

Abstract
The implantation process is complex, requiring reciprocal interactions between implantation-competent blastocysts and the receptive uterus. Because microRNAs (miRNAs) have major roles in regulating gene expression, we speculated that they participate in directing the highly regulated spatiotemporally expressed genetic network during implantation. Here, we show that two miRNAs, mmu-miR-101a and mmu-miR-199a*, are spatiotemporally expressed in the mouse uterus during implantation coincident with expression of cyclooxygenase-2, a gene critical for implantation. More interestingly, our in vitro gain- and loss-of-function experiments show that cyclooxygenase-2 expression is posttranscriptionally regulated by these two miRNAs. We report on miRNA-mediated regulation of uterine gene expression in the context of implantation. We believe that many other critical genes related to this process are also regulated by miRNAs. Thus, elucidating the physiological roles of uterine miRNAs will help us better understand the genetic control of implantation, the gateway to a successful pregnancy.
Keywords: mouse, uterus, blastocyst, decidua
MicroRNAs (miRNAs) are a novel family of small (≈19–22 nt) noncoding RNAs transcribed by genomes of most metazoa. They are diverse in sequence, evolutionary widespread, and involved in sequence-specific posttranscriptional gene regulation by affecting mRNA stability and/or translation (1–3). In mammals, mature miRNAs are generated via a unique biosynthetic cascade involving RNA polymerase II, nuclear and cytosolic RNase III endonucleases, and their dsRNA binding protein partners. miRNA-guided RNA silencing is executed by a RNA-induced silencing complex (RISC) that involves an interaction between a mature miRNA and its binding site located at the 3′ UTR of the target mRNA (1–3). miRNA-mediated gene regulation is classified as “fine-tuning of targets” that provides weak regulation and “switch targets” that confers dramatic down-regulation of gene expression, depending on sequence complementarities between miRNA and its target miRNA responsive element (MRE) and the total number of MREs in a given 3′ UTR (4). The majority of mammalian miRNAs are spatiotemporally regulated and influence a variety of biological processes, including development, tissue morphogenesis, maintenance of tissue identity, cell growth, differentiation, apoptosis, and metabolism (3, 5). Computational and experimental approaches predict that the human genome encodes as many as 1,000 miRNAs, regulating ≈30% of protein-coding genes (6–9). This prediction suggests that potentially all cellular pathways are governed by miRNAs. Emerging evidence also suggests miRNAs' roles in major diseases, including cancer and viral infections (10, 11). Recent reports of profound phenotypic abnormalities in miRNA-knockout mouse models further document their crucial roles as regulators of gene expression (12–15).
The implantation process by which a blastocyst establishes first intimate physical and physiological contact with the uterus is one of the most critical steps in mammalian reproduction and involves a host of endocrine, paracrine, autocrine, and juxtacrine modulators (16). Failure to achieve on-time implantation results in poor pregnancy outcomes and spontaneous pregnancy losses in mice and humans (17–19). Uterine sensitivity to implantation in mice is classified as prereceptive, receptive, and refractory phases (20, 21). Transition from one phase to another is governed by ovarian estrogen and progesterone (P4) as master regulators in collaboration with various lipid mediators, cytokines, growth factors, morphogens, and other signaling molecules (20). The human uterus undergoes similar phases of uterine sensitivity to implantation, and many molecules regulating implantation in mice are also important for human implantation (16).
The embryo–uterine dialogue during implantation is comprised of complex molecular events involving intricate changes in uterine gene expression. For example, cyclooxygenase-2 (Cox-2), critical for implantation in mice (22), is expressed in the uterine luminal epithelium (LE) on day 1 of pregnancy. It is low to undetectable in the uterus on day-4 morning, but exclusively expressed in the LE and subepithelial stromal cells surrounding the blastocyst at the antimesometrial pole on day-4 midnight with blastocyst attachment, persisting through day-5 morning (23). Interestingly, from day 6 onward, Cox-2 expression switches to the mesometrial pole, the presumptive site of placentation (23). Although Cox-2-derived prostaglandins (PGs) are critical to implantation, the underlying mechanism regulating Cox-2's restricted, yet coordinated, expression patterns remains unknown (16). Because miRNAs function as gene regulatory molecules in developmental processes, we speculated that they also impact the implantation process, perhaps by regulating Cox-2. Indeed, a recent report (24) of miRNA expression profiling in human tissues describes that the female reproductive axis (cervix, uterus, and ovary) has the highest levels of specific miRNAs, second to the brain.
Here, we provide evidence that miRNAs potentially regulate gene expression during implantation. Microarray profiling identified preferentially up-regulated miRNAs in the receptive uterus before implantation. Computational algorithms predict Cox-2 as one of the potential targets of two miRNAs, mmu-miR-199a* and mmu-miR-101a. We found that these miRNAs share striking similarity to Cox-2 mRNA in their spatiotemporal expression patterns in periimplantation mouse uteri. In vitro functional assays confirm their roles in controlling Cox-2 protein levels. Considering the absolute requirement of Cox-2-derived PGs in implantation and decidualization (22, 25), our results show potential roles of uterine miRNAs in implantation.
Results and Discussion
The Preimplantation Mouse Uterus Expresses miRNAs.
We hypothesized that miRNAs, known to cause dramatic and subtle changes in gene expression (1–3), are involved in regulating gene expression associated with changes in uterine sensitivity to implantation. To test this hypothesis, we used microarray analysis to identify miRNAs expressed in the preimplantation mouse uterus, especially during the receptive phase when the uterus is permissive to blastocyst implantation (21). We performed expression profiling with oligonucleotide microarrays and compared the expression patterns of ≈380 miRNAs, common to humans, mice, and rats, in day-1 (prereceptive; estrogen dominance) and day-4 (receptive; P4 dominance) pregnant mouse uteri (day 1 = vaginal plug). Each day-1 uterine RNA sample was labeled with cy5 and cy3 dyes separately and hybridized twice with each differently labeled day-4 uterine RNA sample (see Experimental Procedures). Fig 1A represents the combined results from 10 different hybridizations. We found that 32 miRNAs were significantly up-regulated during the receptive phase [supporting information (SI) Table 1]; some of these miRNAs are known to be present at high levels in the human uterus (24). Noticeably, only five miRNAs were expressed at higher levels in day-1 pregnant uteri (SI Table 2). Altogether, these results suggest that miRNAs participate in regulating dynamic changes in uterine gene expression patterns that occur during the transition from the prereceptive to the receptive phase.
Fig. 1.

miRNA biogenesis in periimplantation mouse uteri. (A) miRNA profiling with oligonucleotide microarray analysis of day-1 (D1) and day-4 (D4) pregnant uteri. Yellow spots along the diagonal line represent miRNAs with similar expression levels between days 1 and 4. Orange spots indicate up-regulated miRNAs in prereceptive uteri (day 1), and blue spots indicate up-regulated miRNAs in receptive uteri (day 4). (B) RT-PCR of Dicer1 and Ago1–4 in periimplantation uteri. β-Actin serves as a control. (C) Western blotting of Dicer1 protein. Mouse ovary, positive controls; Actin, loading control. IS, implantation site; IIS, interimplantation site.
Biogenesis of miRNAs Occurs in the Mouse Uterus.
Having identified up-regulated miRNAs in the receptive uterus, our next objective was to confirm that machinery for miRNA biogenesis is operative in periimplantation uteri. We focused on Dicer and Argonaute (Ago) proteins because they are the key components of the miRNA biosynthesis pathway. Dicer is an RNase III endonuclease that converts pre-miRNA to mature miRNA after its transport from the nucleus to the cytosol. Embryonic lethality of Dicer1 null mice confirms its key role during development (26, 27). Conditional inactivation of Dicer1 also shows its roles in stem cell proliferation and differentiation, maintenance of centromeric heterochromatin structure, centromeric silencing, epithelial morphogenesis, and T cell development (28–31). After Dicer-mediated maturation, miRNAs are loaded into the RISC that directs posttranscriptional silencing of target mRNAs (5). Ago proteins are major components of the RISC. Mammals have four closely related Ago proteins, Ago1–Ago4, all of which associate with miRNA; however, only Ago2 is absolutely necessary for embryo development and mediating miRNA-directed target degradation (32, 33). Periimplantation mouse uteri express Dicer1 and Ago1–4 mRNAs (Fig. 1B), and Dicer1 protein is also detected in uteri on day-1, -4, and -5 implantation sites (Fig. 1C). Collectively, the results provide evidence for biogenesis of miRNAs in periimplantation mouse uteri.
Spatiotemporal Expression of mmu-miR-199a* and mmu-miR-101a Closely Matches That of Cox-2 mRNA in Periimplantation Uteri.
The uterus is comprised of distinct cellular compartments, each of which contributes to pregnancy success (21, 34). For example, the luminal epithelium serves as a defensive barrier and is the site of blastocyst attachment, whereas the glandular epithelium secretes factors that support implantation and embryo development. Stromal cells at the site of implanting blastocysts undergo decidualization to provide nutrition to the developing embryo, protect it from maternal immune responses, and regulate trophoblast invasion into the endometrium. Because these various uterine cell types produce distinct sets of gene products that act in concert to allow implantation and decidualization (34), determining the spatiotemporal distribution of miRNAs in the periimplantation uterus is one way of identifying their physiological targets. To select specific miRNAs from 32 potential miRNA candidates relevant to implantation for further studies, we used a publicly accessible miRNA target prediction program, miRGen (35), and found that three miRNAs, hsa-miR-101, hsa-miR-144, and hsa-miR-199a*, are predicted to target Cox-2.
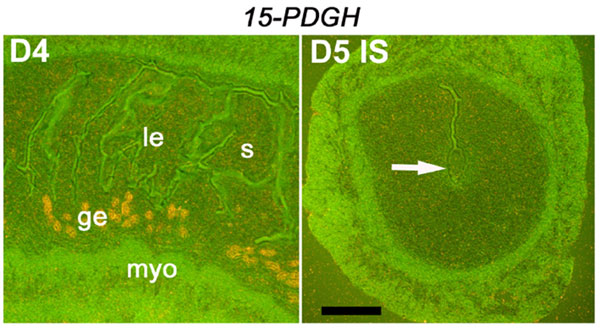
While Cox-2 is critical for implantation (17, 19, 22), Cox-2-derived PGs are also well known for their abilities to promote inflammation and tumorigenesis (36). Because implantation events are highly coordinated, it is conceivable that levels of Cox-2-derived PGs at the site of action are tightly regulated to avoid unregulated growth and inflammation. The question remains how uterine Cox-2 expression is regulated during implantation. Unlike Cox-1, mouse uterine Cox-2 mRNA and protein levels are not regulated by ovarian P4 and estrogen (23). Additionally, uterine expression of 15-hydroxy-PG dehydrogenase gene, which encodes an enzyme that inactivates Cox-2-derived prostaglandin E2 is very low to undetectable at the site of implantation in mice (SI Fig 7). This finding suggests alternative mechanisms for Cox-2 regulation. The fact that Cox-2 is a common predicted target for miRNAs hsa-miR-101, hsa-miR-144, and hsa-miR-199a* led us to speculate that these miRNAs participate in posttranscriptional regulation of uterine Cox-2.
One way to test this hypothesis is to determine whether distribution of these miRNAs correlates with that of Cox-2 mRNA in the uterus. A positive correlation would indicate that Cox-2 messages are accessible for miRNA-mediated posttranscriptional control. Consistent with microarray results, Northern hybridization results show that levels of these miRNAs are higher in day-4 than day-1 pregnant uteri (Figs. 2A and 3A and SI Fig. 8). However, the major uterine cell types are comprised of epithelial cells (≈5–10%), stromal cells (≈30–35%), and myometrial cells (≈60–65%) (37). Thus, RNA or protein analysis in whole uterine extracts provides limited information and gives no indication of which cell types are responding, because of the dilution effect resulting from these heterogeneous cell populations. Thus, we used in situ hybridization to examine the spatiotemporal expression of miR-101a and miR-199a* in periimplantation uteri; miR-144 was not considered for this study because of its low abundance in the uterus (SI Fig. 8).
Fig. 2.

Mature miR-199a* is expressed in periimplantation mouse uteri. (A) Northern hybridization with 32P-labeled probe in total uterine RNA samples on day 1 (D1) and day 4 (D4) of pregnancy. The ethidium bromide-stained gel served as a loading control. (B) In situ hybridization of uterine cross-sections with DIG-labeled LNA probe on days 1, 4, 5, and 6 of pregnancy. Arrows indicate locations of blastocysts. le, luminal epithelium; ge, glandular epithelium; s, stroma; myo, myometrium; M, mesometrial pole; AM, antimesometrial pole; IS, implantation site. (Scale bar: 400 μm.)
Fig. 3.

Mature miR-101a is expressed in periimplantation mouse uteri. (A) Northern hybridization of total uterine RNA samples from day-1 (D1) and day-4 (D4) pregnant uteri. The ethidium bromide-stained gel served as a loading control. (B) In situ hybridization of uterine cross-sections with DIG-labeled LNA probe on days 1, 4, 5, and 6 of pregnancy. Arrows indicate the location of blastocysts. Abbreviations are as in Fig. 2. (Scale bar: 400 μm.)
Both miR-101 and miR-199a* are conserved between human and mouse with the murine homolog of hsa-miR-101 designated as mmu-miR-101a (38, 39). In situ hybridization of these miRNAs was performed with digoxigenin (DIG)-labeled locked nucleic acid (LNA)-modified probes. Localization of these miRNAs on days 1, 5, and 6 of pregnancy (Figs. 2B, 3B, and 4A) overlaps with that of Cox-2 mRNA (23). In day-4 pregnant uteri when Cox-2 is undetectable (23), both of these miRNAs do not show any localized expression; instead they are present at basal levels throughout the uterus (Figs. 2B and 3B). Furthermore, the spatial expression pattern of both miRNAs on days 7 and 8 of pregnancy is similar to that of Cox-2, primarily expressed at the mesometrial pole of the implantation site (data not shown). Hybridization signals for these miRNAs are specific, because a brain-specific miRNA miR-124a (5) did not show any positive signals in the uterus on days 1 and 5 of pregnancy (SI Fig 9).
Fig. 4.

Uterine expression of miR-101a, miR-199a*, and Cox-2. (A) In situ hybridization of Cox-2 with 35S-labeled probe (Left) and protein by immunohistochemistry (Right) in serial cross-sections from day-5 implantation sites (IS). (B) In situ hybridization of miR-199a* with DIG-labeled LNA probe on longitudinal sections from delayed and implanting uteri. Arrows indicate the location of blastocysts. (C) Uterine Cox-2 and miR-101a levels during experimentally induced decidualization. Western blot shows Cox-2 protein levels at time points after intrauterine oil infusion (+); noninfused contralateral uterine horns serve as negative controls (−). Actin is a loading control. The ethidium bromide-stained gels served as loading control for miR-101a Northern blot. (Scale bars: 200 μm, A; 400 μm, B.)
Because miR-101a and miR-199a* expression overlaps with Cox-2 in the pregnant mouse uterus, we assumed that Cox-2 mRNA expression is posttranscriptionally regulated by these miRNAs. If so, it is expected that Cox-2 protein levels will be lower than Cox-2 mRNA. To address this, we performed in situ hybridization and immunohistochemistry on serial sections of day-5 implantation sites. Indeed, stromal cells positive for Cox-2 protein were far fewer than Cox-2 mRNA-positive cells (Fig 4A). However, it is also possible that these miRNAs have more complex functions during implantation, because one miRNA can target so many genes. The distribution of an mRNA detected by a radiolabeled probe and its protein by an antibody cannot be quantitatively compared. In this respect, we also did in situ hybridization to localize Cox-2 mRNA using a DIG-labeled probe, the same detection method for miRNA localization, and found a similar expression pattern (SI Fig. 10). Nonetheless, differential stability and degradation of Cox-2 mRNA and protein may also account for this observation.
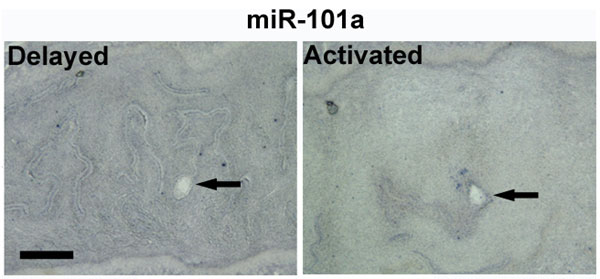
miRNA Expression Is Correlated with That of Cox-2 During Delayed Implantation.
In mice, ovariectomy on day-4 morning before preimplantation ovarian estrogen secretion results in delayed implantation with blastocysts undergoing dormancy. This condition can be maintained for days with continued P4 treatment. However, a single injection of estradiol-17β (E2) results in blastocyst activation with the initiation of implantation (40). We used this model to determine whether miR-101a and miR-199a* expression in the P4-primed delayed uterus is different from the activated (P4+E2) uterus showing implantation. In situ hybridization results show that miR-199a* is undetectable in the uterus around the dormant blastocyst, but is expressed in stromal cells surrounding the activated blastocyst undergoing implantation 24 h post-E2 injection (Fig. 4B). A similar spatial distribution pattern was detected for miR-101a, but at a much lower level (SI Fig. 11). This expression pattern is very similar to that of Cox-2 mRNA, which is induced in the presence of active but not dormant blastocysts (23). Again, a marked similarity between localization of these miRNAs and Cox-2 mRNA strengthens our contention that these small RNAs are positioned to regulate Cox-2 levels in the uterus.
Uterine Cox-2 Protein Levels Are Inversely Correlated with miR-101a Expression During Decidualization.
Concomitant to implantation, uterine stromal cells surrounding the blastocyst proliferate and differentiate to form the deciduum, which supports the developing embryo before the formation of a functional placenta (21). In mice, decidualization can be experimentally induced by intraluminal oil infusion into a receptive uterus (41). Like the initiation of implantation, Cox-2 is also critical for decidualization and is induced as an immediate-early gene after oil infusion (22). We performed a time course experiment to determine how Cox-2 levels are affected after initiation of decidualization in day-4 pseudopregnant uteri. As shown in Fig. 4C, Cox-2 protein is detected within 2 h of oil infusion with further increases at 4 h, with levels then declining over time. As expected, the noninfused uterine horn did not express detectable Cox-2 protein (Fig. 4C). To examine whether Cox-2 expression during decidualization is under the control of miR-199a* and/or miR-101a, we determined the expression pattern of these miRNAs in oil-infused and control uterine horns at the same time points. Interestingly, unlike Cox-2, both miRNAs are present in the uterus in the absence of any decidualization stimulus. More importantly, at each time point after oil infusion, an inverse relationship between Cox-2 protein and miR-101a expression levels was noted (Fig. 4C), reinforcing that this miRNA regulates Cox-2 during decidualization. No obvious correlation between Cox-2 protein and miR-199a* expression levels was detected (data not shown). Further investigation will be needed to sort out whether differential posttranscriptional regulation of Cox-2 by these two miRNAs that bind to overlapping sequences in the Cox-2 3′ UTR depends on either the experimental conditions (in vivo vs. in vitro) and cell types involved or because of their interactions with different proteins/other molecules after association of Ago in the RISC–miRNA complexes (1).
miR-101a and miR-199a* Posttranscriptionally Regulate Cox-2.
Depending on the extent of sequence complementarity between a miRNA and its target gene, miRNA-guided posttranscriptional regulation of gene expression follows two distinct mechanisms: target mRNA cleavage and translational repression. For animal miRNAs, the sequence complementarities with their targets are usually restricted to the 5′ region of a miRNA, termed “seed region” (8, 9, 42, 43). The absence of perfect complementarities between animal miRNAs and their targets leads to translational repression rather than mRNA cleavage (44). Because of the short length of the seed region, an animal miRNA is predicted to target at least 200 genes on average (43, 45). As shown in Fig 5, both miR-101 and miR-199a* bind to a single overlapping MRE. The seed sequences (nucleotides 2–8) and the 3′segment of both miRNAs that are predicted to anneal with Cox-2 mRNA residues are highly conserved from rats to humans. Thermodynamic analysis by RNAhybrid (46) indicates that both are strong sites. Interestingly, the loop structure between the annealed segments of the miRNAs is uniquely exaggerated in primate Cox-2 mRNA by the insertion of 8 nt. This exaggerated loop perhaps plays a role in primate-specific regulation of miR-101 and miR-199a* activity.
Fig. 5.

Cross-species homology of miRNAs targeting COX-2 mRNA. (A and B) The structure and homology of the predicted interactions between Cox-2 mRNA and miR-101 (A) and miR-199a* (B). (C) The predicted annealing sites of the seed and 3′ terminus of these miRNAs are well conserved in Cox-2 mRNA.
Because high-throughput experimental systems for validation of predicted targets are not yet available, major approaches to validate miRNA targets use in vitro gain-of-function and loss-of-function analyses (47). We used two independent gain-of-function and a loss-of-function strategy to show that miR-101a and miR-199a* repress Cox-2 translation. First, a 200-bp region from the mCox-2 3′ UTR containing the putative MRE for miR-101 and miR-199a* was fused downstream of a luciferase (Luc) reporter gene (Fig. 6A). Upon cotransfection of this reporter construct with different concentrations of miR-101a or miR-199a* in HEK293 cells, Luc activity decreased in a concentration-dependent manner, whereas a control (scrambled) miRNA did not show any effect (Fig. 6B). Because transfection of 2′-O-methyl oligoribonucleotides antisense to miRNAs of interest inhibits their functions in cultured cells (48, 49), we used a similar approach to reverse the effects of miR-101a and miR-199a* on mCox-2-3′ UTR-Luc activity. Indeed, miRNA-mediated suppression of Luc activity was reversed up to 50% for miR-101a and 100% for miR-199a* after addition of 5-fold excess of antisense constructs for each miRNA, but not by a control oligonucleotide (Fig. 6C). These results show that both miR-101a and miR-199a* interact with the 3′ UTR of mCox-2 mRNA in vitro, leading to its translational repression.
Fig. 6.

miR-199a* and miR-101a regulate Cox-2 translation. (A) Renilla Luc-mCox-2-3′ UTR reporter construct. (B) Concentration-dependent decreases in reporter activity with increasing doses of precursor miR-199a* or miR-101a in HEK293 cells. A scrambled miRNA had no effect on reporter activity (control). (C) Antisense inhibitors of miR-199a* and miR-101a relieve miRNA-mediated repression of Cox-2-3′ UTR-Luc reporter activity; scrambled antisense sequence (control) had no effect. Each transfection was performed in triplicate. Data are presented as relative Luc activity (mean ± SEM). (D) miR-199a* and miR-101a repress endogenous Cox-2 translation. Concentration-dependent decreases in endogenous Cox-2 protein levels in HeLa cells 72 h after transfection with miR-199a* or miR-101a precursors. Western blotting of Cox-2 (Upper) and corresponding densitometry analyses (Lower) are shown. Actin, loading control.
Because of the lack of a mouse uterine cell line that spontaneously expresses Cox-2 protein, we overexpressed miR-101a and miR-199a* in Cox-2-expressing HeLa cells, originally derived from human cervical carcinoma, and examined whether overexpression of these miRNAs reduces endogenous Cox-2 levels. Indeed, these miRNAs caused dose-dependent decreases in Cox-2 protein levels within 72 h of transfection (Fig. 6D). This result provides further evidence that Cox-2 mRNA is posttranscriptionally controlled by miR-101a and miR-199a*. We found another interesting inverse relationship between Cox-2 protein levels and miR-199a* in two different human cancer cell lines. The colon cancer cell line HCA7 with high Cox-2 protein levels does not express miR-199a*, whereas a uterine cancer cell line AN3CA expresses significant levels of miR-199a*, but no detectable Cox-2 protein (SI Fig. 12). In conclusion, the present investigation provides evidence that miRNAs are potential regulators of Cox-2 expression in the mouse uterus during implantation. Considering the dynamic nature of gene expression that occurs during implantation, other miRNAs are likely to target other genes expressed during this period.
The process of implantation is a powerful system for studying various physiological processes including epithelial–epithelial interaction, epithelial–mesenchymal interaction, cell migration and invasion, vascular permeability and angiogenesis, many of which are dysregulated under pathological conditions, including tumorigenesis (16, 20). Therefore, understanding how miRNAs control implantation events will not only help us to uncover the underlying causes of infertility and gynecological complications, but also the molecular events associated with carcinogenesis. However, developing uterine-specific conditional null mouse models for these miRNAs will be required to establish their definitive roles in uterine biology and implantation.
Experimental Procedures
Animals and Tissue Collection.
Mice were housed at the Vanderbilt University Medical Center Animal Care Facility under the National Institutes of Health and Institutional guidelines for the care and use of laboratory animals. Adult CD-1 females (Charles River, Wilmington, MA) were mated with fertile or vasectomized males to induce pregnancy or pseudopregnancy, respectively. Uteri on days 1 and 4 (day 1 = vaginal plug) were collected for microarray analysis. Implantation sites on mornings of days 5 and 6 were visualized by the blue dye method (50). To experimentally induce decidualization, one uterine horn was infused intraluminally with 25 μl of sesame oil on day 4 of pseudopregnancy; noninfused contralateral horns served as control. The delayed implantation mouse model has been described (23).
Microarray Analysis.
Total RNA was isolated with TRIZOL reagent (Invitrogen, Carlsbad, CA) from day-1 and -4 pregnant uteri pooled from five mice each. Samples were labeled and hybridized at the Specialized Cooperative Centers Program in Reproduction and Infertility Research Array Center (Seattle, WA) with miRNA 400 probe Oligo Set (Ambion, Austin, TX). To eliminate dye bias two slides were hybridized by the dye flip method for each day 1 versus day 4 comparison. Data were analyzed and normalized with GeneSpring 7.2 software (Agilent Technologies, Santa Clara, CA).
RT-PCR.
Total RNA extracted and treated with DNase I was reverse-transcribed and PCR-amplified (LA Taq polymerase; TaKaRa Bio USA, Madison, WI) using mouse-specific primers.
Northern Hybridization for miRNAs.
Total RNA (25–40 μg) was resolved through 12.5% urea-polyacrylamide gel under denaturing conditions, transferred onto GeneScreen Plus membranes (PerkinElmer, Waltham, MA), and UV-cross-linked. Radiolabeling of miRNA antisense oligonucleotides (SI Table 3) was performed with a StarFire labeling kit (IDT, Coralville, IA) according to the manufacturer's instructions. Prehybridization, hybridization, and washing of membranes were performed as described (51).
In Situ Hybridization of mRNAs.
Hybridization using 35S-labeled cRNA probes was performed as described (52).
In Situ Hybridization of miRNAs with DIG-Labeled Probes.
The protocol is described in SI Text.
Luc Reporter Constructs.
A 200-nt region containing the binding site for miR-199a* and miR-101a from the 3′UTR of mouse Cox-2 mRNA (mCox-2) was PCR-amplified with primers shown (SI Table 4). The PCR product was cloned 3′ to the stop codon of the Renilla Luc reporter in pRL-SV40 vector (Promega, Madison, WI).
Cell Culture and Transfection.
HEK293 and HeLa cells (ATCC, Manassas, VA) were maintained in DMEM (Invitrogen) supplemented with 10% heat-inactivated FBS (Sigma–Aldrich, St. Louis, MO) at 37°C under 5% CO2 in air. HEK293 cells were seeded at 6 × 104 cells per well in 24-well plates and transfected with 125 ng of mCox-2-3′ UTR-Luc construct and 25 ng pGL3 control vector (Promega) as internal normalization control in the presence and absence of pre-miRNA precursors (Ambion) with Lipofectamine 2000 reagent (Invitrogen). For in vitro knockdown of miRNA, cells were cotransfected with mCox-2-3′ UTR-Luc construct, miRNA precursors (2.5 pmol, miR-101a; 10 pmol, miR-199a*) and 5-fold molar excess of antisense inhibitors. Cells were harvested 30 h after transfection, and cell lysates were used for dual Luc reporter assays (Promega). HeLa cells were plated at 3 × 104 cells per well in 24-well plates and transfected with miRNA precursors by using Lipofectamine RNAiMax (Invitrogen). Cell lysates were collected 72 h after transfection.
Western Blotting.
Western blotting was performed as described (53). Antibodies are actin (sc-1615; Santa Cruz Biotechnology, Santa Cruz, CA); Dicer1 (ab13502; Abcam, Cambridge, MA); Cox-2 (160112 and 160126; Cayman, Ann Arbor, MI); and HRP-conjugated donkey anti-rabbit, anti-mouse, and anti-goat antibodies (Jackson ImmunoResearch, West Grove, PA). An ECL kit (GE Healthcare, Piscataway, NJ) was used for signal detection.
Immunohistochemistry.
Immunolocalization of Cox-2 in frozen uterine sections has been described (23, 52).
Supplementary Material
Acknowledgments
We thank James Patton (Vanderbilt) for providing DIG-labeled LNA probe for miR-124a and Jiyoung Hong for help in tissue sectioning. This work was supported by National Institutes of Health Grants R37 HD12304 and DA06668 (to S.K.D.) and DA022226 (to H.F.). S.T. is a recipient of National Institutes of Health National Research Service Award Predoctoral Fellowship F31 DA021062. T.D. is a recipient of the Gynecologic Cancer Foundation Awards/Ann Schreiber Ovarian Cancer Research Grant.
Abbreviations
- miRNA
microRNA
- RISC
RNA-induced silencing complex
- MRE
miRNA responsive element
- P4
progesterone
- Cox-2
cyclooxygenase-2
- PG
prostaglandin
- DIG
digoxigenin
- LNA
locked nucleic acid
- Luc
luciferase.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705917104/DC1.
References
- 1.Nilsen TW. Trends Genet. 2007;23:243–249. doi: 10.1016/j.tig.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Du T, Zamore PD. Development (Cambridge, UK) 2005;132:4645–4652. doi: 10.1242/dev.02070. [DOI] [PubMed] [Google Scholar]
- 3.Quellet DL, Perron MP, Gobeil L-A, Plante P, Provost P. J Biomed Biotechnol. 2006 doi: 10.1155/JBB/2006/69616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Song L, Tuan RS. Birth Defects Res. 2006;78:140–149. doi: 10.1002/bdrc.20070. [DOI] [PubMed] [Google Scholar]
- 6.Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 7.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 8.Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindbald-Toh K, Lander ES, Kellis M. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis BP, Burge CB, Bartel DP. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 10.Esau CC, Monia BP. Adv Drug Delivery Rev. 2007;59:101–114. doi: 10.1016/j.addr.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Esquela-Kerscher A, Slack FJ. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 12.Van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, et al. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, et al. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Dey SK. Nat Rev Genet. 2006;7:185–199. doi: 10.1038/nrg1808. [DOI] [PubMed] [Google Scholar]
- 17.Song H, Lim H, Paria BC, Matsumoto H, Swift LL, Morrow J, Bonventre JV, Dey SK. Development (Cambridge, UK) 2002;129:2879–2889. doi: 10.1242/dev.129.12.2879. [DOI] [PubMed] [Google Scholar]
- 18.Wilcox AJ, Baird DD, Weinberg CR. N Engl J Med. 1999;340:1796–1799. doi: 10.1056/NEJM199906103402304. [DOI] [PubMed] [Google Scholar]
- 19.Ye X, Hama K, Contos JJA, Anliker B, Inoue A, Skinner MK, Suzuki H, Amano T, Kennedy G, Arai H, et al. Nature. 2005;435:104–108. doi: 10.1038/nature03505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dey SK, Lim H, Das SK, Reese J, Paria BC, Daikoku T, Wang H. Endocr Rev. 2004;25:341–373. doi: 10.1210/er.2003-0020. [DOI] [PubMed] [Google Scholar]
- 21.Dey SK. In: Reproductive Endocrinology, Surgery, and Technology. Adashi EY, Rock JA, Rosenwaks Z, editors. New York: Lippincott–Raven; 1996. pp. 421–434. [Google Scholar]
- 22.Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Cell. 1997;91:197–208. doi: 10.1016/s0092-8674(00)80402-x. [DOI] [PubMed] [Google Scholar]
- 23.Chakraborty I, Das SK, Wang J, Dey SK. J Mol Endocrinol. 1996;16:107–122. doi: 10.1677/jme.0.0160107. [DOI] [PubMed] [Google Scholar]
- 24.Shingara J, Keiger K, Shelton J, Laosinchai-Wolf W, Powers P, Conrad R, Brown D, Labourier E. RNA. 2005;11:1461–1470. doi: 10.1261/rna.2610405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim H, Gupta RA, Ma W, Paria BC, Moller DE, Morrow JD, DuBois RN, Trzakos JM, Dey SK. Genes Dev. 1999;13:1561–1574. doi: 10.1101/gad.13.12.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang WJ, Yang DD, Na S, Sandusky GE, Zhang Q, Zhao G. J Biol Chem. 2005;280:9330–9335. doi: 10.1074/jbc.M413394200. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 28.Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ. Proc Natl Acad Sci USA. 2005;102:12135–12140. doi: 10.1073/pnas.0505479102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris KS, Zhang Z, MacManus MT, Harfe BD, Sun X. Proc Natl Acad Sci USA. 2006;103:2208–2213. doi: 10.1073/pnas.0510839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. J Exp Med. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson MJ, Song J, Hammond SM, Leemor J, Gregory HJ. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 33.Meister G, Landthaler M, Pathaniowska A, Dorsett Y, Teng G, Tuschl T. Mol Cell. 2004;15:185–197. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Sherwin R, Catalano R, Sharkey A. Reproduction. 2006;132:1–10. doi: 10.1530/rep.1.00355. [DOI] [PubMed] [Google Scholar]
- 35.Megraw M, Sethupathy P, Corda B, Hatzigeorgiou AG. Nucleic Acids Res. 2007;35:D149–D155. doi: 10.1093/nar/gkl904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams CS, Mann M, DuBois RN. Oncogene. 1999;18:7908–7916. doi: 10.1038/sj.onc.1203286. [DOI] [PubMed] [Google Scholar]
- 37.Finn CA, Porter DG. Reproductive Biology Handbook. Vol 1. London: Elek Science; 1975. pp. 18–26. [Google Scholar]
- 38.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griffiths-Jones S. Nucleic Acids Res. 2004;32:D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshinaga K, Adams CE. J Reprod Fertil. 1966;12:593–595. doi: 10.1530/jrf.0.0120593. [DOI] [PubMed] [Google Scholar]
- 41.Rankin JC, Ledford BE, Baggett B. Biol Reprod. 1977;17:549–554. doi: 10.1095/biolreprod17.4.549. [DOI] [PubMed] [Google Scholar]
- 42.Brennecke J, Stark A, Russell RB, Cohen SM. PLoS Biol. 2005;3:E85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 44.Olsen PH, Ambros V. Dev Biol. 1999;216:671–680. doi: 10.1006/dbio.1999.9523. [DOI] [PubMed] [Google Scholar]
- 45.Krek A, Grun D, Poly MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 46.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krützfeldt J, Poy MN, Stoffel M. Nat Genet Suppl. 2006;38:S14–S19. doi: 10.1038/ng1799. [DOI] [PubMed] [Google Scholar]
- 48.Hutvagner G, Simard MJ, Mello CC, Zamore PD. PLoS Biol. 2004;2 doi: 10.1371/journal.pbio.0020098. 0465–0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meister G, Landthaler M, Dorsett Y, Tuschl T. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paria BC, Huet-Hudson YM, Dey SK. Proc Natl Acad Sci USA. 1993;90:10159–10162. doi: 10.1073/pnas.90.21.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sempere LF, Dubrovsky EB, Dubrovskaya VA, Berger EM, Ambros V. Dev Biol. 2002;244:170–179. doi: 10.1006/dbio.2002.0594. [DOI] [PubMed] [Google Scholar]
- 52.Das SK, Wang X, Paria BC, Damm D, Abraham JA, Klagsbrun M, Andrews GK, Dey SK. Development (Cambridge, UK) 1994;120:1071–1083. doi: 10.1242/dev.120.5.1071. [DOI] [PubMed] [Google Scholar]
- 53.Daikoku T, Wang D, Tranguch S, Morrow JD, Orsulic S, DuBois RN, Dey SK. Cancer Res. 2005;65:3735–3744. doi: 10.1158/0008-5472.CAN-04-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}