

Abstract
Complement per se has been shown to play an important role in demyelinating disease but controversy remains regarding the role of C3 in the development and progression of experimental autoimmune encephalomyelitis (EAE), the animal model for multiple sclerosis. In this study we used C3-/- mice to confirm previous findings that C3 is required for full development of EAE. Furthermore, C3+/- mice (with serum C3 levels 50% that of wild type mice) developed EAE with a severity intermediate between wild type and C3-/- mice. Importantly transfer of wild type encephalitogenic T cells to C3-/- mice resulted in attenuated EAE. C3-/- mice with EAE had fewer CD4+ and CD8+ T cells in the CNS and 50% fewer of these cells produced IFN-γ compared to wild type mice. When treated with anti-CD3 antibody, CD4+ T cell from wild type and C3-/- mice had similar activation profiles as judged by IFN-γ production and CD25 and CD69 expression, indicating there is no gross or intrinsic defect in T cells from C3-/- mice. T cells from primed C3-/- mice proliferated comparably to that of control T cells on re-stimulation with MOG peptide. Our results confirm a requirement for C3 for maximal development of EAE and suggest that receptors for C3-derived activation fragments might be a viable therapeutic target for prevention and treatment demyelinating disease.
Keywords: complement, C3, EAE
1. Introduction
A causal role of complement in central nervous system (CNS) health and disease, particularly in the context of demeylinating conditions has become increasingly clear in the last few years (reviewed in Barnum and Szalai, 2006; Gasque et al., 2002; Lucchinetti et al., 2000; Stahel and Barnum, 2006; van Beek et al., 2003). Recent informative studies have demonstrated that C3 and C3-derived fragments (and their associated receptors) play a critical role in complement-mediated pathophysiology in experimental autoimmune encephalomyelitis (EAE), the animal model of the human demyelinating disease multiple sclerosis (MS). Thus deletion of C3, the C3a receptor, or the iC3b receptor (a.k.a. CR3 or Mac-1) attenuates the severity of EAE and reduces leukocyte infiltration into the CNS, while transgenic expression of C3a in the CNS markedly exacerbates EAE (Boos et al., 2004; Bullard et al., 2005; Nataf et al., 2000). In contrast deletion of C5, C5-derived fragments, and their associated receptors has no effect on the course of EAE (Reiman et al., 2002; Reiman et al., 2005; Weerth et al., 2003). Pharmacological inhibition of the C5a receptor also fails to lessen the severity of EAE (Morgan et al., 2004). All of the pertinent studies done to date indicate a clear dichotomy with respect to the role(s) in EAE of C3 and C5, the two proteins essential for propagating most complement-mediated biological functions, in EAE and possibly too in MS.
Despite the overall consensus of the findings regarding the role of complement in the development and progression of EAE, controversy still exists regarding the role of C3. Studies by Nataf and colleagues (Nataf et al., 2000) demonstrated that genetic deletion of C3 resulted in attenuated EAE symptoms, reduced leukocyte infiltration into the spinal cord and substantially less demyelination compared to littermate (C3 sufficient) controls. Shortly thereafter, in a report by Calida and colleagues (Calida et al., 2001) conflicting results were presented indicating that deletion of C3 had no effect on the course of EAE. The reasons for these opposing findings remain unclear. In the current study, we reaffirm our original observation that C3 is indeed critical to full development of EAE. We show here that EAE is attenuated in both C3-/- and C3+/- mice and importantly, that transfer of encephalitogenic T cells from control mice to C3-/- mice results in attenuated disease. We also show that T cells isolated from C3-/- mice have no intrinsic activation defects compared to T cells isolated from C3-sufficient controls, suggesting that the absence of C3 in the CNS during disease development accounts for the protective phenotype of C3-/- mice during EAE.
2. Materials and Methods
2.1. Mice
Mice carrying a null mutation for C3 were generated by gene targeting using 129/Sv/C57BL/6 embryonic stem cells as previously described (Circolo et al., 1999). The C3 mutation was then backcrossed onto the C57BL/6 strain for at least 10 generations (The Jackson Laboratory, Bar Harbor, ME). C57BL/6 lacking C3 and their complement-sufficient littermates (C3+/- and C3-/- genotypes) were used in all experiments. All studies were performed with approval from the UAB IACUC.
2.2. Induction of active and transferred EAE
For active EAE, mice were immunized with MOG peptide35-55 as described (Szalai et al., 2002). MOG peptide was synthesized by standard 9-fluorenyl-methoxycarbonyl chemistry and was >95% pure as determined by reversed phase-HPLC (Biosynthesis, Lewisville, TX). Onset and progression of EAE symptoms was monitored daily using a standard clinical scale ranging from 0 to 6 as follows: 0, asymptomatic; 1, loss of tail tone; 2, flaccid tail; 3, incomplete paralysis of one or two hind limbs; 4, complete hind limb paralysis; 5, moribund; 6, dead. Only mice with a score of at least 2 (flaccid tail) for more than 2 consecutive days were judged to have onset of EAE. For each animal a cumulative disease index (CDI) was calculated from the sum of the daily clinical scores observed between day 7 and day 30. For transferred EAE, spleens of control donors were removed two to three weeks following induction of active EAE, and prepared as previously described (Szalai et al., 2002). Passive EAE was induced by injecting ~5×106 purified T cells derived from wild-type mice into C3-/- mice. Purified T cells derived from wild-type mice were injected in wild-type mice as a control to monitor disease development.
2.3. Isolation and flow cytometric analysis of leukocytes from spinal cords
After perfusion with PBS, spinal cords were removed from control and C3-/- mice with active EAE (day 15 post disease induction). These were then disrupted through a cell strainer, and isolated cells were washed in HBSS, resuspended in 40% Percoll, and layered on 70% Percoll. After centrifugation at 2000 rpm (RT, 25 min.), cells at the interface were removed and washed in HBSS and stained as described. Cells obtained from spinal cords were incubated with anti-CD16/32 (24G2, FcR block) to prevent non-specific staining. Spinal cord leukocytes were stained with anti-CD4-FITC (GK1[CR1].5), antiCD-8-PE (53-6.7), anti-CD45-FITC (30F11), anti-TNF-α-PE (MP6-XT22) and anti-IFN-γ-FITC (XMG1.2), all from eBiosciences, San Diego, CA. Stained cells were analyzed by flow cytometry using a FACSCalibur and the data analyzed using CellQuest software (BD Biosciences, San Jose, CA).
2.4. T cell activation, proliferation and cytokine production
Single cell suspensions prepared from wild type and C3-/- lymph nodes were cultured in 96-well plates (2 × 105 cells/well) coated with anti-CD3 (5μg/ml, clone 145-2C11, eBiosciences). At 24 and 48 hrs, cells were collected and CD3+ T cells (anti-CD3-FITC, clone 145-2C11) were analyzed by flow cytometry for expression of CD25 (anti-CD25-PE, clone PC61.5) and CD69 (anti-CD69-Biotin-Sav-PerCP, clone H1.2F3, all from eBiosciences). IFN-γ production was determined by ELISA performed on culture supernatants collected at 24 hrs, according to the manufacturers instructions (eBiosciences). Surface marker expression by cultures of wild type and C3-/- cells are reported as the percent of wild type levels. The results of pooled data are presented.
Antigen-specific T cell proliferation assays were performed as previously described (Szalai et al., 2002). Single cell suspensions from spleens obtained 14 days after EAE induction were cultured in 96-well plates at 5 × 105 cells/well with increasing concentrations of MOG35-55 peptide in triplicate. After 48 h cultures were pulsed with 3H-thymidine for an additional 18 h and incorporation of thymidine was measured. The results of pooled data from four mice in each group are presented.
2.5. Statistics
Statistical significance of differences in progression of active, transferred EAE and T cell proliferation between control, C3-/- and C3+/- mice was calculated using the Wilcoxon signed rank test using Prism 4 (GraphPad Software, Inc., San Diego, CA).
3. Results and Discussion
We originally reported that deletion of C3 lead to markedly reduced clinical signs of EAE and reduced leukocyte infiltration into the spinal cords of C3-deficient mice compared to C3-sufficient controls (Nataf et al., 2000). In contrast, Calida and colleagues reported that deletion of C3 had essentially no effect on the course of EAE compared to control mice (Calida et al., 2001). Since the publication of both sets of data, a number of other studies from our group have demonstrated that receptors for C3-derived activation fragments, including the C3aR and CR3 (Mac-1; CD18/CD11b), are critical for the development of EAE (Boos et al., 2004; Bullard et al., 2005). These more recent findings prompted us to re-examine the role of C3 in EAE. As we previously reported, we show here that C3-/- mice have attenuated EAE compared to wild type littermates (Fig. 1, Table 1). C3-/- mice usually did not present with disease that progressed beyond tail weakness and the cumulative disease index (CDI) for C3-/- mice was significantly lower than that of wild type (16.7 vs. 56.7, p=0.0002, Wilcoxon signed rank test, see Table 1). Importantly these results confirm our previously reported findings (Nataf et al., 2000). To extend and reinforce these observations, we induced EAE in C3+/- mice to determine if the course of EAE would be altered in mice with lower than normal C3 levels. Serum C3 levels in the C3+/- mice we used were approximately 50% of wild type levels. We observed that the onset and initial course of EAE in C3+/- mice was similar to that of control mice (Fig. 1, Table 1). However the overall course of EAE was significantly reduced in C3+/- mice compared to controls (CDI: 33.9 vs. 56.7, p=0.0003, Wilcoxon signed rank test), particularly in the chronic phase of disease. Progression of EAE in C3+/- mice was intermediate in intensity: less severe than in wild type but more severe than in C3-/- mice (Fig. 1, Table 1). These data indicate that not only is C3 is required for the development and progression of EAE, but that the requirement is C3 gene and C3 protein dose-dependent.
Figure 1.
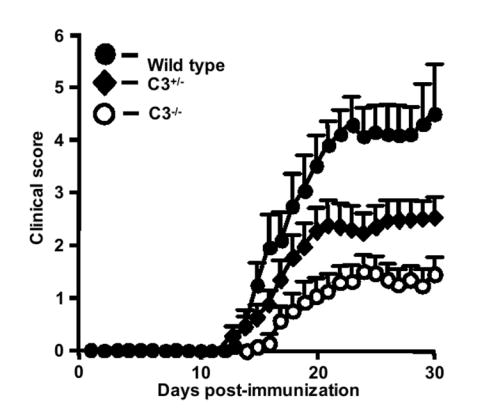
Clinical course of actively induced EAE is attenuated in C3+/- and C3-/- mice. EAE was induced with MOG35-55 peptide and signs of disease scored for 30 days as described in Materials and Methods. Results shown are the daily mean clinical scores (n ± SEM) for wild type (n=11), C3+/- (n=20) and C3-/- (n=16) from three pooled experiments.
Table 1.
Active and passive EAE phenotypes in wild-type (WT) mice versus C3-/- mice
| Strain or Transfer | CDIA | Disease
OnsetB |
Disease
IncidenceC |
|---|---|---|---|
| WT
n=11 |
56.7D | 17 | 100 |
| C3+/-
N=20 |
33.9 | 20.5 | 85 |
| C3-/-
n=16 |
16.7 | - | 75 |
|
| |||
| WT > WT
n=5 |
37.4E | 9 | 100 |
| WT > C3-/-
n=5 |
20.3 | 18 | 100 |
Cumulative Disease Index (CDI) is the mean of the sum of daily clinical scores observed between days 7 and 30.
Disease onset is defined as the first day of two consecutive days with a clinical score of 2 or greater.
Disease incidence is the percent of mice that displayed any clinical signs of disease.
WT vs. C3-/-, p=0.0002; WT vs. C3+/-, p=0.0003, Wilcoxon signed rank test.
WT>WT vs. WT > C3-/-, p=0.0005, Wilcoxon signed rank test.
We also induced EAE passively by adoptive transfer of MOG-sensitized wild type T cells into wild type and C3-/- recipients. In these experiments, we observed delayed onset and significantly attenuated disease in C3-/- recipients compared to wild type recipients (Fig. 2, Table 1, p=0.0005, Wilcoxon signed rank test). These data demonstrate that encephalitogenic T cells require a C3-sufficient environment in order for disease to develop fully. One mechanism that might lead to reduced severity of actively and passively induced EAE in C3-/- mice is reduced leukocyte infiltration into the CNS. In previous studies we observed reduced leukocyte infiltration in C3-/- mice, however those results were based on semi-quantitative analysis of a limited number of spinal cord sections (Nataf et al., 2000). To overcome the limitations of that approach, we performed flow cytometry on single cell suspensions derived from the spinal cords of wild type and C3-/- mice with active EAE (day 15 post-immunization) and analyzed them for expression of CD45, CD4 and CD8. Using these markers, we found that the total leukocyte infiltrate into the spinal cords of C3-/- mice was not significantly different from that observed in controls mice (data not shown). Notably however, spinal cord infiltrates from C3-/- mice had markedly reduced numbers of CD4+ and CD8+ T cells compared to wild type mice (Fig. 3A). These data combined with previous studies demonstrating an important role for C3a and C3aR in EAE (Boos et al., 2004) suggests that C3a, directly or indirectly, fosters T cell CNS infiltration in EAE. In addition, we observed 50% fewer IFN-γ producing CD4+ T cells in C3-/- mice. In contrast, TNF-α production by CD4+ T cells from C3-/- mice was elevated compared to T cells from wild type mice (Fig. 3B). Similarly, we observed fewer IFN-γ producing, but more TNF-α producing, CD8+ T cells in C3-/- mice (Fig. 3C). CD8+ wild type T cells produced five-fold more IFN-γ than in C3-/- mice (data not shown). To determine if the reduced frequency of IFN-γ producing T cells from C3-/- mice was due to an intrinsic defect in T cell activation, we activated T cells in vitro by treatment with anti-CD3 antibody. T cells from both C3-sufficient and deficient genotypes expressed CD25 and CD69 at equal frequency as determined by flow cytometry (Fig. 4A). CD3-stimulated T cells from wild type and C3-/- mice produced similar amounts of IFN-γ (wild type: 195 ng/ml versus C3-/-: 176 ng/ml, pooled samples from three mice from each group). In addition, we performed antigen re-stimulation assays to determine if antigen-specific T cells responses were altered in C3-/- mice. Stimulation of MOG-sensitized T cells from wild type and C3-/- mice with various concentrations of MOG, revealed no overall significant difference in proliferation (Fig. 4B). These data demonstrate the absence of an intrinsic defect in T cell activation in C3-/- mice as a mechanism that might account for attenuated EAE in C3-/- mice.
Figure 2.
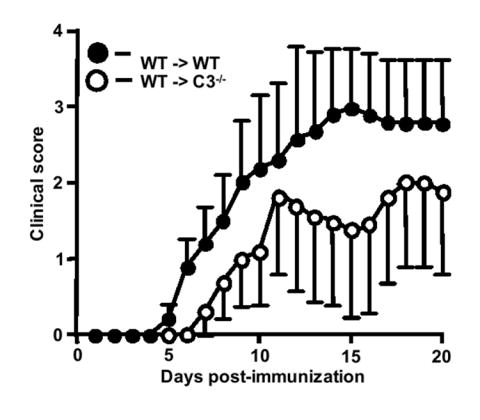
The clinical course of passively induced EAE is attenuated in C3-/- mice. EAE was induced in wild type (n=5) and C3-/- recipients (n=5) by injecting encephalitogenic T cells (~5 × 106) derived from wild-type donors with active EAE. Results shown are the daily mean clinical score (n ± SEM) from two separate experiments.
Figure 3.

Spinal cord infiltrate from C3-/- mice with EAE contains fewer CD4+, CD8+ and IFN-γ– and TNF-α-producing leukocytes. (A) Leukocytes isolated from spinal cords of control (n=8) and C3-/- mice (n=8) as described in Materials and Methods were immunostained for CD4 and CD8. The infiltration of both T cell subsets at day 15 after disease induction was markedly reduced in C3-/- mice compared to controls. The results shown are from a representative experiment using cells pooled within each group of mice. Leukocytes isolated from spinal cords of control (n=8) and C3-/- mice (n=8) as described in Materials and Methods were immunostained for CD4 (B) or CD8 (C) and IFN-γ and TNF-α at day 15 after disease induction. The results shown are from a representative experiment using cells pooled within each group of mice.
Figure 4.
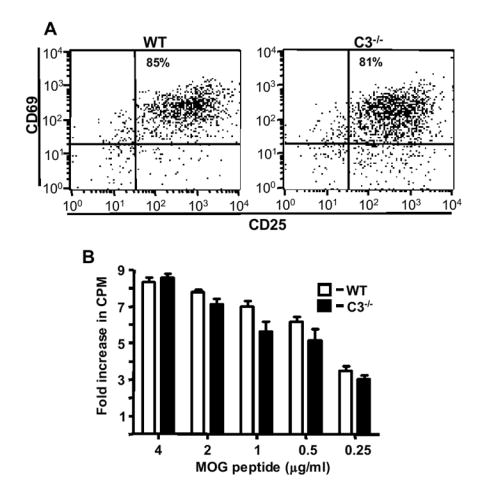
Expression of T cell activation markers CD25 and CD69, production of IFN-γ and T cell proliferation are similar between wild type and C3-/- T cells. (A) Lymph nodes from control and C3-/- mice were stimulated by plating on anti-CD3-coated wells for 48 hrs. Cells were immunostained for CD4, CD25 and CD69 as described in Materials and Methods. The results shown are from a representative experiment using cells from each group of mice. (B) Encephalitogenic T cells enriched by nylon-wool adherence from the spleens of wild type (n=4) or C3-/- mice (n=4) undergoing active EAE, or T cells from healthy controls (naïve cells), were co-cultured with irradiated splenic APCs plus MOG peptide (0.125-4 μg/ml). The cells were pulsed with 3H-thymidine and harvested at 18 h for determination of radioisotope incorporation. The results shown are expressed as the mean of fold-induction of wild type or C3-/- T cell proliferation relative to background proliferation.
The absence of gross differences in T cell effector functions between wild type and C3-/- mice, indicates that other mechanisms must account for the reported differences in EAE phenotypes (Calida et al., 2001; Nataf et al., 2000). Mouse strain-specific differences are possible since the reports in question used mice with different genotypes (C57BL/6 mice versus 129 × C57BL6/F1 mice), but this by itself seems unlikely to explain the paradoxical findings. Lack of information regarding the 129 substrain that was crossed with C57BL/6 by the Calida group, coupled with the wide genetic variation and recognized differential susceptibility to EAE in 129 substrains, makes it difficult to directly address this point (Sechler et al., 1997; Simpson et al., 1997; Threadgill et al., 1997; Willenborg et al., 1996). The exact protocol used to induce disease could also contribute to the reported differences in EAE phenotype, but this too seems an insufficient explanation since in our hands a statistically significant difference in disease severity was retained between wild type and C3-/- or other mutant mice when EAE was induced using reduced amounts of immunogen and/or pertussis toxin (data not shown).
The aforementioned possibilities not withstanding, recent reports of an important role for C3-derived fragments and C3 receptors in EAE provide the most plausible explanation for the altered EAE phenotype in C3-/- mice. The chemoattractant C3a and the phagocyte promoting ligand iC3b appear central to C3-derived functions in EAE. In previous studies we have shown that transgenic expression of C3a in the CNS of C3a/GFAP mice resulted in massive recruitment of T cells and macrophages to the spinal cord during EAE (Boos et al., 2004). However, when C3a/GFAP mice were crossed with C3aR-/- mice, the massive leukocyte infiltration seen in C3aGFAP mice was prevented and EAE was significantly attenuated. Thus C3a either indirectly or directly, clearly does contribute to cellular infiltration in EAE. Indeed C3a may participate in the retention of leukocytes in the CNS, although this has not been experimentally verified. In addition to C3a, iC3b is also likely involved in the EAE disease phenotype. iC3b promotes CR3-mediated phagocytosis, thus blockade of iC3b-CR3 interaction by treatment with anti-CR3 antibodies inhibits demyelination in vitro and in vivo (reviewed in Barnum and Szalai, 2006). Furthermore, deletion of CR3 significantly attenuates EAE, in part through CR3-dependent effector functions on T cells (Bullard et al., 2005). This raises the possibility that iC3b directly modulates T cell function(s) and that the absence of iC3b costimulation during EAE diminishes the potency of encephalitogenic T cells. Our most recent results add to the growing evidence that complement can regulate the immune function of T cell (reviewed in Kemper and Atkinson, 2007). This knowledge could be translated into C3 receptor-based therapeutic approaches for the treatment and/or prevention of demyelinating disease.
Acknowledgments
This work was supported by NIH grant NS46032 to SRB and AJS. The authors thank Kate Kosmac for help with the flow cytometry studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barnum SR, Szalai AJ. Complement and demyelinating disease: No MAC needed? Brain Research Reviews. 2006;52:58–68. doi: 10.1016/j.brainresrev.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Boos L, Campbell IL, Ames R, Wetsel RA, Barnum SR. Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J Immunol. 2004;173:4708–14. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- Bullard DC, Hu X, Schoeb TR, Axtell RC, Raman C, Barnum SR. Critical requirement of CD11b (Mac-1) on T cells and accessory cells for development of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:6327–33. doi: 10.4049/jimmunol.175.10.6327. [DOI] [PubMed] [Google Scholar]
- Calida DM, Constantinescu C, Purev E, Zhang GX, Ventura ES, Lavi E, Rostami A. Cutting edge: C3, a key component of complement activation, is not required for the development of myelin oligodendrocyte glycoprotein peptide-induced experimental autoimmune encephalomyelitis in mice. J Immunol. 2001;166:723–6. doi: 10.4049/jimmunol.166.2.723. [DOI] [PubMed] [Google Scholar]
- Circolo A, Garnier G, Fukuda W, Wang X, Hidvegi T, Szalai AJ, Briles DE, Volanakis JE, Wetsel RA, Colten HR. Genetic disruption of the murine complement C3 promoter region generates deficient mice with extrahepatic expression of C3 mRNA. Immunopharmacology. 1999;42:135–49. doi: 10.1016/s0162-3109(99)00021-1. [DOI] [PubMed] [Google Scholar]
- Gasque P, Neal JW, Singhrao SK, McGreal EP, Dean YD, Van BJ, Morgan BP. Roles of the complement system in human neurodegenerative disorders: pro-inflammatory and tissue remodeling activities. Mol Neurobiol. 2002;25:1–17. doi: 10.1385/mn:25:1:001. [DOI] [PubMed] [Google Scholar]
- Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Morgan BP, Griffiths M, Khanom H, Taylor SM, Neal JW. Blockade of the C5a receptor fails to protect against experimental autoimmune encephalomyelitis in rats. Clin Exp Immunol. 2004;138:430–8. doi: 10.1111/j.1365-2249.2004.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataf S, Carroll SL, Wetsel RA, Szalai AJ, Barnum SR. Attenuation of experimental autoimmune demyelination in complement-deficient mice. J Immunol. 2000;165:5867–73. doi: 10.4049/jimmunol.165.10.5867. [DOI] [PubMed] [Google Scholar]
- Reiman R, Gerard C, Campbell IL, Barnum SR. Disruption of the C5a receptor gene fails to protect against experimental allergic encephalomyelitis. Eur J Immunol. 2002;32:1157–63. doi: 10.1002/1521-4141(200204)32:4<1157::AID-IMMU1157>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Reiman R, Torres AC, Martin BK, Ting JP, Campbell IL, Barnum SR. Expression of C5a in the brain does not exacerbate experimental autoimmune encephalomyelitis. Neurosci Lett. 2005 doi: 10.1016/j.neulet.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Sechler JM, Yip JC, Rosenberg AS. Genetic variation among 129 substrains: practical consequences. J Immunol. 1997;159:5766–8. [PubMed] [Google Scholar]
- Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- Stahel PF, Barnum SR. The role of the complement system in CNS inflammatory diseases. Expert Review in Clinical Immunology. 2006;2:445–456. doi: 10.1586/1744666X.2.3.445. [DOI] [PubMed] [Google Scholar]
- Szalai AJ, Nataf S, Hu XZ, Barnum SR. Experimental allergic encephalomyelitis is inhibited in transgenic mice expressing human C-reactive protein. J Immunol. 2002;168:5792–7. doi: 10.4049/jimmunol.168.11.5792. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Yee D, Matin A, Nadeau JH, Magnuson T. Genealogy of the 129 inbred strains: 129/SvJ is a contaminated inbred strain. Mamm Genome. 1997;8:390–3. doi: 10.1007/s003359900453. [DOI] [PubMed] [Google Scholar]
- van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- Weerth SH, Rus H, Shin ML, Raine CS. Complement C5 in experimental autoimmune encephalomyelitis (EAE) facilitates remyelination and prevents gliosis. Am J Pathol. 2003;163:1069–80. doi: 10.1016/S0002-9440(10)63466-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–7. [PubMed] [Google Scholar]