

Abstract
DNA polymerases achieve accurate DNA replication through a delicate balance between primer elongation and proofreading. While insufficient proofreading results in DNA replication errors, indiscriminate removal of correct along with incorrect nucleotides is wasteful and may prevent completion of DNA synthesis. The transition between polymerization and proofreading modes is proposed to be governed by a kinetic barrier that prevents proofreading unless the rate of primer elongation is significantly reduced by the presence of an incorrect base pair at the primer–terminus. We have used mutational analysis, coupled with a sensitive, fluorescence-based assay to characterize intermediate steps in the proofreading pathway. A highly fluorescent complex forms between the bacteriophage T4 DNA polymerase and DNA primer–templates labeled at the 3′ terminus with the base analog 2-aminopurine. Formation of the fluorescent complex appears to be a rate-determining step in the proofreading pathway and is impaired for several mutator T4 DNA polymerases with amino acid substitutions in the exonuclease domain. Although these mutant DNA polymerases are proficient in hydrolysis, their reduced ability to form the fluorescent complex imposes a higher kinetic barrier. As a consequence, the mutant DNA polymerases proofread less frequently, resulting in more DNA replication errors.
DNA polymerases ensure faithful DNA replication by combining accurate nucleotide incorporation with exonucleolytic proofreading. The status of the primer–terminus determines which pathway, polymerization or proofreading, will be followed. Extension of a correctly base-paired primer–terminus is rapid and precludes proofreading, whereas the reduced rate of extension for a mispair provides an opportunity for the primer–terminus to be transferred to the exonuclease active center, where the incorrect nucleotide is removed (reviewed in refs. 1 and 2).
The reaction steps involved in proofreading include translocation of the primer–terminus from the polymerase to the exonuclease active center, strand separation of at least two nucleotides (3, 4), positioning of the 3′ end of the primer in the exonuclease active center, and finally, hydrolysis of the phosphodiester bond to excise the terminal nucleotide. The hydrolysis reaction has been well characterized by crystallographic (5–7) and genetic (8, 9) studies of the Klenow fragment of Escherichia coli DNA polymerase I, and a conserved mechanism for hydrolysis is implied by the invariant acidic active site residues in DNA polymerases with an associated proofreading activity (10–15).
Although the hydrolysis reaction has been characterized extensively, much less is known about the earlier reaction steps leading to hydrolysis. We have used genetic selections for bacteriophage T4 mutator DNA polymerases to identify amino acid residues important for additional steps of the proofreading pathway (16, 17). One class of mutants, active-site “switching” mutants (18), have reduced ability to transfer DNA from the polymerase to the exonuclease active center, but the mutant enzymes retain the ability to hydrolyze single-stranded DNA. The mutational analyses, combined with pre-steady-state kinetic studies (4, 19) using the fluorescent base analog 2-aminopurine (2AP), have revealed a minimal kinetic scheme for the T4 DNA polymerase proofreading pathway (Fig. 1). According to this scheme, partial melting of the primer–terminus facilitates transfer of the primer strand from the polymerase active center to a pre-exonuclease complex, and then further strand separation permits binding of the primer–terminus in the exonuclease active center, where the hydrolysis reaction is catalyzed (4).
Figure 1.

A minimal kinetic scheme for T4 DNA polymerase proofreading. P corresponds to 2AP and d2APMP is 2-aminopurine deoxynucleoside monophosphate.
2AP can be used as a reporter of proofreading activity because the excision product, 2-aminopurine deoxynucleoside monophosphate (d2APMP), is considerably more fluorescent than the 2AP-labeled DNA substrate (19). 2AP is also highly fluorescent when 2AP-DNA is bound in the exonuclease active center of the T4 DNA polymerase (19). Therefore, the increase in fluorescence intensity detected in pre-steady-state excision reactions with the T4 DNA polymerase and 2AP-DNA can result from fluorescent complex formation, product release, or a combination of both processes. The two fluorescent species can be distinguished by fluorescence anisotropy, which provides an estimate of the size of a fluorescent species. During the time course of reaction, a large decrease in anisotropy is observed that corresponds to 2AP residing initially in the environment of a fluorescent pre-exonuclease complex followed by formation of the much smaller fluorescent excision product, d2APMP (19). The rapid increase in fluorescence intensity when d2APMP is produced coincides with the rapid decrease in anisotropy. These observations suggest that formation of a fluorescent pre-exonuclease complex is an intermediate in the proofreading pathway (Fig. 1).
We have identified mutant DNA polymerases defective in forming a fluorescent pre-exonuclease complex, and these mutants provide the means to equate changes in fluorescence intensity with specific reaction steps. Two protein regions in the exonuclease domain have been implicated in forming the fluorescent complex. One region is a novel protein loop structure and the informative mutant DNA polymerase has a Ser substitution for Gly-255 (4). The second region is near the exonuclease active center and includes the Asp-131 and Tyr-317 residues. A reaction scheme is proposed in which these residues in the exonuclease domain orchestrate the transfer of the primer–terminus from the polymerase active center to form the fluorescent pre-exonuclease complex.
MATERIALS AND METHODS
Enzymes.
Mutations specifying amino acid substitutions in the exonuclease domain were introduced into the cloned T4 DNA polymerase gene (20) by site-directed mutagenesis (21). Wild-type and mutant T4 DNA polymerases were purified as described previously (22). Enzyme concentrations were measured spectrophotometrically, using the experimentally determined molar extinction coefficient at 280 nm (ɛ280) of 1.49 × 105 M−1⋅cm−1 (13).
Mutant Phage Strains.
Mutations engineered into the cloned T4 DNA polymerase gene by site-directed mutagenesis were introduced into the phage genome by homologous recombination and marker rescue (14). The T4 DNA polymerase mRNA transcripts from mutant phage were sequenced to ensure that only the desired mutations were present (23, 24).
Mutator Activity.
Spontaneous mutation frequencies were determined by measuring the number of reversion events at the rIIUV199oc site (14, 18). The reversion frequency was derived from the median reversion frequency of five individual cultures. The reversion frequency determined for wild-type phage T4 was 4 × 10−6, but substantially higher values were detected for mutator DNA polymerases.
3′→5′ Exonuclease Activity (Steady-State Conditions).
Hydrolysis of single-stranded [3H]DNA was assayed as described previously (14, 18). Exonuclease activity was also monitored by using an oligonucleotide d(T)16 DNA substrate that was 5′-end-labeled by using [γ-32P]ATP and T4 polynucleotide kinase. The exonuclease reaction mixture contained 67 mM Tris⋅HCl (pH 8.8), 16.7 mM (NH4)2SO4, 0.5 mM DTT, 167 μg/ml BSA, 6 nM 32P-labeled d(T)16, and a 2-fold excess of T4 DNA polymerase (12 nM). Reactions were initiated by the addition of MgCl2 to a final concentration of 6.67 mM and were terminated by addition of an equal volume of gel loading buffer (94 μl of deionized formamide, 4 μl of 10× Tris/borate/EDTA gel buffer, and 2 μl of 2% bromophenol blue/xylene cyanol blue). Samples were heated at 95°C for 5 min prior to electrophoresis on a 15% polyacrylamide/8 M urea gel. The 32P-labeled products were visualized by autoradiography with a PhosphorImager (Molecular Dynamics).
2AP-DNA Substrates.
2AP-terminated primers and complementary templates were prepared as described previously (19) and with commercially available 2AP phosphoramidite (Glen Research) by using the standard β-cyanoethylphosphoramidite method. The 2AP phosphoramidite was added directly to a 3′-phosphate controlled pore glass support (Glen Research). The 3′-phosphatase activity of bacteriophage T4 polynucleotide kinase (25) was used to remove the 3′-phosphate group.
Annealing reaction mixtures contained a 20% excess of template over primer in buffer consisting of 25 mM Hepes (pH 7.5) and 50 mM NaCl as described (19). The complete sequences of the two primer–templates used were as follows (P designates 2AP and the underlined sequences indicate the relative G+C- or A+T-richness of the primer–terminus):
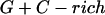 |
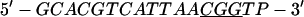 |
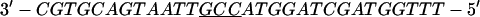 |
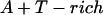 |
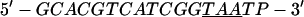 |
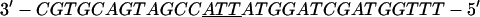 |
Fluorescence-Based Assays.
The increase in fluorescence intensity produced by the rapid mixing of T4 DNA polymerase and 2AP-DNA was detected with a SX.17 MV stopped-flow spectrofluorimeter (Applied Photophysics; Surrey, U.K.). Samples were excited at 310 nm by using a xenon arc source passing through a 2-nm band pass. Fluorescence emission was collected through a 335-nm cut-off filter. The photomultiplier voltage setting was 700 V, and scattered light was subtracted from the reaction by applying an offset voltage.
Experiments were performed in the presence and absence of Mg2+. Enzyme-initiated experiments performed in the presence of Mg2+ allowed the detection of rate-determining steps in the proofreading reaction. Mixtures of reaction components were prepared in two separate syringes. The first syringe contained 1.4 μM T4 DNA polymerase, 2 mM DTT, and 16 mM MgCl2, in a buffer solution of 25 mM Hepes (pH 7.5)/50 mM NaCl, and the second syringe contained 400 nM primer–template DNA (expressed in terms of 3′-primer ends) and 1 mM EDTA in the same buffer. Reactions were initiated in the stopped-flow apparatus by mixing equal volumes (50 μl) of each solution within the instrument dead time of 1.5 ms by pneumatic rams triggered by 140 psi (1 psi = 6.89 kPa) of N2 gas. The final concentrations of reaction components were 700 nM T4 DNA polymerase, 200 nM DNA, 1 mM DTT, and 8 mM MgCl2. Rates of formation of fluorescent complex were measured under the same reaction conditions, except that Mg2+ was omitted to prevent the excision reaction. Rate constants were determined by fitting the fluorescence emission curves to single- or double-exponential equations with the SX.17 MV kinetic software package provided by Applied Photophysics. Single- or double-exponential curve fits were selected, depending on which equation yielded a more uniform distribution of residuals. Multiple runs (six to eight) of each reaction were performed and the mean rate constants were calculated.
RESULTS AND DISCUSSION
In Vivo and in Vitro Characterization of T4 DNA Polymerase Mutator Mutants.
Mutant DNA polymerases with reduced exonucleolytic proofreading but normal hydrolysis activity can provide valuable insight into the reaction steps that occur prior to hydrolysis. The D131N- and D131G-DNA polymerases, which have Asn and Gly substitutions for Asp-131, and the G255S-DNA polymerase, with a Ser substitution for Gly-255, were identified in genetic selections for mutants with high spontaneous mutation frequencies (16, 17). The mutant DNA polymerases increased the spontaneous mutation frequency from 15- to more than 200-fold above the wild-type level (Table 1).
Table 1.
Effect of amino acid substitutions in the exonuclease domain on spontaneous mutation rates and exonuclease activity
| Enzyme | Mutator activity* | Exonuclease activity† (ssDNA) |
|---|---|---|
| Wild type | 1 | 1 |
| G255S | 58 | 0.9 |
| D131N | 15 | 0.7 |
| D131G | 250 | 0.2 |
| D112A + E114A | 300 | 4 × 10−5 |
The mutant DNA polymerases were next assayed in vitro for their ability to hydrolyze single-stranded E. coli [3H]DNA (Table 1). The approximate wild-type level of exonuclease activity was detected for the G255S- and D131N-DNA polymerases, and only a 5-fold reduction in exonuclease activity was observed for the D131G-DNA polymerase, despite the strong mutator phenotype. The relatively high exonuclease activity measured for the D131G-DNA polymerase was surprising, because a similar strong mutator phenotype was seen previously only for mutants with Ala substitutions for one or more essential active site residues, such as the D112A+E114A-DNA polymerase. The D112A+E114A-DNA polymerase, however, has little exonuclease activity (Table 1). Thus, the high level of single-stranded DNA exonuclease activity detected for the G255S-, D131N-, and D131G-DNA polymerases indicates that the mutator activity of these mutant enzymes cannot be attributed to reduced ability to hydrolyze DNA, but must be due instead to a defect in an earlier step in the proofreading pathway.
A more biologically relevant DNA substrate to study DNA polymerase exonucleolytic proofreading is double-stranded DNA. Unlike exonucleolytic degradation of single-stranded DNA substrates, proofreading of duplex DNAs requires partial strand separation and transfer of the primer–terminus from the polymerase to the exonuclease active center. 2AP-labeled duplex DNA substrates were used to study the pre-steady-state kinetics of the proofreading reaction.
2AP as a Spectroscopic Probe of DNA Polymerase Proofreading.
The fluorescence intensity of DNA labeled with 2AP at the 3′ terminus increases when bound in the exonuclease active center of T4 DNA polymerase. Fluorescence is also detected when the excision product, d2APMP, is released (4, 19). Reactions were initiated in a stopped-flow apparatus (see Materials and Methods) by the rapid mixing of a solution of 1.4 μM DNA polymerase and 16 mM Mg2+ with an equal volume of a solution containing 400 nM 2AP-DNA substrate, conditions established previously as pre-steady-state conditions (19). Fluorescence intensity increased with time, but the mutant DNA polymerases displayed slower reaction rates than detected for the wild-type enzyme. The time courses for the wild-type, G255S-, and D131N-DNA polymerases were best fit by double-exponential equations, suggesting that there are two reaction rates (Table 2). For the wild-type enzyme, apparent rates of about 3.6 s−1 (k1) and 20 s−1 (k2) were detected, but the k1 rate was decreased about 10-fold for the G255S- and D131N-DNA polymerases, while a single slow rate of 0.02 s−1 was detected for the D131G-DNA polymerase.
Table 2.
Pre-steady-state kinetic analysis of wild-type and mutant T4 DNA polymerases with double-stranded 2AP-DNA substrate
| Enzyme | Rate-limiting step before hydrolysis*
|
|||
|---|---|---|---|---|
| 1
|
2
|
|||
| k1, s−1 | a1 | k2, s−1 | a2 | |
| Wild type | 3.6 ± 0.2 | 0.8 | 20.1 ± 2.7 | 0.2 |
| G255S | 0.34 ± 0.01† | 0.6 | 14.9 ± 1.8† | 0.4 |
| D131N | 0.5 ± 0.1 | 0.6 | 21.6 ± 2.9 | 0.4 |
| D131G | 0.02 ± 0.01 | 1.0 | ||
Rate constants k1 and k2 were determined from double-exponential curve fits of time courses measuring the increase in fluorescence intensity in proofreading reactions with the wild-type, G255S-, or D131N-DNA polymerase and the G+C-rich 2AP-DNA substrate. The relative amplitudes for the two rates are designated a1 and a2, respectively. The time course for the D131G-DNA polymerase was best described by a single-exponential equation, and the corresponding rate constant is given for comparison in the k2 column.
Data are from Marquez and Reha-Krantz (4).
The k1 rate corresponds to a rate of about 5 s−1 that was measured for the wild-type enzyme by a radioactivity-based rapid-quench method and was equated with formation of a pre-exonuclease complex activated for the hydrolysis reaction (26). The 2AP-fluorescence-based assay has allowed detection of a possible second reaction intermediate, which is formed at the rate of about 20 s−1 (k2) (Table 2). Because the k1 and k2 rates are slower than either the hydrolysis rate of 80 to 100 s−1 (19, 26) or the rate of 400 s−1 measured for extension of a correct base pair (26), formation of proofreading intermediates at the slower rates of about 4–5 s−1 and 20 s−1 provides a kinetic barrier that prevents indiscriminate excision of correctly base-paired primer–termini. The significantly reduced k1 rates detected for the mutant enzymes suggest that the kinetic barrier to initiating the proofreading pathway is raised to a level that increases the probability of extending mismatched primer–termini that would normally be proofread by the wild-type DNA polymerase.
Fluorescence anisotropy experiments (19) demonstrate that the fluorescent d2APMP excision product is formed from a larger fluorescent enzyme–DNA complex. Thus, the k1 and k2 rates may be rates for formation of fluorescent pre-exonuclease complexes as illustrated in Fig. 1. The mutant DNA polymerases, which display reduced k1 rates (Table 2), may be defective in forming fluorescent complexes. This proposal can be tested directly by measuring the rates at which the mutant enzymes form fluorescent enzyme–DNA complexes with 2AP-DNA substrates.
The Fluorescent Pre-Exonuclease Complex.
The rates for formation of the fluorescent complex were measured in the absence of Mg2+ so that fluorescent complex formation could be monitored without interference by production of the fluorescent excision product, d2APMP. Although the rates measured in the absence of Mg2+ may not equal the rates measured with Mg2+, these conditions still allow detection of possible differences in complex formation for the wild-type and mutant enzymes.
The wild-type T4 DNA polymerase formed a fluorescent complex at the apparent rate of about 70 s−1 with a relatively G+C-rich DNA substrate and at a faster rate of about 200 s−1 when the 3′-terminal 2AP was adjacent to four consecutive A⋅T base pairs (Fig. 2, Table 3). Since A+T-richness near the primer–terminus facilitates local melting of the DNA strands (27–29), the faster rate of complex formation with the A+T-rich primer-template suggests a requirement for partial strand separation in forming the fluorescent pre-exonuclease complex. In accordance with this proposal, the rates were highest with single-stranded DNA substrates, about 400 to 500 s−1 (Table 3). Thus, the fluorescent complex likely resembles the partially strand-separated pre-exonuclease intermediates illustrated in Fig. 1.
Figure 2.

Time courses for the formation of fluorescent complexes in the absence of Mg2+ for the wild-type and mutant T4 DNA polymerases with the G+C-rich 2AP-DNA substrate. A single-exponential curve fit is shown superimposed on the data for the wild-type and D131N-DNA polymerases. The time course for the G255S-DNA polymerase was better described by a double-exponential equation. Fluorescence intensity remained at the background level (equivalent to the fluorescence of a mixture of enzyme, DNA, EDTA, and DTT) in the time course with the D131G-DNA polymerase.
Table 3.
Observed rates for formation of fluorescent complexes between wild-type and mutant T4 DNA polymerases and 2AP-DNA substrates
| Enzyme | Fluorescent complex formation rate, s−1
|
|||||
|---|---|---|---|---|---|---|
| G+C-rich DNA substrate
|
A+T-rich DNA substrate
|
|||||
| dsDNA | ssDNA | dsDNA | ssDNA | |||
| Wild type | 71.0 ± 1.4 | 406.0 ± 5.5 | 204.7 ± 4.8 | 530.3 ± 18.9 | ||
| D131N | 61.1 ± 1.2 | 263.0 ± 13.6 | 114.7 ± 1.9 | 322.5 ± 8.6 | ||
| G255S* | 69.7 ± 2.8 (0.3) | 18.6 ± 1.3 (0.7) | 263.5 ± 6.6 | 123.8 ± 21.4 (0.3) | 32.9 ± 2.9 (0.7) | 384.9 ± 13.8 |
Fluorescent complex formation was measured in the absence of Mg2+ as described in the text. The DNA substrates are also described in the text. The single-stranded DNA (ssDNA) is the 2AP-labeled primer strand of the double-stranded DNA (dsDNA) substrates. The reactions for the G+C-rich dsDNA are shown in Fig. 2. The observed rates are independent of enzyme concentration under the conditions used (19). The rates correspond to bimolecular association rates that range from about 0.5 to 5 × 108 M−1⋅s−1.
Two rates were detected for the formation of a fluorescent complex between the G255S-DNA polymerase and the dsDNA substrates. The relative amplitudes of the two phases are indicated in parentheses.
The three mutant enzymes displayed a range of defects in forming the fluorescent complex in the absence of Mg2+ (Fig. 2). Biphasic kinetics were observed for the formation of the fluorescent complex with the G255S-DNA polymerase and the G+C- and A+T-rich primer–templates (Fig. 2, Table 3). While approximately 30% of the fluorescent pre-exonuclease complexes were formed at the wild-type rate, most complexes were formed at the apparent slower rates of about 20 to 30 s−1, depending on the A+T- or G+C-richness of the DNA substrates (Table 3). The steady-state level of fluorescence intensity with the G255S-DNA polymerase approached the wild-type level on the double-stranded DNA substrate (Fig. 2) and equaled the wild-type level of fluorescence on the single-stranded DNA substrate (data not shown). A flexible protein loop structure containing the G255 residue is implicated in facilitating the strand separation required to form a pre-exonuclease complex in the presence of Mg2+ (4). The two rates detected for the formation of a fluorescent complex in the absence of Mg2+ may reflect differences in the conformation of the loop structure. The G255 loop may be less perturbed in some of the mutant DNA polymerases, which would account for the small proportion of fluorescent pre-exonuclease complexes formed at the wild-type rate, while a more severely altered loop structure in the remaining enzymes may impede the strand separation required for fluorescent complex formation.
Amino acid substitutions for the D131 residue also reduced the ability of the DNA polymerase to form a fluorescent complex with 2AP-DNA. The most significant defect observed for the D131N-DNA polymerase was not in the rate of fluorescent complex formation, but in the steady-state level of fluorescence intensity reached (Fig. 2). The D131N-DNA polymerase exhibited an approximate wild-type rate of fluorescent complex formation with the G+C-rich duplex DNA substrate (Table 3), but the steady-state level of fluorescence intensity reached by the mutant enzyme was only 50% of the wild-type level with the double-stranded DNA substrates (Fig. 2). A 20% reduction in the steady-state level of fluorescence intensity was observed with the single-stranded DNA substrates (data not shown). The decreased level of fluorescence observed for the D131N-DNA polymerase may indicate that the complex formed is less fluorescent, that more nonfluorescent than fluorescent complexes are made, or that the D131N-DNA polymerase dissociates more readily from the complex than does the wild-type enzyme. If the complex formed with the D131N-DNA polymerase is less fluorescent, a similar decrease in fluorescence is expected with single- and double-stranded DNA substrates; however, this proposal is unlikely since the level of fluorescence was reduced by a larger amount with double-stranded compared with single-stranded DNA. The dissociation rate measured for the D131N-DNA polymerase is also similar to the rate measured for the wild-type enzyme (data not shown). Thus, the decrease in the steady-state level of fluorescence intensity (Fig. 2) is likely due to increased formation of nonfluorescent complexes. The D131N substitution may affect partitioning of the DNA between fluorescent and nonfluorescent species.
An observation relevant to this hypothesis is that the D131N-DNA polymerase was slower than the wild-type enzyme to degrade short oligonucleotides (Fig. 3). In reactions with 32P-labeled d(T)16, the wild-type and D131N-DNA polymerases displayed similar digestion patterns after incubation for 1 min, with most of the full-length 16-mer degraded to 6- and 5-mers (Fig. 3). Further digestion, however, was slower for the D131N-DNA polymerase. Thus, although the D131N-DNA polymerase is proficient in the hydrolysis of single-stranded DNA (Table 1), the mutant enzyme may have difficulty degrading very short DNA substrates that are stabilized only by amino acid residues in the exonuclease active center.
Figure 3.

Exonucleolytic degradation of 32P-labeled d(T)16 by the wild-type and D131N-DNA polymerases. Reaction time points of 1, 5, 10, and 20 min are indicated above each lane. Full digestion is the dinucleotide product.
Structural data (Fig. 4) show that D131 resides near catalytic residues in the exonuclease active center and may participate in hydrogen bonding with Y317 and T116 (ref. 30; J. Wang and T. A. Steitz, personal communication). Y317 is 4 Å from the catalytic residue, E114, and a water molecule may mediate an indirect interaction between the two amino acids in the active enzyme–DNA complex (J. Wang and T. A. Steitz, personal communication). The hydrogen bond between D131 and Y317 may be important for maintaining the proper conformation of the exonuclease active center by positioning Y317 for interaction with E114. Mutational analysis points to a biological significance of the D131–Y317 interaction, since genetic selections for mutator DNA polymerases that yielded the D131N-, D131G-, and G255S-DNA polymerases also identified the Y317C-DNA polymerase (16, 17). Thus, we propose that the protein region occupied by the D131 and Y317 residues is involved in positioning the primer–terminus correctly in the pre-exonuclease complex.
Figure 4.

Magnified view of the T4 DNA polymerase exonuclease active center. Residues D112 and E114 reside within the exonuclease active center. Proposed hydrogen bonds between D131 and Y317 and between D131 and T116 are illustrated. The Y317 and E114 residues are separated by 4 Å and may interact indirectly through an ordered water molecule (J. Wang and T. A. Steitz, personal communication).
The less conservative D131G amino acid substitution resulted in a DNA polymerase that was severely defective in forming the fluorescent pre-exonuclease complex in the absence of Mg2+. Although fluorescence intensity did not rise above the background level of fluorescence in reactions with either a duplex (Fig. 2) or a single-stranded DNA substrate (data not shown) in the absence of Mg2+, a gradual increase in fluorescence intensity was observed in the presence of Mg2+ because of formation of the fluorescent excision product, d2APMP. Excision product was produced at the rate of 0.02 s−1 in reactions with the duplex DNA substrate (Table 2) and at 2 s−1 with the single-stranded 2AP-labeled DNA substrate (data not shown). The 100-fold faster excision rate detected with the single-stranded DNA substrate suggests that the rate-limiting step for production of d2APMP is not cleavage of the phosphodiester bond, but a reaction step required to prepare double-stranded DNA for hydrolysis. Thus, proofreading by the D131G-DNA polymerase is substantially reduced by the slow rate for forming an activated pre-exonuclease complex.
SUMMARY
Mutant DNA polymerases with G255S, D131N, and D131G amino acid substitutions are defective in forming fluorescent enzyme–DNA complexes (Fig. 2). The severity of the defect in forming fluorescent complexes correlates with the fidelity of DNA replication: mutant DNA polymerases that produce the largest reductions in DNA replication fidelity (Table 1) also have the most difficulty in forming fluorescent complexes (Fig. 2). These results are consistent with the reduced excision rates detected for the mutant DNA polymerases with double-stranded DNA substrates (Table 2). Thus, a defect in preparing the primer–terminus for the hydrolysis reaction can reduce proofreading activity in vivo as effectively as an alteration that prevents the hydrolysis reaction because these preparatory steps form a kinetic barrier to the hydrolysis reaction. Our results, combined with the crystallographic data (4, 30), suggest that the G255 loop plays a role in the initial strand separation required to transfer the primer–terminus from the polymerase active center to a pre-exonuclease complex and that subsequent stabilizing interactions between the primer–terminus and residues in the exonuclease active center rely on a region of the DNA polymerase that includes the D131 and Y317 residues (Fig. 4). The biphasic nature of the excision reaction (Table 2) suggests the possibility of two pre-exonuclease intermediates, and one or both may be fluorescent (Fig. 1). The fluorescence of pre-exonuclease complexes formed with 2AP-labeled DNA substrates, combined with mutational analysis, provides the means to gain additional insight into the mechanism by which the primer–terminus is prepared for the hydrolysis reaction.
Acknowledgments
We thank E. Elisseeva and L. Marquez for helpful discussions and J. Wang and T. Steitz for providing the structural data and Fig. 4. We also thank Y. Hsuanyu, R. Nonay, and Z. Ozum for technical assistance and B. Dunford and S. Dunn for the use of spectrofluorimeters. This work was supported by a Natural Sciences and Engineering Research Council of Canada Collaborative Research Program grant (to L.J.R.-K.) and by graduate fellowships (to R.P.B.) from the Natural Sciences and Engineering Research Council of Canada and the Alberta Heritage Foundation for Medical Research. L.J.R.-K. is a Scientist of the Alberta Heritage Foundation for Medical Research.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: 2AP, 2-aminopurine; d2APMP, 2-aminopurine deoxynucleoside monophosphate.
References
- 1.Johnson K A. Annu Rev Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 2.Goodman M F, Creighton S, Bloom L B, Petruska J. Crit Rev Biochem Mol Biol. 1993;28:83–126. doi: 10.3109/10409239309086792. [DOI] [PubMed] [Google Scholar]
- 3.Cowart M C, Gibson K J, Allen D J, Benkovic S J. Biochemistry. 1989;28:1975–1983. doi: 10.1021/bi00431a004. [DOI] [PubMed] [Google Scholar]
- 4.Marquez L A, Reha-Krantz L J. J Biol Chem. 1996;271:28903–28911. doi: 10.1074/jbc.271.46.28903. [DOI] [PubMed] [Google Scholar]
- 5.Ollis D L, Brick P, Hamlin R, Xuong N G, Steitz T A. Nature (London) 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 6.Freemont P S, Friedman J M, Beese L S, Sanderson M R, Steitz T A. Proc Natl Acad Sci USA. 1988;85:8924–8928. doi: 10.1073/pnas.85.23.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beese L S, Steitz T A. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derbyshire V, Freemont P S, Sanderson M R, Beese L, Friedman J M, Joyce C M, Steitz T A. Science. 1988;240:199–201. doi: 10.1126/science.2832946. [DOI] [PubMed] [Google Scholar]
- 9.Derbyshire V, Grindley N D F, Joyce C M. EMBO J. 1991;10:17–24. doi: 10.1002/j.1460-2075.1991.tb07916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrison A, Bell J B, Kunkel T A, Sugino A. Proc Natl Acad Sci USA. 1991;88:9473–9477. doi: 10.1073/pnas.88.21.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanco L, Bernad A, Salas M. Gene. 1992;112:139–144. doi: 10.1016/0378-1119(92)90316-h. [DOI] [PubMed] [Google Scholar]
- 12.Joyce C M, Steitz T A. Annu Rev Biochem. 1994;63:777–822. doi: 10.1146/annurev.bi.63.070194.004021. [DOI] [PubMed] [Google Scholar]
- 13.Reha-Krantz L J, Stocki S, Nonay R L, Dimayuga E, Goodrich L D, Konigsberg W H, Spicer E K. Proc Natl Acad Sci USA. 1991;88:2417–2421. doi: 10.1073/pnas.88.6.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reha-Krantz L J, Nonay R L. J Biol Chem. 1993;268:27100–27108. [PubMed] [Google Scholar]
- 15.Frey M W, Nossal N G, Capson T L, Benkovic S J. Proc Natl Acad Sci USA. 1993;85:8924–8928. doi: 10.1073/pnas.90.7.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reha-Krantz L J, Liesner E M, Parmaksizoglu S, Stocki S. J Mol Biol. 1986;189:261–272. doi: 10.1016/0022-2836(86)90508-5. [DOI] [PubMed] [Google Scholar]
- 17.Reha-Krantz L J. J Mol Biol. 1988;202:711–724. doi: 10.1016/0022-2836(88)90552-9. [DOI] [PubMed] [Google Scholar]
- 18.Stocki S A, Nonay R L, Reha-Krantz L J. J Mol Biol. 1995;254:15–28. doi: 10.1006/jmbi.1995.0595. [DOI] [PubMed] [Google Scholar]
- 19.Bloom L B, Otto M R, Eritja R, Reha-Krantz L J, Goodman M F, Beechem J M. Biochemistry. 1994;33:7576–7586. doi: 10.1021/bi00190a010. [DOI] [PubMed] [Google Scholar]
- 20.Lin T-C, Rush J R, Spicer E K, Konigsberg W H. Proc Natl Acad Sci USA. 1987;84:7000–7004. doi: 10.1073/pnas.84.20.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunkel T A, Roberts J D, Zakour R A. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 22.Reha-Krantz L J, Nonay R L, Stocki S. J Virol. 1993;67:60–66. doi: 10.1128/jvi.67.1.60-66.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McPheeters D S, Christensen A, Young E T, Stormo G, Gold L. Nucleic Acids Res. 1986;14:5813–5826. doi: 10.1093/nar/14.14.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reha-Krantz L J. Mol Gen Genet. 1987;209:90–93. doi: 10.1007/BF00329841. [DOI] [PubMed] [Google Scholar]
- 25.Cameron V, Uhlenbeck O C. Biochemistry. 1977;16:5120–5126. doi: 10.1021/bi00642a027. [DOI] [PubMed] [Google Scholar]
- 26.Capson T L, Peliska J A, Kaboord B F, Frey M W, Lively C, Dahlberg M, Benkovic S J. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 27.Bessman M J, Reha-Krantz L J. J Mol Biol. 1977;116:115–123. doi: 10.1016/0022-2836(77)90122-x. [DOI] [PubMed] [Google Scholar]
- 28.Eritja R, Kaplan E, Mhaskar D, Sowers L C, Petruska J, Goodman M F. Nucleic Acids Res. 1986;14:5869–5884. doi: 10.1093/nar/14.14.5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carver T E, Jr, Hochstrasser R A, Millar D P. Proc Natl Acad Sci USA. 1994;91:10670–10674. doi: 10.1073/pnas.91.22.10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Yu P, Lin T C, Konigsberg W H, Steitz T A. Biochemistry. 1996;35:8110–8119. doi: 10.1021/bi960178r. [DOI] [PubMed] [Google Scholar]