl ester formation[26] with phenyl diazomethane[27] (Table 1, entry 9). A small amount (~10%) of ester 22 was also isolated from the reaction mixture. In addition to pyranosides, furanoside 23[28] was successfully transformed into carboxylic acid 24 in quantitative yield (Table 1, entry 10).
Table 1.
Oxidation of primary alcohols and monosaccharides.
| Alcohol | Carboxylic acids/esters | Yield [%] | |
|---|---|---|---|
| 1 |
|
|
100 |
| 2 |
|
|
95 |
| 3 |
|
|
90 |
| 4 |
|
|
90 |
| 5 |
|
|
82 |
| 6 |
|
|
85 |
| 7 |
|
|
86 |
| 8 |
|
|
92 |
| 9 |
|
|
75[a] |
| 10 |
|
|
100 |
Ester 22 (10%) was isolated from the reaction mixture.
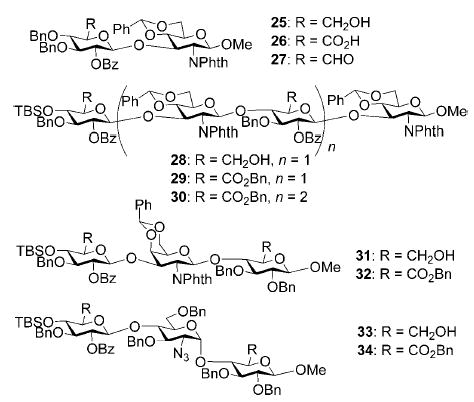
Next we examined the application of this new protocol in GAG syntheses. A hyaluronic acid disaccharide 26 was obtained in 95% yield by oxidizing disaccharide 25[29] (Table 2, entry 1). The oxidation was highly efficient even for large oligosaccharides, converting the tetrasaccharide 28[29] and hexasaccharide 1[29] to oligocarboxylic acids, which were subsequently benzylated[26] with phenyl diazomethane[27] to produce hyaluronic acid tetrasaccharide diester 29 and hexasaccharide triester 30 in 86 and 82% overall yields, respectively (Table 2, entries 2 and 3). Trisaccharide 31[29] was also oxidized and benzylated to produce chondroitin trisaccharide 32 in 76% yield (Table 2, entry 4). In addition, a heparin trisaccharide 34 was obtained in a similar manner in 81% yield from diol 33[29] (Table 2, entry 5). Glycosidic linkages were not affected during oxidation and no epimerization or elimination products were identified with these sensitive substrates. The successful introduction of carboxyl groups after glyco-assembly makes this alternative strategy for GAG synthesis highly attractive.
Table 2.
Oxidation of primary alcohols for GAG synthesis.
| Alcohol | GAG derivative | Yield [%] | |
|---|---|---|---|
| 1 | 25 | 26 | 95 |
| 2 | 28 | 29 | 86 |
| 3 | 1 | 30 | 82 |
| 4 | 31 | 32 | 76 |
| 5 | 33 | 34 | 81 |
The fact that the previously reported TEMPO/NaOCl procedure[14–16] failed to produce desired carboxylic acids is most likely due to hydrophobicities of our substrates. It has been proposed that aldehydes are intermediates in TEMPO-catalyzed NaOCl oxidation of primary alcohols to carboxylic acids.[14] With a hydrophilic aldehyde, the carbonyl group can be hydrated to form a vicinal diol under phase-transfer conditions, which is subsequently oxidized by TEMPO/NaOCl to produce a carboxylic acid (Scheme 1). However, due to the hydrophobic nature of fully protected oligosaccharides, hydration of newly-formed aldehydes is presumably slow even under phase-transfer conditions. The direct conversion of an aldehyde to the carboxylic acid without going through vicinal diol was apparently difficult with TEMPO/NaOCl and prolonged treatment did not yield the desired carboxylic acid. The major product isolated from TEMPO/NaOCl oxidation of disaccharide 25 under phase-transfer conditions was aldehyde 27. Attempt to enhance aldehyde hydration by performing TEMPO/NaOCl oxidation in an acetonitrile and water mixture did not alleviate the problem. In contrast, the aldehyde was quickly converted to the carboxylic acid with addition of NaClO2 to the reaction mixture following TEMPO/NaOCl oxidation. A one-step procedure that involved the used of a combination of TEMPO, NaOCl, and NaClO2 together was not successful, probably due to the instability of NaClO2/NaOCl mixture.[19]
Scheme 1.
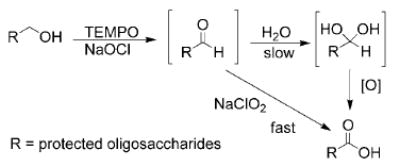
Proposed intermediates for our new oxidation method.
The excellent yields and extraordinary functional group compatibility achieved by using our oxidation procedure are related with the relatively high loading of TEMPO employed (0.3 equiv per hydroxyl group), as slower reaction was observed with less TEMPO. Furthermore, the two-phase condition (methylene chloride and water) for the TEMPO/NaOCl oxidation is crucial. When oxidation of thioglycoside 13 was carried out in a homogeneous acetonitrile/water mixture with TEMPO/NaOCl, multiple side products without the thiotolyl moieties were obtained with no desired carboxylic acid 14 or the corresponding aldehyde. This indicates that the single-phase homogenous condition for TEMPO/NaOCl oxidation is less selective and can affect sensitive functional groups.
The formation of side product ester 22 in oxidation of diol 20 (Table 1, entry 9) can be explained with the intermediacy of aldehyde as well. Upon generation of aldehyde 35 from 20, intermolecular nucleophilic attack of the carbonyl group by the free secondary hydroxyl group produced hemiacetal 36, oxidation and benzylation of which led to carboxylic ester 22 (Scheme 2).
Scheme 2.
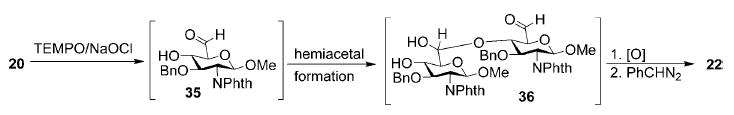
Proposed mechanism for the formation of ester 22.
Conclusion
In summary, we have established a new two-step, one-pot oxidation protocol, which efficiently converts a wide range of primary alcohols to carboxylic acids. This convenient procedure does not require an inert atmosphere, anhydrous solvents, or an extremely low reaction temperature. It does not generate toxic heavy-metal or malodorous side products; this fact is important with respect to increasing environmental awareness. A remarkably wide range of sensitive functional groups, such as electron-rich aromatic rings, acid-labile isopropylidene ketal and glycosidic linkages, and oxidation-prone thioacetal, PMB, and allyl moieties are little affected by the oxidation. Several GAGs, including hyaluronic acid, chondroitin, and heparin oligosaccharides were prepared in high yields by using this procedure. Further applications of this new protocol in solution and solid-phase syntheses of complex oligosaccharides are ongoing.
Experimental Section
General conditions
Chemicals used were reagent grade as supplied except where noted. Analytical thin-layer chromatography was performed using silica gel 60 F254 glass plates (EM Science); compound spots were visualized by UV light (254 nm) and/or by staining with a solution containing Ce(NH4)2(NO3)6 (0.5 g) and (NH4)6Mo7O24·4H2O (24.0 g) in 6% H2SO4 (500 mL) or a solution of KMnO4 (3 g), K2CO3 (20 g) and NaOH (0.25 g) in water (300 mL). Flash column chromatography was performed on silica gel 60 (230–400 Mesh, EM Science). 1H NMR and 13C NMR spectra were recorded on a Varian VXRS-400 or Inova-600 instrument and were referenced to Me4Si (0 ppm), residual CHCl3 (1H NMR δ = 7.26 ppm) CDCl3 (13C NMR δ = 77.0 ppm), residual CH2Cl2 (1H NMR δ = 5.32 ppm), and CD2Cl2 (13C NMR δ = 54.0 ppm). ESI mass spectra were recorded on an ESQUIRE LC-MS operated in both positive and negative ion mode. High-resolution mass spectra were recorded on a Micromass electrospray Tof™ II (Micromass, Wythenshawe, UK) mass spectrometer equipped with an orthogonal electrospray source (Z-spray) operated in positive ion mode, which is located at the Mass Spectrometry and Proteomics Facility at the Ohio State University.
General procedure for oxidation
An aqueous solution of NaBr (1M, 25 μL), an aqueous solution of tetrabutylammonium bromide (1M, 50 μL), TEMPO (2.2 mg, 0.014 mmol, 0.3 equiv per hydroxyl group) and a saturated aqueous solution of NaHCO3 (125 μL) were added to a solution of alcohol (0.045 mmol) in CH2Cl2 (1 mL) and H2O (170 μL) in an ice–water bath. The resulted mixture was treated with an aqueous solution of NaOCl (150 μL, chlorine content not less than 4%) and continuously stirred for 1 hour as the temperature increased from 0°C to RT. The reaction media was neutralized with HCl (1N, about 50 μL) to pH 6–7. It is important to keep the acidity of the reaction close to neutral as lower pH resulted in formation of large amount of hemiacetal side product in oxidation of diol 20. After neutralization, tBuOH (0.7 mL), 2-methylbut-2-ene in THF (2M, 1.4 mL)[30] and a solution of NaClO2 (50 mg, 0.44 mM) and NaH2PO4 (40 mg, 0.34 mM) in water (200 μL) were added. The reaction mixture was kept at room temperature for 1–2 h, diluted with saturated aqueous NaH2PO4 solution (5 mL), and extracted with EtOAc (3010 mL). The organic layers were combined and dried over MgSO4. After removal of the solvent, the desired compound was purified by flash column chromatography.
General procedure for benzyl ester formation
The crude product from the oxidation reaction was dissolved in dichloromethane (5 mL) and treated with phenyl diazomethane solution in diethyl ether (~2 equiv per acid)[27] for 2–3 h until the disappearance of all starting material as judged by TLC. The residue after evaporation was purified by flash column chromatography to provide the benzyl ester.
General procedure for regioselective opening of 4,6-O-benzylidene
[31]Bu2BOTf in dichloromethane (1 M, 1 equiv) was added to a solution of 4,6-O-benzylidene containing glycoside in borane in THF (1M, 10 equiv) in a flame dried flask at 0°C under N2. The reaction mixture was stirred for 3 h. Triethylamine (~0.5 mL) was added followed by careful addition of methanol until the evolution of gas had ceased. All solvents were evaporated and the desired product was isolated by flash column chromatography.
Benzoic acid (4)
Compound 4 was obtained from benzyl alcohol (0.100 g, 0.92 mmol) following general oxidation procedure in 100% yield. Comparison of the 1H and 13C NMR data with the Aldrich NMR library confirmed the identity of benzoic acid 4. 1H NMR (600 MHz, CDCl3): δ = 7.45–7.49 (m, 1H), 7.58–7.62 (m, 2H), 8.09–8.13 ppm (m, 2H); 13C NMR (100 MHz, CDCl3): δ = 128.75, 129.54, 130.47, 134.09, 172.75 ppm; ESI-MS: m/z calcd for C7H6NaO2 [M+Na]+: 145.1; found: 145.0.
p-Anisic acid (6)
Compound 6 was obtained from 4-methoxy benzyl alcohol (0.100 g, 0.72 mmol) following general oxidation procedure in 95% yield. Comparison of the 1H and 13C NMR data with the Aldrich NMR library confirmed the identity of compound 6. 1H NMR (600 MHz, CDCl3): δ = 3.87 (s, 3H), 6.92–6.96 (m, 2H), 8.03–8.07 ppm (m, 2H); 13C NMR (100 MHz, CDCl3): δ = 55.73, 113.97, 121.85, 132.59, 164.27, 171.90 ppm; ESI-MS: cm/z calcd for C8H8NaO3 [M+Na]+: 175.2; found: 175.4.
Decanoic acid (8)
Compound 8 was obtained from 1-decyl alcohol (0.100 g, 0.63 mmol) following general oxidation procedure in 90% yield. Comparison of the 1H and 13C NMR data with the Aldrich NMR library confirmed the identity of compound 8. 1H NMR (400 Hz, CDCl3): δ = 0.88 (t, 3J(H,H) = 6.8 Hz, 3H), 1.24–1.32 (m, 12H), 1.61–1.66 (m, 2H), 2.35 ppm (t, 3J(H,H) = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 14.33, 22.89, 24.92, 29.29, 29.49, 29.62, 32.09, 34.37, 180.52 ppm; ESI-MS: m/z calcd for C10H20NaO2 [M+Na]+: 195.3; found: 195.5.
1,2,3,4-Di-O-isopropylidene-α-D-galacturonic acid (10)
Compound 10 was obtained from 1,2,3,4-di-O-isopropylidene-α-D-galactopyranose (0.100 g, 0.38 mmol) following the general oxidation procedure in 90% yield. Comparison of the 1H NMR data with literature[32] confirmed the identity of compound 10. 1H NMR (600 MHz, CDCl3): δ = 1.33 (s, 6H), 1.44 (s, 3H), 1.52 (s, 3H), 4.39 (dd, 3J(H,H) = 3.0, 4.2 Hz, 1H), 4.45 (d, 3J(H,H) = 1.8 Hz, 1H), 4.61–4.68 (m, 3H), 5.63 ppm (d, 3J(H,H) = 4.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 24.65, 25.00, 26.06, 26.21, 68.43, 70.65, 70.77, 71.89, 96.59, 109.60, 110.38, 172.01 ppm; ESI-MS: m/z calcd for C12H18NaO7 [M+Na]+: 297.3; found: 297.1.
p-Tolyl 2,3,4-tri-O-benzoyl-1-thio-β-D-glucopyranosyluronic acid (12)
Compound 12 was synthesized from compound 11[21] (50 mg, 84 μmol) according to the general oxidation procedure in 82% yield. 1H NMR (400 MHz, CDCl3): δ = 2.33 (s, 3H; p-MePh), 4.34 (d, 3J(H,H) = 9.6 Hz, 1H), 5.01 (d, 3J(H,H) = 9.6 Hz, 1H), 5.48 (t, 3J(H,H) = 9.6 Hz, 1H), 5.66 (t, 3J(H,H) = 9.6 Hz, 1H), 5.90 (t, 3J(H,H) = 9.6 Hz, 1H), 7.11–7.98 ppm (m, 19H); 13C NMR (100 MHz, CDCl3): δ = 21.47, 70.11, 70.32, 73.74, 76.35, 86.87, 127.48–139.30 (aromatic carbon atoms), 165.15, 165.71, 165.91, 170.65 ppm; ESI-MS: m/z calcd for C34H27O9S [M−1]−: 611.2; found: 611.3; HRMS: m/z calcd for C34H28NaO9S [M+Na]+: 635.1352; found: 635.1376.
p-Tolyl 2-deoxy-2-N-phthalimido-3-O-p-methoxybenzyl-4-O-benzyl-1-thio-β-D-glucopyranoside (13)
Compound 13 was synthesized from p-tolyl 2-deoxy-2-N-phthalimido-3-O-p-methoxybenzyl-4,6-O-benzylidene-1-thio-β-D-glucopyranoside[33] (62 mg, 100 μmol) following the general procedure of regioselective opening of benzylidene ring in 68% yield. 1H NMR (400 MHz, CDCl3): δ = 1.92 (t, 3J(H,H) = 6.8 Hz, 3H), 2.29 (s, 3H), 3.52–3.58 (m, 1H), 3.58 (s, 3H), 3.66 (t, 3J(H,H) = 9.6 Hz, 1H), 3.72–3.78 (m, 1H), 3.88–3.96 (m, 1H), 4.14 (t, 3J(H,H) = 10.2 Hz, 1H), 4.34 (dd, 3J(H,H) = 8.8, 10.0 Hz, 1H), 4.38 (d, 3J(H,H) = 12.4 Hz, 1H), 4.70 (d, 3J(H,H) = 12.4 Hz, 1H), 4.72 (d, 3J(H,H) = 12.4 Hz, 1H), 4.88 (d, 3J(H,H) = 12.4 Hz, 1H), 5.50 (d, 3J(H,H) = 10.8 Hz, 1H), 6.33–6.39 (m, 2H), 6.86–6.92 (m, 2H), 7.02–7.06 (m, 2H), 7.20–7.25 (m, 2H), 7.28–7.38 (m, 5H), 7.58–7.84 ppm (m, 4H); 13C NMR (100 MHz, CDCl3): δ = 21.07, 54.74, 55.00, 61.92, 74.44, 75.08, 79.21, 79.35, 79.66, 83.31, 113.32, 123.05, 123.29, 127.73–133.71, 137.75, 138.32, 158.73, 167.27, 167.90 ppm; ESI-MS: m/z calcd for C36H35NNaO7S [M+Na]+: 648.2; found: 648.3.
p-Tolyl 2-deoxy-2-N-phthalimido-3-O-p-methoxybenzyl-4-O-benzyl-1-thio-β-D-glucopyranosyluronic acid (14)
Compound 14 was synthesized from compound 13 (40 mg, 66 μmol) according to the general oxidation procedure in 85% yield with 6% of the glycosyl lactone 15 isolated. 1H NMR (400 MHz, CDCl3): δ = 2.33 (s, 3H; p-MePh), 3.57 (s, 3H), 3.86 (t, 3J(H,H) = 9.2 Hz, 1H), 4.10 (d, 3J(H,H) = 10 Hz, 1H), 4.32 (t, 3J(H,H) = 10.2 Hz, 1H), 4.30–4.37 (m, 2H), 4.68–4.74 (m, 3H), 5.52 (d, J = 10.8 Hz, 1H), 6.33–7.81 ppm (m, 17H; ArH); 13C NMR (100 MHz, CDCl3): δ = 21.36, 54.65, 55.00, 74.72, 75.48, 79.29, 81.09, 84.65, 113.58–158.99 (aromatic carbon atoms), 167.39, 168.10, 177.56 ppm; HRMS: m/z calcd for C36H33NaO8S [M+Na]+: 662.1825; found: 662.1826.
2-Deoxy-2-N-phthalimido-3-O-p-methoxybenzyl-4-O-benzyl-β-D-glucopyranosidurono-6,1-lactone (15)
1H NMR (400 MHz, CDCl3): δ = 3.56 (s, 3H), 3.91 (d, 3J(H,H) = 6.8 Hz, 1H), 4.22 (d, 3J(H,H) = 9.6 Hz, 1H), 4.31 (dd, 3J(H,H) = 7.2, 10 Hz, 1H), 4.42 (d, 3J(H,H) = 12.0 Hz, 1H), 4.61 (d, 3J(H,H) = 12.0 Hz, 1H), 4.64 (s, 1H), 4.68 (d, 3J(H,H) = 11.6 Hz, 1H), 4.82 (d, 3J(H,H) = 11.6 Hz, 1H), 5.96 (s, 1H), 6.32–7.72 ppm (m, 13H); ESI-MS: m/z calcd for C29H25NaNO8 [M+Na]+: 538.1; found: 538.2; HRMS: m/z calcd for C29H25NaNO8 [M+Na]+: 538.1478; found: 538.1501.
p-Tolyl 2-deoxy-2-N-phthalimido-3-O-tert-butyldimethylsilyl-4-O-benzyl-1-thio-β-D-glucopyranoside (16)
Compound 16 was synthesized from p-tolyl 2-deoxy-2-N-phthalimido-3-O-tert-butyldimethylsilyl-4,6-O-benzylidene-1-thio-β-D-glucopyranoside[33] (62 mg, 100 μmol) following the general procedure of regioselective opening of benzylidene ring in 77% yield. 1H NMR (600 MHz, CDCl3): δ = −0.44 (s, 3H), −0.06 (s, 3H), 0.71 (s, 9H), 1.95 (t, 3J(H,H) = 6.6 Hz, 3H), 2.28 (s, 3H), 3.48–3.58 (m, 2H), 3.64–3.72 (m, 1H), 3.83–3.90 (m, 1H), 4.22 (t, 3J(H,H) = 10.2 Hz, 1H), 4.48 (dd, 3J(H,H) = 8.4, 9.6 Hz, 1H), 4.64 (d, 3J(H,H) = 12.0 Hz, 1H), 4.81 (d, 3J(H,H) = 12.0 Hz, 1H), 5.55 (d, 3J(H,H) = 10.8 Hz, 1H), 7.00–7.06 (m, 2H), 7.21–7.36 (m, 7H), 7.72–7.92 ppm (m, 4H); ESI-MS: m/z calcd for C34H31NNaO6SSi [M+Na]+: 642.2; found: 642.7.
p-Tolyl 2-deoxy-2-N-phthalimido-3-O-tert-butyldimethylsilyl-4-O-benzyl-1-thio-β-D-glucopyranosyluronic acid (17)
Compound 17 was synthesized from compound 16 (55 mg, 89 μmol) according to the general oxidation procedure in 86% yield. 1H NMR (600 MHz, CDCl3): δ = −0.44 (s, 3H), −0.09 (s, 3H), 0.71 (s, 9H), 2.28 (s, 3H), 3.67 ?t, 3J(H,H) = 8.8 Hz, 1H), 4.11 (d, 3J(H,H) = 9.6 Hz, 1H), 4.31 (t, 3J(H,H) = 10.2 Hz, 1H), 4.47 (t, 3J(H,H) = 9.2 Hz, 1H), 4.58 (d, 3J(H,H) = 10.8 Hz, 1H), 4.64 (d, 3J(H,H) = 10.8 Hz, 1H), 5.57 (d, 3J(H,H) = 10.2 Hz, 1H), 7.01–7.89 ppm (m, 13H; ArH); 13C NMR (150 MHz, CDCl3): δ = −4.48, −3.69, 17.84, 21.40, 25.88, 56.40, 73.24, 75.25, 77.89, 81.13, 84.71, 127.93–138.76 ppm (aromatic carbon atoms); HRMS: m/z calcd for C34H39NaO7SSi [M+Na]+: 656.2114; found: 656.2072.
Allyl 2,3,4-tri-O-benzoyl-1-thio-α-D-mannopyranosyluronic acid (19)
Compound 19 was synthesized from compound 18[22] (50 mg, 94 μmol) according to the general oxidation procedure in 92% yield. 1H NMR (400 MHz, CDCl3): δ = 4.18 (dd?J = 6.0, 12.8 Hz, 1H), 4.35 (dd, 3J(H,H) = 4.2, 12.8 Hz, 1H), 4.68 (d, 3J(H,H) = 9.2 Hz, 1H), 5.28–5.30 (m, 2H), 5.40 (dd, 3J(H,H) = 1.2, 17.2 Hz, 1H), 5.68 (t, 1H; 3J(H,H) = 4.2 Hz, 1H), 5.91–6.03 (m, 3H), 7.26–8.09 ppm (m, 15H; ArH); 13C NMR (100 MHz, CDCl3): δ = 67.83, 69.64, 69.69, 69.86, 70.25, 97.05, 119.05, 128.60–133.84 (aromatic carbon atoms), 165.53, 165.77, 165.86, 177.74 ppm; HRMS: m/z calcd for C30H26NaO10 [M+Na]+: 569.1424; found: 569.1431.
Benzyl 1-methoxy-2-deoxy-2-N-phthalimido-3-O-benzyl-β-D-glucopyranosyluronate (21)
Compound 21 was synthesized from compound 20[25] (26 mg, 63 μmol) in 75% yield according to the general procedures of oxidation and benzyl ester formation, along with compound 22 (~10%). 1H NMR (600 MHz, CDCl3): δ = 3.08?s, 1H; 4-OH), 3.39 (s, 3H; MeO), 4.0–4.05 (m, 2H), 4.16(dd, 3J(H,H) = 8.4, 9.0 Hz, 1H), 4.23 (dd, 3J(H,H) = 7.2, 10.8 Hz, 1H), 4.53 (d, 3J(H,H) = 12.6 Hz, 1H; PhCH), 4.44 (d, 3J(H,H) = 12.6 Hz, 1H; PhCH), 5.09 (d, 3J(H,H) = 8.4 Hz, 1H), 5.26 (d, 3J(H,H) = 12.6 Hz, 1H; PhCH), 5.30 (d, 3J(H,H) = 12.6 Hz, 1H; PhCH), 6.89–7.67 ppm (m, 14H; ArH); 13C NMR (150 MHz, CDCl3): δ = 55.11, 57.22, 67.69, 73.94, 74.37, 74.68, 77.66, 99.77, 127.66–138.14 (aromatic carbon atoms), 169.53 ppm; HRMS: m/z calcd for C29H27NNaO8+ [M+Na]+: 540.1634; found: 540.1628.
Benzyl 4-O-(benzyl-1-methoxy-2-deoxy-2-N-phthalimido-3-O-benzyl-β-D-glucopyranosyluronate)-1-methoxy-2-deoxy-2-N-phthalimido-3-O-benzyl-β-D-glucopyranosyluronate (22)
1H NMR (400 MHz, CDCl3): δ = 3.35s, 3H; OMe), 3.41 (s, 3H; OMe), 3.75 (t, 3J(H,H) = 5.4 Hz, 1H), 3.86 (dt, 3J(H,H) = 3.6, 8.0 Hz, 1H), 4.10–4.22 (m, 3H), 4.31 (dd, 3J(H,H) = 8.4, 10.4 Hz, 1H), 4.39 (d, 3J(H,H) = 12.4 Hz, PhCH, 1H), 4.55 (dd, 3J(H,H) = 8.8, 10.8 Hz,1H), 4.58 (d, 3J(H,H) = 12.4 Hz, PhCH, 1H), 4.68 (d, 3J(H,H) = 12.4 Hz, PhCH, 1H), 4.86 (d, 3J(H,H) = 12.4 Hz, PhCH, 1H), 5.03 (d, 3J(H,H) = 8.0 Hz, 1H), 5.10 (d, 3J(H,H) = 8.8 Hz, 1H), 5.20 (d, 3J(H,H) = 12.4 Hz, PhCH, 1H), 5.26 (d, 3J(H,H) = 12.4 Hz, PhCH, 1H), 5.42 (dd, 3J(H,H) = 8.8, 10.0 Hz, 1H), 6.89–7.80 ppm (m, 23H; ArH); 13C NMR (150 MHz, CDCl3): δ = 55.03, 55.17, 57.01, 57.37, 68.65, 72.92, 73.84, 74.32, 74.45, 74.71, 74.81, 76.15, 99.44, 99.61, 127.56–138.29 (aromatic carbon atoms), 168.75, 169.51 ppm; HRMS: m/z calcd for C51H46N2NaO15+ [M+Na]+: 949.2796; found: 949.2794.
1-Methoxy-2,3-O-isopropylidene-β-D-ribofuranosyl-5-carboxylic acid (24)
Compound 24 was synthesized from compound 23[28] (40 mg, 96 μmol) according to the general oxidation procedure in 100% yield. 1H NMR (600 MHz, CDCl3): δ = 1.31 (s, 3H), 1.47 (s, 3H), 3.40 (s, 3H), 4.55 (d, 3J(H,H) = 5.4 Hz, 1H), 4.63 (s, 1H), 5.04 (s, 1H), 5.17 ppm (d, 3J(H,H) = 5.4 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ = 25.15, 26.56, 55.89, 82.41, 84.33, 109.91, 113.11, 175.41 ppm; HRMS: m/z calcd for C30H26NaO10 [M+Na]+: 569.1424; found: 569.1431.
Methyl (2-O-benzoyl-3,4-di-O-benzyl-β-D-glucopyranosyluronic acid-(1→3)-(2-deoxy-2-N-phthalimido-4,6-O-benzylidene-β-D-glucopyranoside) (26)
Compound 26 was synthesized from compound 25[29] (28 mg, 33 μmol) following the general oxidation procedure in 95% yield. 1H NMR (400 MHz, CDCl3): δ = 3.37 (s, 3H; OMe), 3.56 (dd, 3J(H,H) = 4.2, 6.4 Hz, 1H), 3.65 (m, 1H), 3.80–3.89 (m, 3H), 3.94 (d, 3J(H,H) = 6.0 Hz, 1H), 4.30 (dd, 3J(H,H) = 8.4, 10.4 Hz, 1H), 4.37–4.51 (m, 5H), 4.73 (t, 3J(H,H) = 11.2 Hz, 1H), 4.96–5.01 (m, 2H), 5.08 (d, 3J(H,H) = 8.8 Hz, 1H), 5.57 (s, 1H), 7.01–7.53 ppm (m, 24H; ArH); 13C NMR (100 MHz, CDCl3): δ = 55.45, 57.23, 66.46, 68.95, 73.56, 73.69, 73.89, 74.28, 76.41, 79.45, 81.08, 99.49, 99.78, 102.42, 126.54–137.34 (aromatic carbon atoms), 164.77, 169.45 ppm (carbonyl groups); HRMS: m/z calcd for C49H45NaO14 [M+Na]+: 894.2738; found: 894.2701.
Methyl (2-O-benzoyl-3,4-di-O-benzyl-β-D-glucohexodialdo-1,5-pyranosyl)-(1→3)-(2-deoxy-2-N-phthalimido-4,6-O-benzylidene-β-D-glucopyranoside) (27)
Compound 27 was obtained from compound 25[29] following the general oxidation procedure without the NaClO2 oxidation step. 1H NMR (600 MHz, CDCl3): δ = 3.35 (s, 3H; OMe), 3.46 (d, 3J(H,H) = 7.8 Hz, 1H), 3.58 (t, 3J(H,H) = 7.8 Hz, 1H), 3.62–3.64 (m, 2H), 3.84–3.87 (m, 2H), 4.28 (dd, 3J(H,H) = 8.4, 10.2 Hz, 1H), 3.73 (dd, 3J(H,H) = 4.8, 10.8 Hz, 1H), 4.39–4.51 (m, 5H), 4.72 (t, 3J(H,H) = 9.0 Hz, 1H), 4.83 (d, 3J(H,H) = 7.2 Hz, 1H), 4.97 (t, 3J(H,H) = 7.2 Hz, 1H), 5.05 (d, 3J(H,H) = 8.4 Hz, 1H), 5.51 (s, 1H; PhCH), 6.95–7.55 (m, 24H; ArH), 9.39 ppm (s, 1H; CHO); ESI-MS: m/z calcd for C50H49NNaO14 [M+Na+MeOH]+: 910.3; found: 910.6.
Methyl (benzyl-2-O-benzoyl-3-O-benzyl-4-O-tert-butyldimethylsilyl-β-D-glucopyranosyluronate)-(1→3)-(2-deoxy-2-N-phthalimido-4,6-O-benzylidene-β-D-glucopyranosyl)-(1→4)-(benzyl-2-O-benzoyl-3-O-benzyl-β-D-glucopyranosyluronate)-(1→3)-2-deoxy-2-N-phthalimido-4,6-O-benzylidene-β-D-glucopyranoside (29)
Compound 29 was synthesized from compound 28[29] (136 mg, 84 μmol) following the general procedures of oxidation and benzyl ester formation in 86% overall yield. 1H NMR (600 MHz, CD2Cl2): δ = −0.24 (s, 3H; CH3Si), −0.13 (s 3H; CH3Si), 0.75 (s, 9H; (CH3)3CSi), 2.25 (m, 1H), 3.28 (s, 3H; CH3O), 3.37 (d, 3J(H,H) = 9.6 Hz, 1H), 3.41 (t, 3J(H,H) = 7.8 Hz, 1H), 3.44-.3.49 (m, 3H), 3.58–3.61 (m, 2H), 3.67 (d, 3J(H,H) = 8.4 Hz, 1H), 3.93–4.13 (m, 5H), 4.18 (dd, 3J(H,H) = 5.4, 9.6 Hz, 1H), 4.32–4.39 (m, 3H), 4.52 (d, 3J(H,H) = 10.8 Hz, 1H), 4.51 (dd, 3J(H,H) = 9.0, 10.2 Hz, 1H), 4.61 (d, 3J(H,H) = 8.4 Hz, 1H), 4.66 (dd, 3J(H,H) = 8.4, 10.2 Hz, 1H), 4.68 (d, 3J(H,H) = 11.4 Hz, 1H), 4.79 (t, 3J(H,H) = 7.8 Hz, 1H), 4.80 (d, 3J(H,H) = 8.4 Hz, 1H), 4.92 (dd, 3J(H,H) = 8.4, 10.2 Hz, 1H), 4.94 (s, 2H), 5.03 (d, 3J(H,H) = 12.0 Hz, 1H), 5.05 (d, 3J(H,H) = 8.4 Hz, 1H), 5.06 (d, 3J(H,H) = 12.0 Hz, 1H), 5.11 (s, 1H; PhCH), 5.32 (s, 1H; PhCH), 6.92–7.81 ppm (m, 48H; ArH); 13C NMR (100 MHz, CD2Cl2): δ = −5.20, −4.29, 19.22, 25.72, 55.16, 55.69, 56.98, 65.85, 66.32, 67.31, 68.43, 68.55, 71.91, 72.99, 73.96, 74.24, 74.68, 75.80, 76.12, 76.96, 77.56, 79.88, 80.96, 81.71, 82.20, 98.30, 99.57, 99.59, 99.84, 101.15, 101.48, 123.19–138.38 (aromatic carbon atoms), 164.53, 164.66, 166.93, 168.11 ppm (carbonyl groups); HRMS: m/z calcd for C103H100NaN2O27Si [M+Na]+: 1847.6180; found: 1847.6165.
Methyl (benzyl-2-O-benzoyl-3-O-benzyl-4-O-tert-butyldimethylsilyl-β-D-glucopyranosyluronate)-(1→3)-(2-deoxy-2-N-phthalimido-4,6-O-benzylidene-β-D-glucopyranosyl)-(1→4)-(benzyl-2-O-benzoyl-3-O-benzyl-β-D-glucopyranosyluronate)-(1→3)-(2-deoxy-2-N-phthalimido-4,6-O-benzylidene-β-D-glucopyranosyl)-(1→4)-(benzyl-2-O-benzoyl-3-O-benzyl-β-D-glucopyranosyluronate)-(1→3)-(2-deoxy-2-N-phthalimido-4,6-O-benzylidene-β-D-glucopyranoside) (30)
Compound 30 was synthesized from compound 1[29] (70 mg, 30 μmol) following the general procedures of oxidation and benzyl ester formation in 82% overall yield. 1H NMR (600 MHz, CD2Cl2): δ = −0.14 (s, 3H; CH3Si), −0.26 (s, 3H; CH3Si), 0.72 (s, 9H; (CH3)3CSi), 3.04 (t, 3J(H,H) = 10.2 Hz, 1H), 3.11–3.15 (m, 2H), 3.21–3.31 (m, 5H), 3.27 (s, 3H; MeO), 3.38–3.47 (m, 6H), 3.56–3.61 (m, 2H), 3.67 (t, 3J(H,H) = 8.4 Hz, 1H), 3.84 (dd, 3J(H,H) = 4.2, 10.2 Hz, 1H), 3.93–4.04 (m, 6H), 4.07–4.12 (m, 2H), 4.18 (dd, 3J(H,H) = 4.2, 9.6 Hz, 1H), 4.27–4.38 (m, 6H), 4.45 (d, 3J(H,H) = 12 Hz, 1H), 4.48 (dd, 3J(H,H) = 8.4, 10.2 Hz, 1H),4.52–4.58 (m, 3H), 4.62–4.68 (m, 3H), 4.97 (d, 3J(H,H) = 8.4 Hz, 1H), 4.99 (s, 1H; PhCH), 5.01 (s, 1H; PhCH), 5.03 (d, 3J(H,H) = 8.4 Hz, 1H), 5.10 (d, 3J(H,H) = 12 Hz, 1H), 5.12 (s, 1H; PhCH), 5.17 (d, 3J(H,H) = 12 Hz, 1H), 4.91–4.93 (m, 3H), 6.86–7.73 ppm (m, 72H; ArH); 13C NMR (100 MHz, CD2Cl2): δ = −5.20, −4.30, 17.91, 25.71, 53.59, 53.86, 54.13, 55.14, 55.55, 55.65, 56.96, 65.70, 65.87, 66.31, 67.28,67.31, 68.21, 68.43, 68.53, 71.89, 72.92, 73.94, 72.18, 74.19, 74.59, 74.74, 75.77, 76.11, 76.96, 77.47, 79.83, 79.87, 80.66, 80.96, 81.15, 82.18, 98.18, 99.54, 99.58, 99.71, 99.79, 100.99, 101.13, 101.47, 123.17–138.33 (aromatic carbon atoms), 164.49, 164.55, 164.64, 166.86, 166.92, 168.10 ppm (carbonyl groups); HRMS: m/z calcd for C151H141NaN3O40Si [M+Na]+: 2686.8758; found: 2686.8855.
Methyl (benzyl-2-O-benzoyl-3-O-benzyl-4-O-tert-butyldimethylsilyl-β-D-glucopyranosyluronate)-(1→3)-(2-deoxy-2-N-phthalimido-4,6-O-benzylidene-β-D-galactopyranosyl)-(1→4)-(benzyl-2-O-benzoyl-3-O-benzyl-β-D-glucopyranosyluronate) (32)
Compound 32 was synthesized from compound 31[29] (29 mg, 24 μmol) following the general procedures of oxidation and benzyl ester formation in 76% overall yield. 1H NMR (600 MHz, CDCl3): δ = −0.16 (s, 3H; CH3Si), −0.13 (s 3H; CH3Si), 0.77 (s, 9H; (CH3)3CSi), 2.89 (s, 1H), 3.31 (t, 3J(H,H) = 9.0 Hz, 1H), 3.36 (s, 3H; MeO), 3.52–3.55 (m, 3H), 3.59 (d, 3J(H,H) = 10.8 Hz, 1H), 4.01–4.16 (m, 6H), 4.46 (s, 2H), 4.55–4.58 (d, 2H), 4.66 (dd, 3J(H,H) = 3.6, 10.8 Hz, 1H), 4.72 (d, 3J(H,H) = 10.2 Hz, 1H), 4.75 (d, 3J(H,H) = 10.2 Hz, 1H), 4.84 (d, 3J(H,H) = 7.2 Hz, 1H), 5.03–5.16 (m, 6H), 5.24 (s, 1H; PhCH), 6.81–7.55 ppm (m, 39H; ArH); 13C NMR (150 MHz, CDCl3): δ = −4.99, −4.04, 18.07, 25.93, 52.39, 57.38, 66.73, 67.19, 67.44, 68.86, 71.73, 73.66, 73.97, 74.46, 74.55, 74.89, 75.41, 75.54, 76.84, 81.51, 82.41, 82.64, 98.29, 100.66, 101.06, 104.88, 122.92–139.17 (aromatic carbon atoms), 164.54, 167.84, 168.40, 169.41 ppm (carbonyl groups); ESI-MS: m/z calcd for C82H85NNaO20Si [M+Na]+: 1454.5; found: 1454.6; HRMS: m/z calcd for C82H85NNaO20Si [M+Na]+: 1454.5170; found: 1454.5332.
Methyl (benzyl-2-O-benzoyl-3-O-benzyl-4-O-tert-butyldimethylsilyl-β-D-glucopyranosyluronate)-(1→4)-(2-deoxy-2-azido-3,6-O-dibenzyl-α-D-glucopyranosyl)-(1→4)-benzyl-2,3-O-dibenzyl-β-D-glucopyranosyluronate (34)
Compound 34 was synthesized from compound 33[29] (40 mg, 33 μmol) following the general procedures of oxidation and benzyl ester formation in 81% overall yield. 1H NMR (600 MHz, CDCl3): δ = −0.04 (s, 3H; CH3Si), −0.04 (s 3H; CH3Si), 0.85 (s, 9H; (CH3)3CSi), 3.15 (dd, 3J(H,H) = 4.2, 10.8 Hz, 1H), 3.24 (d, 3J(H,H) = 10.2 Hz, 1H), 3.29 (t, 3J(H,H) = 9.0 Hz, 1H), 3.39–3.42 (m, 2H), 3.45 (s, 3H; OMe), 3.61–3.67 (m, 2H), 3.73 (d, 3J(H,H) = 10.8 Hz, 1H), 3.76 (d, 3J(H,H) = 9.0 Hz, 1H), 3.79 (d, 3J(H,H) = 10.2 Hz, 1H), 3.95–4.00 (m, 2H), 4.06 (t, 3J(H,H) = 9.0 Hz, 1H), 4.26–4.29 (m, 3H), 4.42 (d, 3J(H,H) = 10.8 Hz, 1H), 4.48 (d, 3J(H,H) = 10.8 Hz, 1H), 4.56 (d, 3J(H,H) = 10.8 Hz, 1H), 4.59–4.63 (m, 3H), 4.68–4.70 (m, 2H), 4.83–4.85 (m, 2H), 4.92 (d, 3J(H,H) = 10.8 Hz, 1H), 5.01 (d, 3J(H,H) = 12.6 Hz, 1H), 5.01–5.08 (m, 2H), 5.27 (t, 3J(H,H) = 8.4 Hz, 1H), 5.53 (d, 3J(H,H) = 3.6 Hz, 1H), 7.06–7.65 ppm (m, 40H; ArH); 13C NMR (150 MHz, CDCl3): δ −4.77, −3.81, 18.17, 26.06, 57.55, 62.67, 65.66, 66.94, 67.17, 67.42, 70.68, 72.41, 73.86, 73.91, 74.12, 74.16, 74.86, 75.19, 75.63, 75.71, 76.67, 76.87, 77.55, 82.13, 82.94, 84.19, 97.36, 100.45, 105.04, 137.73–141.06 (aromatic carbon atoms), 164.73, 167.79, 168.44 ppm (carbonyl groups); HRMS: m/z calcd for C81H89N3NaO18Si [M+Na]+: 1442.5808; found: 1442.5804.
Supplementary Material
Acknowledgments
This work was supported by the University of Toledo and the National Institutes of Health (R01-GM-72667).
Footnotes
Dedicated to Prof. Koji Nakanishi on the occasion of his 80th birthday
Supporting information (copies of 1H NMR and 13C NMR spectra) for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Larock RC. Comprehensive Organic Transformations. Wiley-VCH; New York: 1999. pp. 1646–1650. [Google Scholar]
- 2.Yeung BKS, Chong PYC, Petillo PA. In: Glycochemistry. Principles, Synthesis, and Applications. Wang PG, Bertozzi CR, editors. Marcel Dekker; New York: 2001. pp. 425–492. [Google Scholar]
- 3.a) Gama CI, Hsieh-Wilson LC. Curr Opin Chem Biol. 2005;9:609–619. doi: 10.1016/j.cbpa.2005.10.003. [DOI] [PubMed] [Google Scholar]; b) Raman R, Sasisekharan V, Sasisekharan R. Chem Biol. 2005;12:267–277. doi: 10.1016/j.chembiol.2004.11.020. [DOI] [PubMed] [Google Scholar]; c) Linhardt RJ, Toida T. Acc Chem Res. 2004;37:431–438. doi: 10.1021/ar030138x. [DOI] [PubMed] [Google Scholar]; d) Koeller KM, Wong CH. Nat Biotechnol. 2000;18:835–841. doi: 10.1038/78435. [DOI] [PubMed] [Google Scholar]
- 4.Petitou M, van Boeckel CAA. Angew Chem. 2004;116:3180–3196. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]
- 5.a) Karst NA, Linhardt RJ. Curr Med Chem. 2003;10:1993–2031. doi: 10.2174/0929867033456891. [DOI] [PubMed] [Google Scholar]; b) Poletti L, Lay L. Eur J Org Chem. 2003:2999–3024. [Google Scholar]
- 6.Palmacci ER, Seeberger PH. Tetrahedron. 2004;60:7755–7766. [Google Scholar]
- 7.Oscarson S, Svahnberg P. J Chem Soc Perkin Trans 1. 2001:873–879. [Google Scholar]
- 8.a) Seeberger PH. Chem Commun. 2003:1115–1121. doi: 10.1039/b210230g. [DOI] [PubMed] [Google Scholar]; b) Seeberger PH. Principles, Synthesis, and Applications. In: Wang PG, Bertozzi CR, editors. Glycochemistry. Marcel Dekker; New York: 2001. pp. 1–32. [Google Scholar]
- 9.Vermeer HJ, Halkes KM, van Kuik JA, Kamerling JP, Vliegenthart JFG. J Chem Soc Perkin Trans 1. 2000:2249–2263. [Google Scholar]
- 10.a) Kramer S, Nolting B, Ott AJ, Vogel C. J Carbohydr Chem. 2000;19:891–921. [Google Scholar]; b) La Ferla B, Lay L, Guerrini M, Poletti L, Panza L, Russo G. Tetrahedron. 1999;55:9867–9880. [Google Scholar]
- 11.Halkes KM, Slaghek TM, Hypponen TK, Kruiskamp PH, Ogawa T, Kamerling JP, Vliegenthart JFG. Carbohydr Res. 1998;309:161–174. doi: 10.1016/s0008-6215(98)00116-5. [DOI] [PubMed] [Google Scholar]
- 12.a) Garegg PJ, Olsson L, Oscarson S. J Org Chem. 1995;60:2200–2204. [Google Scholar]; b) Slaghek T, Nakahara Y, Ogawa T, Kamerling JP, Vliegenthart JFG. Carbohydr Res. 1994;255:61–85. doi: 10.1016/s0008-6215(00)90971-6. [DOI] [PubMed] [Google Scholar]; c) Slaghek T, Hypponen TK, Ogawa T, Kamerling JP, Vliegenthart JFG. Tetrahedron: Asymmetry. 1994;5:2291–2301. [Google Scholar]
- 13.Allanson NM, Liu D, Chi F, Jain RK, Chen A, Ghosh M, Hong L, Sofia MJ. Tetrahedron Lett. 1998;39:1889–1892. [Google Scholar]
- 14.de Nooy AEJ, Besemer AC, van Bekkum H. Synthesis. 1996:1153–1174. [Google Scholar]
- 15.a) Litjens REJN, Heeten RD, Timmer MSM, Overkleeft HS, van der Marel GA. Chem Eur J. 2005;11:1010–1016. doi: 10.1002/chem.200400862. [DOI] [PubMed] [Google Scholar]; b) Chauvin AL, Nepogodiev SA, Field RA. J Org Chem. 2005;70:960–966. doi: 10.1021/jo0482864. [DOI] [PubMed] [Google Scholar]
- 16.a) Lee JC, Lu XA, Kulkarni SS, Wen YS, Hung SC. J Am Chem Soc. 2004;126:476–477. doi: 10.1021/ja038244h. [DOI] [PubMed] [Google Scholar]; b) Haller M, Boons GJ. J Chem Soc Perkin Trans 1. 2001:814–822. [Google Scholar]; c) Yeung BKS, Hill DC, Janicka M, Petillo PA. Org Lett. 2000;2:1279–1282. doi: 10.1021/ol0057075. [DOI] [PubMed] [Google Scholar]; d) Baisch G, Ohrlein R. Carbohydr Res. 1998;312:61–72. doi: 10.1016/s0008-6215(98)00229-8. [DOI] [PubMed] [Google Scholar]; e) Li K, Helm RF. Carbohydr Res. 1995;273:249–253. [Google Scholar]; f) Davis NJ, Flitsch SL. Tetrahedron Lett. 1993;34:1181–1184. [Google Scholar]
- 17.van den Bos LJ, Codee JDC, van der Toorn JC, Boltje TJ, van Boom JH, Overkleeft HS, van der Marel GA. Org Lett. 2004;6:2165–2168. doi: 10.1021/ol049380+. [DOI] [PubMed] [Google Scholar]
- 18.van den Bos LJ, Litjens REJN, van den Berg RJBHN, Overkleeft HS, van der Marel GA. Org Lett. 2005;7:2007–2010. doi: 10.1021/ol050491y. [DOI] [PubMed] [Google Scholar]
- 19.Zhao M, Li J, Mano E, Song Z, Tschaen DM, Grabowski EJJ, Reider PJ. J Org Chem. 1999;64:2564–2566. [Google Scholar]
- 20.Clausen MH, Madsen R. Chem Eur J. 2003;9:3821–3832. doi: 10.1002/chem.200204636. [DOI] [PubMed] [Google Scholar]
- 21.Huang X, Huang L, Wang H, Ye XS. Angew Chem. 2004;116:5333–5336. [Google Scholar]; Angew Chem Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 22.Heng L, Ning J, Kong F. J Carbohydr Chem. 2001;20:285–296. [Google Scholar]
- 23.Garegg PJ. Adv Carbohydr Chem Biochem. 1997;52:179–205. doi: 10.1016/s0065-2318(08)60091-8. [DOI] [PubMed] [Google Scholar]
- 24.Gilewski T, Ragupathi G, Bhuta S, Williams LJ, Musselli C, Zhang XF, Bencsath KP, Panageas KS, Chin J, Hudis CA, Norton L, Houghton AN, Livingston PO, Danishefsky SJ. Proc Natl Acad Sci USA. 2001;98:3270–3275. doi: 10.1073/pnas.051626298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz DA, Lee HH, Carver JP, Krepinsky JJ. Can J Chem. 1985;63:1073–1079. [Google Scholar]
- 26.Benzylation was carried out for easier product characterization. The purities of the carboxylic acids obtained prior to benzylation were high, which can be used directly for further synthetic manipulations.
- 27.Creary X. Org Synth. 1986;64:207. [Google Scholar]
- 28.Barrett AGM, Lebold SA. J Org Chem. 1990;55:3853–3857. [Google Scholar]
- 29.Syntheses of these compounds will be published elsewhere.
- 30.2-Methyl-but-2-ene was added to as a chlorine scavenger to prevent potential side reactions and tert-butanol presumably kept the reaction medium more homogeneous. See Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091–2096.
- 31.Jiang L, Chan TH. Tetrahedron Lett. 1998;39:355–358. [Google Scholar]
- 32.Chapman TM, Davies IG, Gu B, Block TM, Scopes DIC, Hay PA, Courtney SM, McNeill LA, Schofield CJ, Davis BG. J Am Chem Soc. 2005;127:506–507. doi: 10.1021/ja043924l. [DOI] [PubMed] [Google Scholar]
- 33.Huang L, Wang Z, Li X, Huang X. Carbohydr Res. 2006 doi: 10.1016/j.carres.2006.01.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.