

Introduction
Background and significance
With as many as 1 infant per 500–650 live births being born with a permanent and significant sensorineural hearing loss, congenital hearing loss represents the most common neurological birth defect in the United States (2000; Mehl and Thomson, 1998; Mehl and Thomson, 2002). A genetic etiology is estimated to be responsible for 50–60% of these cases of congenital hearing impairment (Grundfast and Lalwani, 1992; Tomaski and Grundfast, 1999). Accordingly, understanding the molecular and genetic processes governing development of the inner ear is very relevant to congenital hearing loss.
Morphologically, the study of inner ear development begins with the otic placodes that originate as ectodermal thickenings on either side of the early hindbrain. These placodes invaginate to form the primitive otocyst that eventually separates from the surface ectoderm. Neuroblasts delaminate from the otic placode generating the vestibulocochlear ganglion that innervates the auditory and vestibular sensory organs of the inner ear (Schlosser, 2006; Streit, 2001). Subsequently, the otocyst undergoes a complex morphogenesis that yields three semicircular canals (that detect angular acceleration), the utricle and saccule (that detect linear acceleration), the endolymphatic duct and sac (that help regulate endolymph homeostasis) and the cochlea (the auditory end organ).
A growing body of literature suggests that the morphogenesis of the various inner ear structures is specified very early and is likely determined at least to some extent by developmental programs that are outlined at the early otocyst stage (Brigande et al., 2000a, 2000b; Lang and Fekete, 2001; Wu et al., 1998). Intriguingly, and central to this review of inner ear development, the adjacent neural tube appears to play a significant role in patterning the otocyst. The literature strongly implicates rhombomeres 4, 5 and 6 (r4, r5 and r6) of the early hindbrain as being critical to formation of the inner ear and the molecular mechanisms underlying these hindbrain effects on the inner ear are just now becoming clearer (Barrow et al., 2000; Choo et al., 2006; Frohman et al., 1993; Giudicelli et al., 2003; Irving et al., 1996; Kwak et al., 2002; Leger and Brand, 2002; Li et al., 2002; Lin et al., 2005; Mansour et al., 1993; Manzanares et al., 2002; Marin and Charnay, 2000; McKay et al., 1994; McKay et al., 1996). This review will focus on this very restricted aspect of otic development starting with several of the early advances leading up to our current understanding of hindbrain effects on the otocyst, the likely signaling mechanisms involved in hindbrain patterning of the otocyst and likely avenues for future investigation.
Historical and current perspectives
Perhaps the earliest interest in the role of the hindbrain on inner ear morphogenesis arose from reports focusing more on early induction of the inner ear (otocyst induction) rather than otocyst patterning (Detwiler and Van Dyke, 1950; Fritzsch et al., 1998; Schlosser, 2006; Torres and Giraldez, 1998; Yntema, 1950; Yntema, 1955; Zwilling, 1940). However, tracing the advances in this field reflects the evolution of our understanding and provides some underpinnings for the study of hindbrain patterning of the otocyst that we focus upon in this review.
The data identifying the primitive hindbrain as relevant to inner ear development comes from multiple species. As early as 1926, investigators recognized that ectoderm from different areas of salamander embryos (Amblystoma punctatum) were competent to generate an inner ear (otocyst) when transplanted next to an appropriate hindbrain-inducing region within an appropriate temporal window (Kaan, 1926; Yntema, 1950; Yntema, 1955). Similar findings were also reported in avian models where sections of ectoderm could be transplanted from quail embryos to various paraxial regions along the rostrocaudal axis of chick hosts. These data narrowed the apparent ear-inducing region of the primitive hindbrain to be restricted somewhere between rhombomeres 2 and 3 (r2–r3, rostrally) and the first pair of somites (caudally). Ectoderm transplanted within this roughly defined region between the 3 and 10 somite stage was consistently shown to induce Pax2-expressing otocysts in these locations (Groves and Bronner-Fraser, 2000). Notably, the same quail epiblast ectoderm transplanted into older embryos (11–21 somite stage) yielded dramatically fewer morphologically or molecularly distinguishable otocysts, indicating the importance of certain temporal windows.
In mice, early reports supporting a key role of the hindbrain in development of the otocyst were produced by studies of kreisler mice (Deol, 1964; Hertwig, 1944; Ruben, 1973). Kreisler (german for “circler”) mice were initially generated by irradiation of founder males with the offspring found to have primary hindbrain defects (lacking rhombomeres 5 and 6) and gross malformation of the inner ear (Hertwig, 1944; Frohman et al., 1993; McKay et al., 1994; Ruben, 1973). Later, targeted gene inactivation studies of key hindbrain genes such as Hox1.6 (i.e. Hoxa1), Hoxb1 and Gbx2 supported the observation that disruption of normal hindbrain development also perturbed inner ear patterning (Chisaka et al., 1992; Gavalas et al., 1998; Lin et al., 2005; Mark et al., 1993). Some of these studies were confounded by the fact that the targeted genes were also expressed in the developing otocyst. As a result, some of the hindbrain-mediated effects versus direct otocyst-mediated effects were not discernible. However, a growing body of evidence seemed to suggest that the hindbrain does, indeed, have a significant patterning effect on the early otocyst.
These data identifying key portions of the rhombencephalon critical to otocyst induction were then combined with reports that int-2 (i.e. Fgf3) expression was noted in the appropriate temporospatial pattern in the hindbrain to make it a reasonable candidate as the otocyst-inducing signal (Wilkinson et al., 1989). Consistent with this hypothesis, Represa et al. (1991) reported on an in vitro model of the chick hindbrain and adjacent ectoderm that allowed them to test whether int-2 was indeed the developmental signal that induced otocyst formation. These experiments suggested that int-2 antisense oligonucleotides and blocking antibodies could both inhibit otocyst formation in this chick explant system and that basic FGF protein could mimic the inductive signal in the absence of the rhombencephalon. However, conflicting data from studies of an Fgf3 null mutant mouse showed that Fgf3 mutant mice formed grossly normal otocysts and ultimately produced ears that lacked an endolymphatic duct and sac but, overall, appeared grossly normal (Mansour et al., 1993). Mansour et al. (1993) concluded that while Fgf3 was not required for induction of the inner ear, it was necessary to properly pattern the otocyst and specifically, the dorsal region of the otocyst that gives rise to the endolymphatic duct and sac.
More recently, additional investigations on the developing chick inner ear have demonstrated that anterior–posterior rotation of r4–r7 (including the notochord) was nonessential for proper anterior–posterior patterning, but was critical for dorso-ventral patterning of the otocyst (Bok et al., 2005). In these studies, 10–13 somite stage embryos underwent surgical manipulation that rotated the hindbrain and notochord segment either vertically (yielding a dorsoventral rotation), or horizontally (yielding an anterior–posterior rotation) in ovo. Interestingly, the authors concluded that the hindbrain is crucial in specifying a dorsoventral axis for the otocyst but that temporal differences between placode induction and axial specification in the chick inner ear suggest two distinct regulatory mechanisms for these two processes (Bok et al., 2005).
Collectively, these earlier reports lay the foundation for reviewing the developmental role(s) of the hindbrain in patterning the otocyst. This discussion will examine first the evidence from mouse and zebrafish mutants that demonstrate both hindbrain and inner ear defects and explore insights into the links between the two developing organ systems provided by these mutant models. Second, the literature on candidate signaling molecules from the hindbrain such as Fibroblast growth factors (Fgfs), hedgehog and Wnt genes will be reviewed. Finally, discussion will focus on a few of the otic targets of hindbrain signaling that have only recently begun to be identified. By reviewing these topics as a group, we begin to discern the complex embryonic interactions occurring between the hindbrain and inner ear that help pattern a normal otocyst and ultimately, a normal functional ear.
Hindbrain mutants with inner ear phenotypes
Early during development of the vertebrate hindbrain, several metameric segments called rhombomeres (Fraser et al., 1990; Keynes et al., 1990; Keynes and Lumsden, 1990; Lumsden and Keynes, 1989) can be identified both morphologically as well as molecularly. This segmentation reflects an embryologic differentiation that, in turn, reflects their function, serving as a source of patterned neural crest cells that innervate the branchial arches (e.g. cranial nerves V, VII and IX). For the most part, neural crest cells that migrate into the 1st, 2nd and 3rd branchial arches originate from r2, r4 and r6, respectively. In this manner, the rhombomeres can influence non-hindbrain development and patterning.
Yet the hindbrain also plays a role in inner ear patterning, not necessarily by providing a source of patterned cells but (in all likelihood) also by producing/secreting morphogens (e.g. fibroblast growth factors) or other important developmental cues that then affect nearby targets (e.g. otocyst).
One approach to identifying the potential effects of the hindbrain on inner ear development is to examine mutants with hindbrain as well as inner ear defects. This analysis clearly does not present any causal mechanisms that might be involved between then developing hindbrain and the otocyst, but it does begin linking defects or abnormalities of the hindbrain with maldevelopment of the inner ear. Table 1 summarizes those murine and zebrafish mutants with both phenotypes.
Table 1.
Mutants with hindbrain and inner ear phenotypes
| Mutant | Species | Hindbrain phenotype | Inner ear phenotype | Reference |
|---|---|---|---|---|
| Kreisler (kr/mafB) | Mouse | Loss of r5 and r6 | Spectrum of cochlear and vestibular malformations | Choo et al. (2006); Cordes and Barsch (1994) |
| Gbx2 | Mouse | Upregulation of Fgf3 and kreisler expression in r5 and r6 | Spectrum of cochlear and vestibular malformations | Lin et al., 2005 |
| Hoxa1 | Mouse | Severe reduction of r4 and absence of r5 | Spectrum of cochlear and vestibular malformations | Mark et al. (1993); Pasqualetti et al. (2001 |
| Wheels (unidentified mutation, possibly EphA7) | Mouse | Disrupted rhombomere boundaries | Semicircular canal and cristae defects | Alavizadeh et al. (2001) |
| Hoxa1/b1 | Mouse | Loss of r4 and r5 | Severe cochlear and vestibular malformations | Gavalas et al. (1998) |
| Valentino (kr/mafB) | Zebrafish | Loss of r5 and r6, upregulation of FGF3 | Ectopic hair cells, spectrum of malformations | Kwak et al. (2002) |
| Acerebellar (fgf8) | Zebrafish | Midbrain and hindbrain defects | Smaller ears, one otolith, semicircular canal defects | Leger and Brand (2002) |
| Vhn1 | Zebrafish | Caudal hindbrain defects | Smaller otocysts, spectrum of malformations | Lecaudey et al. (Electronic publication ahead of print) |
Overall, the data summarized in Table 1 show a variety of defects in the hindbrain that appear to be relevant to otocyst patterning and morphogenesis. In considering the listed mutants, a few common threads seemed apparent in the analysis. One interesting commonality is that despite significant hindbrain anomalies (including absence of one or more rhombomeres), otic development is not completed arrested or ablated. In several of the mutants listed, inner ears (though mispatterned) still form in roughly the appropriate region. Another common trend observed in these hindbrain mutants is that the inner ear defects display significant variability in severity of malformation. In several examples, a spectrum of cochlear and vestibular malformations is observed (see Table 1). Such observations suggest that the hindbrain effects on the inner ear are not simple stoichiometric phenomenon, but more intricate interactions that may involve several signals from the hindbrain as well as other relevant regions (surface ectoderm, mesoderm and notochord) as suggested by many investigators and as discussed further in detail below. From the ear perspective, a third common feature in these mutant mice and zebrafish is that the otocysts retain a significant amount of autonomous patterning as reflected by their ability to form ear-specific structures such as otoliths and hair cell patches despite gross patterning and morphologic abnormalities.
Examining the data from each mutant more closely, the more caudal rhombomeres (r4, r5 and r6) are particularly strongly implicated in otic development. Taking into consideration the developmental proximity of the otic placode and otocyst to r5 and r6, it is certainly plausible that these caudal rhombomeres could have bearing on the developing otocyst. It is also noteworthy that early during otic placode and otocyst stages, the otic and hindbrain epithelium are physically connected until later in the otocyst stage (Hilfer et al., 1989).
Consider, for example, the Hoxa1 null mutant. Disruption of Hoxa1 results in r5 being entirely absent (Carpenter et al., 1993) or at least severely reduced (Mark et al., 1993) along with a defective r4 that is smaller and consists of a different population of neural crest cells than is normally observed. The inner ears of Hoxa1 homozygous mutant mice show defects of the semicircular canals and the cochlea that suggest that overall otocyst patterning has been disrupted. Given the reductions in r5 as well as the altered composition of r4, the data suggest that both a normal r4 and r5 are requisite for normal inner ear formation. Similarly, the double mutant, Hoxa1/b1, completely lacks r4 and r5 and generates a malformed inner ear comparable to the Hoxa1 null (Gavalas et al., 1998).
Looking then at the kreisler and valentino mutants, the loss of r5 and r6 can be associated with severely malformed inner ears that are mispatterned with respect to both cochlear and vestibular components (Choo et al., 2006; Kwak et al., 2002). Along those same lines, recently published work on the Gbx2 mutant mouse also point to kreisler-like inner ear malformations being related to perturbation of Fgf3 and kr/mafB expression in r5 and r6.
It is relevant to point out that the Krox20 mutant is not included in Table 1 because ear development is normal. However, the Krox20 null mutant fails to develop r3 and r5; suggesting that the absence of r5 (in concert with the absence of r3) is not requisite for normal otic patterning (Swiatek and Gridley, 1993). An important caveat and potential explanation reconciling these data with that from other mutants, is that r5 actually seems to form in Krox20 mutants but is subsequently lost (Schneider-Maunoury et al., 1993). Therefore, it is feasible that in Krox20 mutants, the transient r5 provides the necessary hindbrain cues for normal otic development.
Lastly, evidence from studies of the zebrafish Vhnf1 mutant indicate that altered caudal hindbrain development affects otocyst patterning in terms of expanding anterior gene expression domains, reducing dorsal gene expression domains and reorganizing sensory regions of the inner ear (Lecaudey et al., 2006). These data are consistent with that from other species and also fit neatly in terms of suggesting the interaction of Vhnf1 with other significant hindbrain patterning genes such as MafB/kr, Krox20, Fgfs and the retinoid signaling system (Aragon et al., 2005; Hernandez et al., 2004; Kim et al., 2005; Lecaudey et al., 2006; Maves and Kimmel, 2005).
Additional details about specific rhombomere contributions to the guidance of otic patterning may be gleaned from the study of other hindbrain mutants (such as the Wheels mutant) in which disruption of normal rhombomere boundaries are associated with defects of the vestibular portions of the inner ear (Alavizadeh et al., 2001). The Wheels mutation may possibly be related to defects of EphA7 (a tyrosine kinase receptor) specifically expressed in the developing hindbrain. Finally, studies of Acerebellar, a zebrafish mutant with midbrain and hindbrain defects also show abnormal semicircular canal development and a single otolith (vs. the normal 2) (Leger and Brand, 2002).
Compiling all of these observations and experimental models of hindbrain and inner ear maldevelopment, the data strongly support the importance of a combination of signals or cues from r4, r5 and r6 in normal patterning of the developing inner ear.
Candidate molecules in hindbrain-ear signaling
In the quest to discover signaling molecules that might be involved in guiding otic induction, patterning and development, the Fibroblast growth factor (Fgf) family of proteins has been consistently implied.
The Fgfs are highly conserved, diffusible proteins whose developmental roles (in almost every organ system) range from induction, to differentiation, cell proliferation and survival (for reviews, see Capdevila and Izpisua Belmonte, 2001; Nie et al., 2006; Torres and Giraldez, 1998). Many Fgfs are known to play different roles in a given developing organ system at different developmental time points. More than 23 different Fgf members have been identified whose effects are mediated by binding to at least 4 well-characterized receptors (Fibroblast growth factor receptors 1–4, Fgfr1–4) (Nie et al., 2006). Each Fgfr has 3 extracellular immunoglobulin-like loops that demonstrate variability generated by splice variation (Pickles and Chir, 2002). Most Fgfs display differential binding affinities for Fgfrs with some showing near exclusivity in binding (e.g. Fgf8 to Fgfr3c and Fgr4) (Ornitz et al., 1996; Pickles and Chir, 2002). Further complicating the Fgf-Fgfr interactions is the role of heparan sulphate proteoglycans (HSPGs) that can enhance Fgf-binding to the respective receptor(s) (Ornitz et al., 1996).
The first experiments pointing towards Fgfs playing a role in otocyst patterning implicated Fgf3 and were reported by Represa et al. (1991) and Mansour et al. (1993) (as mentioned above). Continued work in this field has subsequently generated further data indicating the importance of various Fgfs in inner ear induction and patterning. However, the apparent mechanisms continue to become increasingly complex with multiple tissue sources (endoderm, paraxial mesoderm, ectoderm as well as the neural tube) all demonstrating some signaling activity guiding inner ear development via several anticipated signaling molecules such as Fgfs and Wnts (Ladher et al., 2000, 2005; Wright et al., 2003; Wright and Mansour, 2003).
As an illustration, recent studies have revealed that specific Fgfs are potent inducers of the inner ear and vital for proper patterning (Nie et al., 2006; Pickles and Chir, 2002; Wilkinson et al., 1989; Wright et al., 2003, 2004; Wright and Mansour, 2003). In one of the sentinel reports, Ladher et al. (2000) concluded from chick experiments that Fgf19 secreted by the mesoderm underlying the otic epithelium was essential (with neural Wnt8c) for proper otic induction and expression of normal otic markers. Somewhat confusingly, data from murine studies did not confirm the similar requirement for Fgf15 (the mouse ortholog of chick Fgf19) but did point towards Fgf3 and Fgf10 as being the key molecules for mouse otocyst induction and early differentiation (Wright et al., 2003; Wright and Mansour, 2003). In the developing mouse, the caudal hindbrain produces Fgf3 just prior to and concomitant with early inner ear formation. Concurrently, the mesenchyme underlying the presumptive otic placode expresses Fgf10. In mice lacking both Fgf3 and Fgf10, the inner ears do not form at all while mutants with 3 out of 4 mutant alleles for Fgf3 and Fgf10 (e.g. Fgf3−/−Fgf10+/− or Fgf3+/−Fgf10−/−), display smaller ears with more ventral positioning and altered expression patterns of Pax2 and Dlx5 (Wright and Mansour, 2003). Such data suggest that a certain dosage or level of Fgf signaling is required for normal otic induction and development.
Another strong line of evidence from zebrafish mutants and morpholino experiments implicate neural fgf3 and fgf8 as important cues for early otic patterning. In Ace mutants that lack fgf8, there is absence of the cerebellum associated with smaller otocysts that lack the normal complement of otoliths and semicircular canals (Leger and Brand, 2002; Reifers et al., 1998). Confirmatory evidence identifying fgf8 as the key gene is provided by knockdown experiments in which fgf8 morpholinos effectively suppress fgf8 protein synthesis and reproduce the cerebellar and otic defects seen in the ace mutants (Leger and Brand, 2002).
Finally, more recent reports demonstrate that yet another tissue source (endoderm) also plays a vital role in inner ear development using another Fgf ligand (Fgf8) to mediate the effects. Initially, the investigators showed that the ablation of cranial endoderm in stage 5 chick embryos, resulted in absent or markedly hypoplastic otic vesicles in the majority of specimens (Ladher et al., 2005). The investigators then hypothesized that since endoderm is never in contact with the pre-otic ectoderm during embryogenesis, that an indirect phenomenon must be involved. Accordingly, Ladher et al. (2005) devised a series of avian explant and mouse experiments, that demonstrated that quail endoderm in concert with chick mesoderm explants could induce the normal mesodermal Fgf19 expression that was required for chick inner ear development. In mice, the pathways appear to be more redundant and complex. Fgf8 is expressed in the appropriate temporospatial patterns in early mouse embryos to be potentially involved in early otic induction and development. Muddying the picture though is the finding that Fgf8 is expressed not only in the cranial endoderm, but in all three germ layers in areas potentially relevant to otic induction and differentiation. Unfortunately, Fgf8 null mutants die too early in embryogenesis to allow any meaningful study of inner ear development (Sun et al., 1999). Accordingly, hypomorphic Fgf8 mutants were used to study the role of endodermal Fgf8 in inner ear development. Further complicating these experiments were the redundancies observed in mouse Fgf signaling. As a result, the investigators examined Fgf8 hypomorphs in the background of Fgf3 null mutant mice (Ladher et al., 2005). From these complex studies, the authors concluded that in mouse, neural Fgf3 and mesodermal Fgf10 act redundantly to induce the otic placode. Fgf8 from one of the germ layers acts upstream to induce the mesodermal Fgf10 expression.
Considering these representative studies together, it is apparent that simple models of single-source Fgf-mediated signaling that guide inner ear development are not applicable. Despite this caveat, our focus on the hindbrain component of this complex system continues to support a potential role of Fgfs in mediating the hindbrain effects on the developing inner ear. The data discussed above from mouse and zebrafish models consistently point towards key Fgfs (e.g. Fgf3, Fgf8, Fgf10) as being important for normal inner ear patterning and formation. The challenge will be to continually dissect this system in more creative methods to discern the roles of specific Fgfs from specific sources in steering the intricate developmental process.
It is also important to acknowledge other important cues from the hindbrain that are not Fgf mediated. For example, Riccomagno et al. (2005) reported on the role of Wnt signals from the dorsal hindbrain that guide patterning and morphogenesis of the dorsal regions of the otocyst. In their report, the authors used otic explant methods to study this Wnt signaling pathway and demonstrated the dependence of otic Dlx5 and Gbx2 expression upon Wnt1 and Wnt3a from the dorsal hindbrain. In order to confirm the proposed role of Wnts in this process, the investigators also used otic explants carrying a Topgal reporter (an indicator of Wnt pathway activation) to shown marked down regulation of Wnt-dependent activity in the dorsal otocyst when the dorsal hindbrain was removed. Furthermore, the addition of 30 mM LiCl to the culture media (an agonist for Wnt pathway activation) was shown to rescue the Topgal activity in the dorsal otocyst and restore normal expression of Dlx5 and Gbx2 in the dorsal otocyst even in the absence of the dorsal hindbrain.
On a related topic, reports have also indicated the importance of Sonic Hedgehog (Shh) secreted by the notochord in promoting ventral otic fates and development of a normal cochlea via regulation of Pax2 and Ngn1 (Liu et al., 2002; Riccomagno et al., 2005). The interaction of dorsal neural tube signals (such as Wnt1 and Wnt3a) with Shh appear to regulate the dorsomedial expression of Gbx2 in the otocyst and inhibit the more ventral expression of dorsal markers such as Dlx5 (Riccomagno et al., 2005). As a result, it is evident that ventral signals from the notochord as well as Fgf and Wnt signals from the neural tube are pertinent to otic patterning and deserve further investigation.
Molecular mechanisms guiding otocyst patterning
An intriguing patterning theory based upon compartments and boundaries (such as those described in the fly wing and vertebrate hindbrain; Dahmann and Basler, 1999) suggest that similar compartment boundaries are also relevant in the developing otocyst (Brigande et al., 2000b; Fekete and Wu, 2002). A growing body of literature supports the concept that gene expression domains define compartments of the otocyst that will ultimately determine identity within the inner ear (e.g. cochlea, semicircular canal, endolymphatic duct, etc.). The boundaries of these compartments may then be critical in the positioning of specific structures (e.g. sensory hair cell patches, the endolymphatic apparatus) and may be mediated by molecular signals transmitted across boundaries (Brigande et al., 2000a, 2000b) (see Figs. 1 and 2).
Fig. 1.

Schematic representation of the mouse otic vesicle, showing the expression domains of several genes (top row) and the phenotypes that result when the gene is knocked out (bottom row). Dark lines encircling the vesicle indicate theoretical boundaries that segregate the vesicle into compartments. In the bottom row, structures reported missing in the knockout are left off the drawings, while those that have been reported to have a variable phenotype either by a single group or by two different groups are shaded gray. asc, anterior semicircular canal; ac, anterior crista; cc, common crus; csd, cochlear-saccular duct; ed, endolymphatic duct; es, endolymphatic sac; lc, lateral crista; lsc, lateral semicircular canal; oC, organ of Corti; pc, posterior crista; psc, posterior semicircular canal; s, saccular macula; u, utricular macula; usd, utricular-saccular duct (from “Molecular genetics of pattern formation in ear: do compartment boundaries play a role?” PNAS October 24, 2000. 97(22): 11700–11706. John V. Brigande, Amy E. Kiernan, Xiaoying Gao, Laurie E. Iten, and Donna M. Fekete).
Fig. 2.
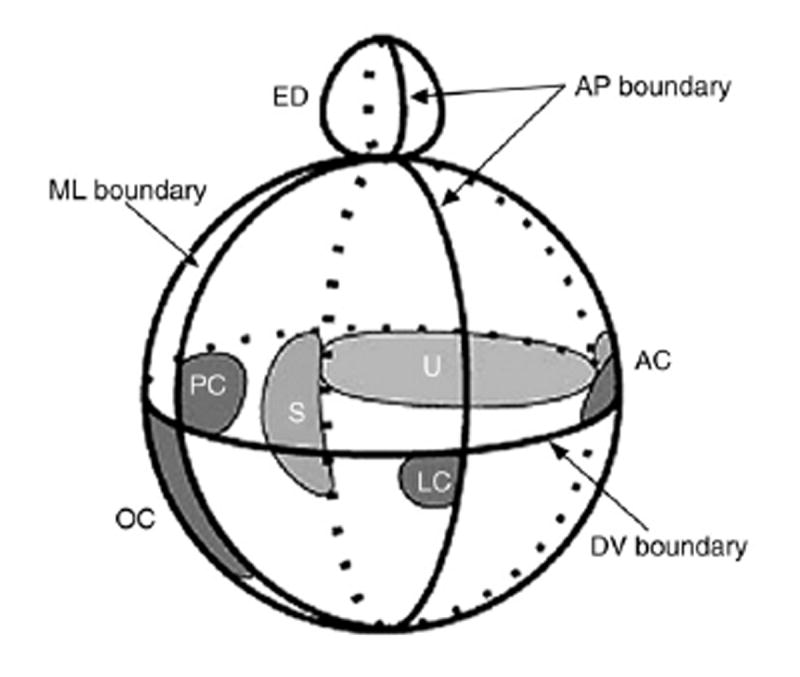
Model showing one possible arrangement of sensory primordia with respect to the theoretical compartment boundaries. Both chicken and mouse data were used to derive the model, which is viewed from the side. Several sensory organ primordial arise on the edge of a SOHo boundary. The lateral crista primordium is located on the edge of the Otx2 domain. The endolymphatic duct also arises on one side of the medial-lateral (ML) boundary at the dorsal pole of the otocyst. AC, anterior crista; ED, endolymphatic duct; LC, lateral crista; OC, organ of Corti; PC, posterior crista; S, saccular macula; U, utricular macula. (from “Molecular genetics of pattern formation in ear: do compartment boundaries play a role?” PNAS October 24, 2000. 97(22): 11700-11706. John V. Brigande, Amy E. Kiernan, Xiaoying Gao, Laurie E. Iten, and Donna M. Fekete).
Extrapolating from this theory, it is then reasonable to hypothesize that disruption of the normal gene expression boundaries would lead to mispatterning of the otocyst. Such a process would lend itself to potential perturbation by neighboring organs or cellular environments (such as the hindbrain or paraxial mesoderm, for example).
The prior discussion reviewing the hindbrain-inner ear mutants helps set the stage for potential genes or molecules involved in such a process.
Consider, for example, the MafB/kr mutant. The hindbrain phenotype in kreisler mutant mice consists of the complete absence of r5 and r6 (Deol, 1964; Hertwig, 1944; Ruben, 1973). Associated ear abnormalities include the consistent absence of the endolymphatic duct and sac, and variable but severe malformations of the semicircular canals and the cochlea (Choo et al., 2006). The search for molecular mechanisms underlying such maldevelopment of the inner ear led to the examination of expression of molecular markers for specific otic structures (e.g. genes with specific expression compartments or boundaries). As specific examples, the normal expression of Wnt2b, Gbx2 and Dlx5, all demonstrated an early restricted expression pattern in the very dorsal otocyst at E9–E10; a region shown by fate mapping to ultimately give rise to the endolymphatic duct and sac (Brigande et al., 2000a; Choo et al., 2006). In kreisler, the expression of these endolymphatic duct and sac markers are consistently absent in the otocyst between E9.5 and E10.5 (Choo et al., 2006). In the framework of a compartment boundary model of otocyst patterning, it is plausible to speculate that the hindbrain defects caused by MafB/kr mutation, alter hindbrain cues resulting in perturbation of the otocyst gene expression patterns that ultimately give rise to the kreisler inner ear phenotype.
Additional supportive data for such a process come from studies of the Gbx2 null mutant. Collaborative experiments were instigated by the remarkable similarities in phenotype as well as gene expression perturbations noted between kreisler and Gbx2 mutant mice. Notably, the Gbx2 mutants display similar caudal hindbrain anomalies and the same endolymphatic duct and sac absence as kreisler mice and also show the same down regulation of Wnt2b and Dlx5 that were observed in MafB/kr mutants.
Taking the kreisler and Gbx2 data together, the data suggest that hindbrain defects involving r5 and r6 are capable of influencing the overall patterning of the otocyst by perturbing the gene expression compartments and/or boundaries. Furthermore, some of the early otic targets of hindbrain signaling include several logical candidate genes (including those from the Wnt and Dlx family of genes).
Additional conclusions gleaned from the study of kreisler and Gbx2 mutants show that the hindbrain abnormalities in these mice also perturb the otic patterning that leads to proper cochlear morphogenesis. In the midbrain–hindbrain junction, Gbx2 and Otx2 display an antagonistic interaction that sharply demarcates the morphologic junction. In the normal developing (ventral) otocyst, Gbx2 and Otx2 similarly display an appositional relationship (Choo et al., 2006; Lin et al., 2005). However, in both kreisler as well as Gbx2 mutants, the hindbrain perturbation, and in turn, the loss of otic Gbx2 expression leads to expansion of the ventral Otx2 domain (Choo et al., 2006; Lin et al., 2005). In the context of a compartment boundary model of the developing otocyst, this expanded Otx2 expression domain disrupts the normal cochlear compartment boundary and can plausibly be associated with the cochlear phenotype consistently observed in both mouse mutants.
On a related note, recent reports on Tbx1 indicate that this T-box transcription factor is involved in regulating cell fate determination in the otocyst and that loss of Tbx1 appears to switch otic epithelial cells from a non-neurogenic to neurogenic fate (Friedman et al., 2005; Xu et al., 2007). In several elegant experiments using Tbx1 null mutant mice as well as timed ablation and fate mapping techniques, the investigators demonstrated that Tbx1 normally identifies a large portion of the otocyst epithelium that specifically excludes the endolymphatic duct and the sensory precursors. However, the timed deletion of Tbx1 produces an expansion of a Delta-like 1-Notch activation domain (that is typically associated with cells of a neurogenic fate) and results in some cells being reassigned to a sensory fate. It seems plausible and worthy of study to determine whether loss of hindbrain kr/Mafb and/or Gbx2 impacts Tbx1 activity in the otocyst and how this molecular pathway might be involved in mediating the abnormal otic patterning events observed in kreisler and Gbx2 mutants. In trying to reconcile theories of hindbrain effects on the developing otocyst and a boundary/compartment model of otocyst patterning, it would be helpful to identify key molecular regulators of critical differentiation steps (such as sensory vs. non-sensory) and then test specific hypotheses based upon those key developmental regulators.
Very recent studies of the zebrafish vhnf1 mutant also provide neatly supportive data for such a hindbrain-triggered mechanism being responsible for otic malformations. In a report by Lecaudey et al. (2006), the vhnf1 mutants displayed an expanded anterior gene expression domain along with an absent or markedly reduced dorsal gene expression compartment. The investigators also report that as a sequela of the reduced dorsal domain, the ventral compartment is grossly expanded dorsally. These overall otocyst patterning perturbations are then related to the disruption of the normal formation of sensory hair cell patches (both vestibular and cochlear) in the zebrafish inner ear.
In summary, there are several lines of evidence that support two developmental processes involved in otocyst patterning and inner ear development. First, evidence from several animal models (chick, mouse and zebrafish) indicate that normal hindbrain development and cues from the caudal hindbrain (r4, r5 and r6) are necessary for proper inner ear development. Second, a wealth of gene expression data now support the importance of gene expression compartments and boundaries that guide development of the various inner ear structures. Lastly, it seems reasonable to merge these two concepts (or processes) to explain how the hindbrain influences the patterning and morphogenesis of the inner ear. In this framework, the caudal rhombomeres, along with the normal expression of MafB/kr, Gbx2, Fgf3 and numerous other genes, result in a concerted signal from the hindbrain to the adjacent otocyst that helps trigger the normal expression of genes within specific compartments that also yield specific gene expression boundaries in the primitive otocyst. However, several key gaps in the details of such a proposed system still require investigation and clarification in order to validate and solidify the system.
Future research in otocyst patterning
At the level of the caudal hindbrain, a cohesive explanation of the complex molecular developmental interactions is beginning to be elucidated but needs greater detail and clarification. For example, the data from vhnf1 zebrafish mutants suggest that retinoic acid signals from the paraxial mesoderm in concert with Fgf signals from r4 and vhnf1, are necessary to induce valentino (val, zebrafish ortholog of MafB/kr) expression, normal r5 and r6 development as well as the induction of the normal hox and ephrin gene cascades in the developing caudal hindbrain (Hernandez et al., 2004). Such reports greatly enhance our understanding of how well-described molecular pathways and gene families in the rhombomeres (such as the Hox, Fgf, Ephrin and retinoid signaling genes) might interact to more globally pattern and guide morphogenesis of the hindbrain.
A second area that warrants future investigation is a more detailed examination of the developmental interactions between the hindbrain, ectoderm and paraxial mesoderm that are relevant to inner ear patterning and morphogenesis. As a starting point and as discussed earlier, Ladher et al. (2000) described an intricate system in which chick Fgf19 in the paraxial mesoderm induces or maintains Wnt8c in the competent neuroectoderm. Wnt8c expression, in turn, induces Fgf3 in the presumptive otic placode where Fgf3 acts in concert with Fgf19 to induce a battery of genes that are typically expressed in the chick inner ear (e.g. Nkx5.1, Pax-2, SOHo-1 and Dlx-5) (Ladher et al., 2000). Additional roles of endodermal Fgf8 have also been described above. Areas that appear fertile for future research include examining the other indirect interactions between hindbrain and mesoderm, or reciprocal signaling between the hindbrain and inner ear that may act as feedback loops in the developmental process.
In order to complete our understanding of how the hindbrain impacts otocyst development, it is necessary to identify the actual molecules or cells that serve as the signal between the two developing organ systems. Mouse and zebrafish mutants have identified key regions of the caudal hindbrain (e.g. r4–r6) and relevant hindbrain genes (Hoxa1, Hoxb1, MafB/kr, Gbx2, Fgf3, Fgf8) that are likely involved in the neural portion of the hindbrain-inner ear interaction. Data have also been reviewed that indicate that certain otocyst genes (Wnt2b, Gbx2, Dlx5, Otx2) represent several of the early otic targets of hindbrain signaling that respond to the cues received from the hindbrain. However, the exact molecular signals that mediate the hindbrain signals to the developing ear remain ambiguous. Evidence of Fgf involvement in this process remains suggestive but far from conclusive. As discussed above, Fgf signaling may indeed be requisite from several sources (endoderm, neural, ectodermal) in order to properly pattern and develop an inner ear. Therefore, the challenge will be to dissect the different temporal and spatial roles of various Fgfs in this complex system. However, by defining the signaling molecules involved in hindbrain-inner ear interactions, a greater understanding of the developmental relationship between these key organ systems can be achieved.
References
- Year 2000 position statement: principles and guidelines for early hearing detection and intervention programs. Joint Committee on Infant Hearing, American Academy of Audiology, American Academy of Pediatrics, American Speech-Language-Hearing Association, and Directors of Speech and Hearing Programs in State Health and Welfare Agencies. Pediatrics. 2000;106:798–817. doi: 10.1542/peds.106.4.798. [DOI] [PubMed] [Google Scholar]
- Alavizadeh A, Kiernan AE, Nolan P, Lo C, Steel KP, Bucan M. The Wheels mutation in the mouse causes vascular, hindbrain, and inner ear defects. Dev Biol. 2001;234:244–260. doi: 10.1006/dbio.2001.0241. [DOI] [PubMed] [Google Scholar]
- Aragon F, Vazquez-Echeverria C, Ulloa E, Reber M, Cereghini S, Alsina B, Giraldez F, Pujades C. vHnf1 regulates specification of caudal rhombomere identity in the chick hindbrain. Dev Dyn. 2005;234:567–576. doi: 10.1002/dvdy.20528. [DOI] [PubMed] [Google Scholar]
- Barrow JR, Stadler HS, Capecchi MR. Roles of Hoxa1 and Hoxa2 in patterning the early hindbrain of the mouse. Development. 2000;127:933–944. doi: 10.1242/dev.127.5.933. [DOI] [PubMed] [Google Scholar]
- Bok J, Bronner-Fraser M, Wu DK. Role of the hindbrain in dorsoventral but not anteroposterior axial specification of the inner ear. Development. 2005;132:2115–2124. doi: 10.1242/dev.01796. [DOI] [PubMed] [Google Scholar]
- Brigande JV, Iten LE, Fekete DM. A fate map of chick otic cup closure reveals lineage boundaries in the dorsal otocyst. Dev Biol. 2000a;227:256–270. doi: 10.1006/dbio.2000.9914. [DOI] [PubMed] [Google Scholar]
- Brigande JV, Kiernan AE, Gao X, Iten LE, Fekete DM. Molecular genetics of pattern formation in the inner ear: do compartment boundaries play a role? Proc Natl Acad Sci U S A. 2000b;97:11700–11706. doi: 10.1073/pnas.97.22.11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila J, Izpisua Belmonte JC. Patterning mechanisms controlling vertebrate limb development. Annu Rev Cell Dev Biol. 2001;17:87–132. doi: 10.1146/annurev.cellbio.17.1.87. [DOI] [PubMed] [Google Scholar]
- Carpenter EM, Goddard JM, Chisaka O, Manley NR, Capecchi MR. Loss of Hox-A1 (Hox-1.6) function results in the reorganization of the murine hindbrain. Development. 1993;118:1063–1075. doi: 10.1242/dev.118.4.1063. [DOI] [PubMed] [Google Scholar]
- Chisaka O, Musci R, Capecchi M. Developmental defects of the ear, cranial nerves and hindbrain resulting from targeted disruption of the mouse homeobox gene Hox-1.6. Nature. 1992;355:516–520. doi: 10.1038/355516a0. [DOI] [PubMed] [Google Scholar]
- Choo D, Ward J, Reece A, Dou H, Lin Z, Greinwald J. Molecular mechanisms underlying inner ear patterning defects in kreisler mutants. Dev Biol. 2006;289:308–317. doi: 10.1016/j.ydbio.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Cordes SP, Barsh GS. The mouse segmentation gene kr encodes a novel basic domain-leucine zipper transcription factor. Cell. 1994;79(6):1025–1034. doi: 10.1016/0092-8674(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Dahmann C, Basler K. Compartment boundaries: at the edge of development. Trends Genet. 1999;15:320–326. doi: 10.1016/s0168-9525(99)01774-6. [DOI] [PubMed] [Google Scholar]
- Deol M. The abnormalities of the inner ear in kreisler mice. J Embryol Exp Morph. 1964;12:475–490. [PubMed] [Google Scholar]
- Detwiler SR, Van Dyke RH. The role of the medulla in the differentiation of the otic vesicle. J Exp Zool. 1950;113:179–199. [Google Scholar]
- Fekete DM, Wu DK. Revisiting cell fate specification in the inner ear. Curr Opin Neurobiol. 2002;12:35–42. doi: 10.1016/s0959-4388(02)00287-8. [DOI] [PubMed] [Google Scholar]
- Fraser S, Keynes R, Lumsden A. Segmentation in the chick embryo hindbrain is defined by cell lineage restrictions. Nature. 1990;344:431–435. doi: 10.1038/344431a0. [DOI] [PubMed] [Google Scholar]
- Friedman RA, Makmura L, Biesiada E, Wang X, Keithley EM. Eya1 acts upstream of Tbx1, Neurogenin 1, NeuroD and the neurotrophins BDNF and NT-3 during inner ear development. Mech Dev. 2005;122:625–634. doi: 10.1016/j.mod.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Barald K, Lomax M. Early embryology of the vertebrate ear. In: Rubel E, Popper A, Fay R, editors. Development of the Auditory System. Springer; New York: 1998. [Google Scholar]
- Frohman MA, Martin GR, Cordes SP, Halamek LP, Barsh GS. Altered rhombomere-specific gene expression and hyoid bone differentiation in the mouse segmentation mutant, kreisler (kr) Development. 1993;117:925–936. doi: 10.1242/dev.117.3.925. [DOI] [PubMed] [Google Scholar]
- Gavalas A, Studer M, Lumsden A, Rijli FM, Krumlauf R, Chambon P. Hoxa1 and Hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development. 1998;125:1123–1136. doi: 10.1242/dev.125.6.1123. [DOI] [PubMed] [Google Scholar]
- Giudicelli F, Gilardi-Hebenstreit P, Mechta-Grigoriou F, Poquet C, Charnay P. Novel activities of Mafb underlie its dual role in hindbrain segmentation and regional specification. Dev Biol. 2003;253:150–162. doi: 10.1006/dbio.2002.0864. [DOI] [PubMed] [Google Scholar]
- Groves AK, Bronner-Fraser M. Competence, specification and commitment in otic placode induction. Development. 2000;127:3489–3499. doi: 10.1242/dev.127.16.3489. [DOI] [PubMed] [Google Scholar]
- Grundfast KM, Lalwani AK. Practical approach to diagnosis and management of hereditary hearing impairment (HHI) Ear Nose Throat J. 1992;71:479–484. 487–493. [PubMed] [Google Scholar]
- Hernandez RE, Rikhof HA, Bachmann R, Moens CB. vhnf1 integrates global RA patterning and local FGF signals to direct posterior hindbrain development in zebrafish. Development. 2004;131:4511–4520. doi: 10.1242/dev.01297. [DOI] [PubMed] [Google Scholar]
- Hertwig P. Die Genese der Hirn- und GehorgangsmiBbildungen bei Rontgen-mutierten Kreisler-Mausen. Z Menschl Vererb Konstitutionsl. 1944;28:327. [Google Scholar]
- Hilfer SR, Esteves RA, Sanzo JF. Invagination of the otic placode: normal development and experimental manipulation. J Exp Zool. 1989;251:253–264. doi: 10.1002/jez.1402510213. [DOI] [PubMed] [Google Scholar]
- Irving C, Nieto M, DasGupta R, Charnay P, Wilkinson D. Progressive spatial restriction of sek-1 and krox-20 gene expression during hindbrain segmentation. Dev Biol. 1996;171:26–38. doi: 10.1006/dbio.1996.0004. [DOI] [PubMed] [Google Scholar]
- Kaan HW. Experiments on the development of the ear of Amblystoma punctatum. J Exp Zool. 1926;46:13–61. [Google Scholar]
- Keynes R, Cook G, Davies J, Lumsden A, Norris W, Stern C. Segmentation and the development of the vertebrate nervous system. J Physiol (Paris) 1990;84:27–32. [PubMed] [Google Scholar]
- Keynes R, Lumsden A. Segmentation and the origin of regional diversity in the vertebrate central nervous system. Neuron. 1990;4:1–9. doi: 10.1016/0896-6273(90)90438-l. [DOI] [PubMed] [Google Scholar]
- Kim FA, Sing lA, Kaneko T, Bieman M, Stallwood N, Sadl VS, Cordes SP. The vHNF1 homeodomain protein establishes early rhombomere identity by direct regulation of Kreisler expression. Mech Dev. 2005;122:1300–1309. doi: 10.1016/j.mod.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Kwak SJ, Phillips BT, Heck R, Riley BB. An expanded domain of fgf3 expression in the hindbrain of zebrafish valentino mutants results in mis-patterning of the otic vesicle. Development. 2002;129:5279–5287. doi: 10.1242/dev.129.22.5279. [DOI] [PubMed] [Google Scholar]
- Ladher RK, Anakwe KU, Gurney AL, Schoenwolf GC, Francis-West PH. Identification of synergistic signals initiating inner ear development. Science. 2000;290:1965–1967. doi: 10.1126/science.290.5498.1965. [DOI] [PubMed] [Google Scholar]
- Ladher RK, Wright TJ, Moon AM, Mansour SL, Schoenwolf GC. FGF8 initiates inner ear induction in chick and mouse. Genes Dev. 2005;19:603–613. doi: 10.1101/gad.1273605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang H, Fekete DM. Lineage analysis in the chicken inner ear shows differences in clonal dispersion for epithelial, neuronal, and mesenchymal cells. Dev Biol. 2001;234:120–137. doi: 10.1006/dbio.2001.0248. [DOI] [PubMed] [Google Scholar]
- Lecaudey V, Ulloa E, Anselme I, Stedman A, Schneider-Maunoury S, Pujades C. Role of the hindbrain in patterning the otic vesicle: a study of the zebrafish vhnf1 mutant. Dev Biol. 2006;303(1):134–143. doi: 10.1016/j.ydbio.2006.10.041. [DOI] [PubMed] [Google Scholar]
- Leger S, Brand M. Fgf8 and Fgf3 are required for zebrafish ear placode induction, maintenance and inner ear patterning. Mech Dev. 2002;119:91–108. doi: 10.1016/s0925-4773(02)00343-x. [DOI] [PubMed] [Google Scholar]
- Li JY, Lao Z, Joyner AL. Changing requirements for Gbx2 in development of the cerebellum and maintenance of the mid/hindbrain organizer. Neuron. 2002;36:31–43. doi: 10.1016/s0896-6273(02)00935-2. [DOI] [PubMed] [Google Scholar]
- Lin Z, Cantos R, Patente M, Wu DK. Gbx2 is required for the morphogenesis of the mouse inner ear: a downstream candidate of hindbrain signaling. Development. 2005;132:2309–2318. doi: 10.1242/dev.01804. [DOI] [PubMed] [Google Scholar]
- Liu W, Li G, Chien JS, Raft S, Zhang H, Chiang C, Frenz DA. Sonic hedgehog regulates otic capsule chondrogenesis and inner ear development in the mouse embryo. Dev Biol. 2002;248:240–250. doi: 10.1006/dbio.2002.0733. [DOI] [PubMed] [Google Scholar]
- Lumsden A, Keynes R. Segmental patterns of neuronal development in the chick hindbrain. Nature. 1989;337:424–428. doi: 10.1038/337424a0. [DOI] [PubMed] [Google Scholar]
- Mansour S, Goddard J, Capecchi M. Mice homozygous for a targeted disruption of the proto-oncogene int-2 have developmental defects in the tail and inner ear. Development. 1993;117:13–28. doi: 10.1242/dev.117.1.13. [DOI] [PubMed] [Google Scholar]
- Manzanares M, Nardelli J, Gilardi-Hebenstreit P, Marshall H, Giudicelli F, Martinez-Pastor MT, Krumlauf R, Charnay P. Krox20 and kreisler co-operate in the transcriptional control of segmental expression of Hoxb3 in the developing hindbrain. EMBO J. 2002;21:365–376. doi: 10.1093/emboj/21.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin F, Charnay P. Hindbrain patterning: FGFs regulate Krox20 and mafB/kr expression in the otic/preotic region. Development. 2000;127:4925–4935. doi: 10.1242/dev.127.22.4925. [DOI] [PubMed] [Google Scholar]
- Mark M, Lufkin T, Vonesch JL, Ruberte E, Olivo JC, Dolle P, Gorry P, Lumsden A, Chambon P. Two rhombomeres are altered in Hoxa-1 mutant mice. Development. 1993;119:319–338. doi: 10.1242/dev.119.2.319. [DOI] [PubMed] [Google Scholar]
- Maves L, Kimmel CB. Dynamic and sequential patterning of the zebrafish posterior hindbrain by retinoic acid. Dev Biol. 2005;285:593–605. doi: 10.1016/j.ydbio.2005.07.015. [DOI] [PubMed] [Google Scholar]
- McKay I, Muchamore I, Krumlauf R, Maden M, Lumsden A, Lewis J. The Kreisler mouse: a hindbrain segmentation mutant that lacks two rhombomeres. Development. 1994;120:2199–2211. doi: 10.1242/dev.120.8.2199. [DOI] [PubMed] [Google Scholar]
- McKay IJ, Lewis J, Lumsden A. The role of FGF-3 in early inner ear development: an analysis in normal and kreisler mutant mice. Dev Biol. 1996;174:370–378. doi: 10.1006/dbio.1996.0081. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. Newborn hearing screening: the great omission. Pediatrics. 1998;101:E4. doi: 10.1542/peds.101.1.e4. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. The Colorado newborn hearing screening project, 1992–1999: on the threshold of effective population-based universal newborn hearing screening. Pediatrics. 2002;109:E7. doi: 10.1542/peds.109.1.e7. [DOI] [PubMed] [Google Scholar]
- Nie X, Luukko K, Kettunen P. FGF signalling in craniofacial development and developmental disorders. Oral Dis. 2006;12:102–111. doi: 10.1111/j.1601-0825.2005.01176.x. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Pasqualetti M, Neun R, Davenne M, Rijli FM. Retinoic acid rescues inner ear defects in Hoxa1 deficient mice. Genet. 2001;29(1):34–39. doi: 10.1038/ng702. [DOI] [PubMed] [Google Scholar]
- Pickles JO, Chir B. Roles of fibroblast growth factors in the inner ear. Audiol Neurootol. 2002;7:36–39. doi: 10.1159/000046861. [DOI] [PubMed] [Google Scholar]
- Reifers F, Bohli H, Walsh EC, Crossley PH, Stainier DY, Brand M. Fgf8 is mutated in zebrafish acerebellar (ace) mutants and is required for maintenance of midbrain–hindbrain boundary development and somitogenesis. Development. 1998;125:2381–2395. doi: 10.1242/dev.125.13.2381. [DOI] [PubMed] [Google Scholar]
- Represa J, Leon Y, Miner C, Giraldez F. The int-2 proto-oncogene is responsible for induction of the inner ear. Nature. 1991;353:561–563. doi: 10.1038/353561a0. [DOI] [PubMed] [Google Scholar]
- Riccomagno MM, Takada S, Epstein DJ. Wnt-dependent regulation of inner ear morphogenesis is balanced by the opposing and supporting roles of Shh. Genes Dev. 2005;19:1612–1623. doi: 10.1101/gad.1303905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben R. Development and cell kinetics of the kreisler (kr/kr) mouse. Laryngoscope. 1973;83:1440–1468. doi: 10.1288/00005537-197309000-00006. [DOI] [PubMed] [Google Scholar]
- Schlosser G. Induction and specification of cranial placodes. Dev Biol. 2006;294:303–351. doi: 10.1016/j.ydbio.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Schneider-Maunoury S, Topilko P, Seitandou T, Levi G, Cohen-Tannoudji M, Pournin S, Babinet C, Charnay P. Disruption of Krox-20 results in alteration of rhombomeres 3 and 5 in the developing hindbrain. Cell. 1993;75:1199–1214. doi: 10.1016/0092-8674(93)90329-o. [DOI] [PubMed] [Google Scholar]
- Streit A. Origin of the vertebrate inner ear: evolution and induction of the otic placode. J Anat. 2001;199:99–103. doi: 10.1046/j.1469-7580.2001.19910099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatek PJ, Gridley T. Perinatal lethality and defects in hindbrain development in mice homozygous for a targeted mutation of the zinc finger gene Krox20. Genes Dev. 1993;7:2071–2084. doi: 10.1101/gad.7.11.2071. [DOI] [PubMed] [Google Scholar]
- Tomaski SM, Grundfast KM. A stepwise approach to the diagnosis and treatment of hereditary hearing loss. Pediatr Clin North Am. 1999;46:35–48. doi: 10.1016/s0031-3955(05)70079-1. [DOI] [PubMed] [Google Scholar]
- Torres M, Giraldez F. The development of the vertebrate inner ear. Mech Dev. 1998;71:5–21. doi: 10.1016/s0925-4773(97)00155-x. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG, Bhatt S, McMahon AP. Expression pattern of the FGF-related proto-oncogene int-2 suggests multiple roles in fetal development. Development. 1989;105:131–136. doi: 10.1242/dev.105.1.131. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Mansour SL. Fgf3 and Fgf10 are required for mouse otic placode induction. Development. 2003;130:3379–3390. doi: 10.1242/dev.00555. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Hatch EP, Karabagli H, Karabagli P, Schoenwolf GC, Mansour SL. Expression of mouse fibroblast growth factor and fibroblast growth factor receptor genes during early inner ear development. Dev Dyn. 2003;228:267–272. doi: 10.1002/dvdy.10362. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Ladher R, McWhirter J, Murre C, Schoenwolf GC, Mansour SL. Mouse FGF15 is the ortholog of human and chick FGF19, but is not uniquely required for otic induction. Dev Biol. 2004;269:264–275. doi: 10.1016/j.ydbio.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Wu DK, Nunes FD, Choo D. Axial specification for sensory organs versus non-sensory structures of the chicken inner ear. Development. 1998;125:11–20. doi: 10.1242/dev.125.1.11. [DOI] [PubMed] [Google Scholar]
- Xu H, Viola A, Zhang Z, Gerken CP, Lindsay-Illingworth EA, Baldini A. Tbx1 regulates population, proliferation and cell fate determination of otic epithelial cells. Dev Biol. 2007;302:670–682. doi: 10.1016/j.ydbio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yntema CL. An analysis of induction of the ear from foreign ectoderm in the salamander embryo. J Exp Zool. 1950;113:211–244. [Google Scholar]
- Yntema CL. Ear and nose. In: Hamburger V, editor. Analysis of Development. Saunders; Philadelphia: 1955. pp. 415–428. [Google Scholar]
- Zwilling E. The determination of the otic vesicle in Rana Pipiens. J Exp Zool. 1940;86:333–343. [Google Scholar]