

Abstract
Precision-cut tissue slices mimic specific organ toxicity because normal cellular heterogeneity and organ architecture are retained. To optimize the use of the smaller tissues of the mouse and to establish easy assays for tissue viability, a tissue chip based system was used to generate large numbers of samples from a single organ. Iodoacetamide (IAM), was used as a model toxicant, and assays for intracellular potassium (normalized to DNA content) were used to establish viability and toxicant susceptibility. Thereafter, assays that were more rapid and specific were pursued. Lysates from tissues incubated in 6-carboxyfluorescein fluoresced proportionately to concentrations of IAM, indicating disruption of cellular membranes. Similarly, FURA-2, a probe applied to lysates to measure calcium levels, fluoresced proportionately to IAM dosage. Monobromobimane, a fluorescent sulfhydryl probe, displayed a decrease in fluorescent intensity at higher IAM challenge; a finding confirmed with an absorbance assay with Ellman’s reagent. Importantly, the number of samples per organ/mouse was increased at least 3-fold and a significant time reduction per analysis was realized.
1. INTRODUCTION
Precision-cut tissue slices provide an excellent model for the study of specific organ toxicity (Brandon et al. 2003; Groneberg et al. 2002; Lerche-Langrand and Toutain 2000). Since a limited number of tissue slices can be generated from any given organ, minimizing the size of the tissue slices would increase the number of experiments available from a single animal. In this manner, the ability to create a large data set can be greatly increased, thus allowing multiple cellular endpoints to be studied within one organ from a single animal. Moreover, the use of multiple tissue chips from different organs from the same animal would further augment the amount of data generated. This would greatly decrease the number of animals required to complete a study. The attributes of the liver (relatively firm, largest organ in the mouse, most prevalent organ in tissue slicing) make it the ideal candidate to determine methods for a reduction in total biomass.
Tissue slices are typically 12 − 14 cell layers deep (about 250 μm), which is optimal for nutrient replenishment and oxygenation since the thickness of tissue slices is limited by the number of cell layers through which oxygen can penetrate (Drobner et al. 2000; Gahwiler et al. 1997). Tissue slices less than twelve cell layers thick detach during roller culture incubations, leading to a loss in viability (Vickers and Fisher 2004). In tissue slices more than fourteen cell layers thick, the innermost cells become oxygen-deprived, thus causing necrosis within this region. Since the optimal tissue slice thickness has been extensively described, the diameter of the tissue slice remains the sole manipulable variable. By decreasing the slice diameter, a significant conservation in both the number of animals used, as well as the associated costs, could be realized and is the basis of this investigation.
The significant decrease in total biomass of a tissue chip, as compared to a tissue slice, demands assays with more sensitivity than those currently in use. In previous studies with tissue slices, numerous viability indicators have been tested: the standard is intracellular potassium content (standardized to either wet weight or to total DNA content) (Gandolfi et al. 1996; Olinga et al. 1997). Extracellular release of lactate dehydrogenase and alanine aminotransferase has been well studied, since overt toxicity causes release of these enzymes into the medium (Fisher et al. 1991; Groneberg et al. 2002). Intracellular glutathione content has also been used as another marker of toxicant perturbation (Wijeweera et al. 1998). Such chemistry-based spectrophotometric assays are time-consuming compared to fluorescent-probe studies, where simple lysis of the cells releases analyte for sample determination. Compared to fluorescence, absorbance assays also have poor detection limits, which may become crucial with mass-limited samples from smaller tissue slices. Techniques to improve toxicant evaluation—such as a transition from chemistry-based absorption assays to faster, more sensitive fluorescent-probe assays—must rely on the use of proven methods for comparison.
These studies aim to establish precision-cut tissue chips as a method to greatly increase the number of possible samples per animal. Additionally, fluorescent probes for specific cellular processes can be used in rapid and simple assays as indicators of cellular insult.
2. MATERIALS AND METHODS
2.1 Materials
Fluorescent probes were purchased from Molecular Probes, Inc. (Eugene, OR). All other chemicals were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO), unless otherwise noted.
2.2 Tissue Chip Generation
C57BL/6 mice (Harlan Sprague-Dawley, Indianapolis, IN) were housed in the Animal Care Facility at the University of Arizona College of Medicine. Mice were anesthetized by CO2 asphyxiation and killed by cervical dislocation. Tissues were excised and immediately placed into oxygenated Krebs-bicarbonate slicing buffer (pH 7.4, 4 °C, 95:5 O2:CO2). Liver cores, obtained via an 8-mm biosy punch, were placed into an 8-mm insert and sliced approximately 250-μm thick with a Brendel/Vitron tissue slicer (Vitron, Inc., Tucson, AZ). Tissue slices were placed on a piece of dental wax, and three 4-mm samples—each comprising a “tissue chip”—were generated from a single 8-mm tissue slice, using a 4-mm biopsy punch (VWR).
2.3 Tissue Chip Incubation
A number of different incubation methods previously used for tissue slices were attempted with tissue chips: submersion in flasks, gyratory-agitated 24-well plates, and roller cultures (Olinga et al. 1997). A 1-h preincubation time was allowed during all experiments to allow sloughing of cells damaged during the slicing procedure. Tissue chips then were incubated in a model toxicant, iodoacetamide (IAM), at concentrations ranging 0 − 1 mM for up to 6 h.
For roller culture incubation, tissue chips were placed onto titanium mesh rollers, then into scintillation vials containing 1.7 ml of Dulbecco’s Modified Eagle Medium/F-12 (containing 2.24 g/l sodium bicarbonate and 50 μg/ml gentamicin), and were incubated in a dynamic roller culture incubator (Vitron, Inc.), which was gassed with humidified 95:5 O2:CO2 at 37 °C.
For the gyratory-agitated well plates (100 rpm), studies used a bacterial shaker incubator (Lab-Line Instruments, Inc., Model 3527, Melrose Park, IL) (37 °C, 95:5 O2:CO2). In order to humidify the chamber, 500-ml Erlenmeyer flasks filled with water were placed in the outer corners of the platform. Three types of 24-well plate incubation methods were explored in these experiments; normal plates, plates with a screen support and plates with 1-mm oxygenation holes drilled in the lid. Screens were supported within the well by steel washers, with a tissue chip placed directly on top of the metal screen, approximately 2 mm from the air:media interface Additionally, to be certain that the media was properly agitated, a ball bearing was placed below the screen to provide a constant stirring for fresh nutrients to the tissue chip. To ensure that the 24-well plates received proper oxygenation, 1-mm holes were drilled into plates at various locations to ensure that the airflow across the plate was sufficient to oxygenate the tissue chips (inlet hole drilled in the center of the plate lid, with vent holes drilled on each corner of the lid). Gas (95:5 CO2:O2) was directed to the plate inlet via a 27-gauge needle at a flow rate of approximately 1.5 l/min.
Another manner in which tissue slices had been previously incubated was within directly-oxygenated Erlenmeyer flasks (Olinga et al. 1997). In these studies, 6 − 12 tissue chips were placed directly into the media during incubation in the orbital shaker, and experiments were performed concurrent with corresponding roller incubations, to rule out tissue chip variability.
2.4 Traditional Viability Indicators
2.4.1 Tissue Chip Potassium Content
Precision-cut tissue chips were collected at the assigned time points by placing them in 1 ml doubly-distilled water. Tissue chips were homogenized by sonication for 10 s using a Kontes Micro Ultrasonic Cell Disrupter (Vineland, NJ) at power level 8. Tissue homogenate (800 μl) was added to a 10 mm × 100 mm plastic tube. To this, 50 μl bovine serum albumin (5 mg/ml) and 20 μl concentrated perchloric acid were added. Tubes were briefly vortexed and centrifuged at 1400 g for 10 min in a swinging bucket centrifuge (RT6000 Refrigerated Centrifuge, Savant Instruments Inc., Farmingdale, NY). Standard curves of potassium (0 − 0.25 μM) and digested calf thymus DNA (0 − 10 μg) were prepared in doubly-distilled water and were treated identically to samples. Potassium concentrations were measured by analyzing supernatants with a Cole-Parmer model 2655−00 Digital Flame Analyzer. Standards were plotted using the linear regression analysis tool in Microsoft Excel (Version 97). The concentrations of the samples were then calculated from this line.
The pellet was resuspended in ice-cold ethanol (3.65 ml) with hydrochloric acid (0.1 ml) and shaken for 30 min to dissociate any salts. DNA then was precipitated at −20 °C, and centrifuged at 1400 g for 15 min. The supernatant was decanted and the pellets were dried overnight. One hundred microliters of 3,5-diaminobenzoic acid (DABA) dihydrochloride (3 g/10 ml ddH2O) was added to each tube, and the tube was then heated for 30 min at 72 °C to allow DABA to intercalate into the DNA strands. After incubation, 200 μl of 1 N HCl was added to each tube. Two hundred microliters of this mixture was added to one well of a 96-well black plate and the fluorescence was determined using a fluorescent plate reader (Gemini XS, Molecular Devices, Sunnyvale, CA) with the excitation wavelength set at 410 nm and emission 500 nm (Azri et al. 1991).
2.4.2 MTT
For this assay, tissue chips were incubated for 45 min with 50 μl (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT, 1 mg/ml in fresh medium) within the scintillation vials. Medium was removed and the tissue chips were washed in PBS to remove residual MTT reagent. The tissue chips were then removed, placed into 500 μl isopropranol:HCl (0.4 M in absolute isopropanol), and briefly sonicated to release the formazan crystals from within the chip. Cellular debris was removed via centrifugation at 3000 g for 5 min. Two hundred microliters of the supernatant from the samples was then analyzed for absorbance at 562 nm on a Molecular Devices VersaMax plate reader using the 96-well format.
2.4.3. Ellman’s Reagent
Chips were homogenized in 200 μl 10% trichloroacetic acid, of which 10 μl of lysate was added to 50 μl of Ellman’s Reagent (50 mM sodium acetate, 2 mM 5,5'-dithiobis-(2-nitrobenzoic acid) in H2O) and 140 μl of Tris (pH 8.0), mixed thoroughly, and analyzed at 412 nm on an absorbance plate reader.
2.5 Fluorescent Probe Viability Indicators
2.5.1 Monobromobimane
Tissue chips were briefly sonicated in 500 μl phosphate-buffered saline (pH 7.4). The insoluble fraction was removed via centrifugation (3000 g, 5 min) and then incubated with 10 μM monobromobimane (MBB) for 15 min. Supernatants (200 μl) were then analyzed for MBB-sulfhydryl conjugates at excitation/emission of 392/492 nm in a 96-well plate.
2.5.2 FURA-2
In these studies, tissue chips were briefly sonicated in 500 μl PBS, and cellular debris was removed via centrifugation (3000 g, 5 min). FURA-2 was then incubated in the supernatant to a final concentration of 1 μM, allowed to bind calcium for 30 min, and 200 μl of the supernatant was analyzed for excitation/emission at 335/505 nm in a 96-well format.
2.5.3 6-Carboxyfluorescein (6-CF)
Chips were incubated in fresh culture medium containing 10 μM 6-carboxyfluorescein for 45 min, with a brief subsequent sonication of the tissue chip in 500 μl PBS (pH 7.4). After a 5 min centrifugation at 3000 g to remove any insoluble proteins, 200 μl of the supernatant was analyzed via fluorimetry (excitation/emission 492/517 nm) in a 96-well plate.
2.6 Statistics
Statistical methods were applied using SAS Version 9.0 (SAS Institute Inc., Cary, NC). Unless otherwise noted, one-way Analysis of Variance (ANOVA) tests were performed followed by Tukey’s multiple comparison test (P < 0.05). All N-values are in the corresponding figure legends.
3. RESULTS
3.1 Core Diameter, Tissue Chip Diameter, Incubation Technique
Smaller (2 mm) and larger tissue (20 mm) core sizes were examined to determine if these could successfully produce smaller tissue slices. However, both the larger and smaller diameter cores were shredded during the tissue slice procedure. The most consistent method was using a 4-mm biopsy punch on 8-mm diameter tissue slices, as shown in Figure 1. This procedure also allowed more selectivity for optimal tissue appearance; that is, the tissue slice procedure typically cuts areas with blood vessels, which can hinder tissue slice uniformity. With the use of this procedure, the means to generate more similar sample sets can be realized. Of the various incubation methods investigated, the dynamic roller culture incubation system enhanced viablity the most, as shown in Figure 2. This was especially true at earlier time points. In the 24-well plate format with screen support, tissue chips were torn apart on the underside, as the tissue chip was rotated and constantly rubbed against the metal screen. Therefore, the roller incubation was used for all subsequent experiments.
Figure 1.

Picture of tissue chip production. A. Whole tissue slice (8-mm). B. Remnants of the tissue slice. C. Three tissue chips.
Figure 2.
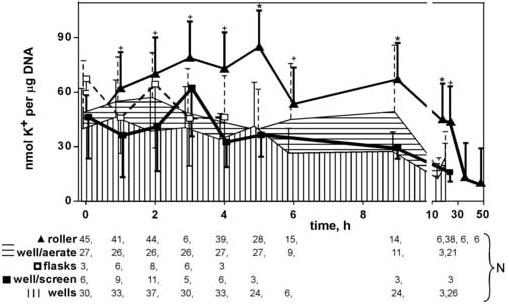
Comparison of incubation methods to enhance viability of tissue chips. Each data point represents the average of N samples (given in legend below the chart), with error bars representing standard deviation (shown unidirectional for clarity). Note break in x-axis. Asterisks mark roller incubations that are statistically different from the best alternative method (P < 0.05 by Tukey’s multiple comparison test). Crosses mark roller incubations that were not statistically different from best of the alternatives, but were statistically different from the second-best alternative (P <0.05, Tukey’s).
3.2 Model Toxicant - Iodoacetamide
The tissue chips thus produced were treated with iodoacetamide, a model toxicant that readily alkylates sulfhydryls without bioactivation, creating an S-carbamidomethyl cysteine. The alkylation rapidly results in cellular dysfunction, and—at higher concentrations—eventually, in general necrosis (van De Water et al. 1999). The 100 μM iodoacetamide dosage demonstrated obvious toxicity at 6 h, as determined by intracellular potassium content (Figure 3). Increasing the concentration of IAM to 1 mM caused almost total loss of potassium content from the tissue chip. Similarly, the MTT assay, which measures activity of mitochondrial reductases, revealed a dose-dependent response to IAM, with toxicity evident after 6 h at 10 μM IAM, as shown in Figure 4. Similar IAM dosages, therefore, were used for subsequent studies, and all toxicities were allowed to develop for 6 h.
Figure 3.
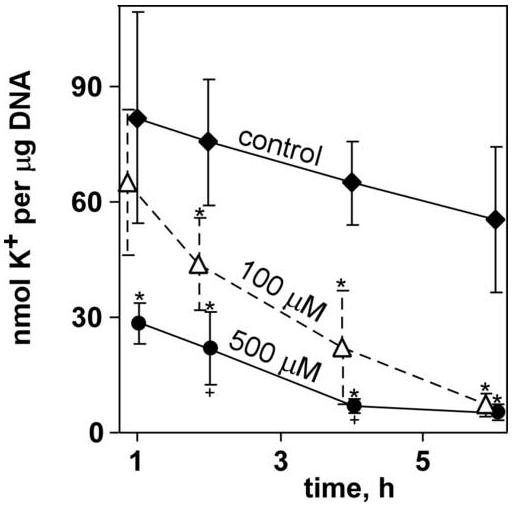
Effect of selected dosages of IAM on tissue chips over time. Data points represent averages (N = 6 to 15 for each data point) with error bars representing standard deviation; an exception is the control 6-h timepoint, for which N = 60. Asterisks mark data points that were statistically different from control; crosses mark 500 μM treatments that were statistically different from 100 μM treatment (P < 0.05, Tukey’s).
Figure 4.
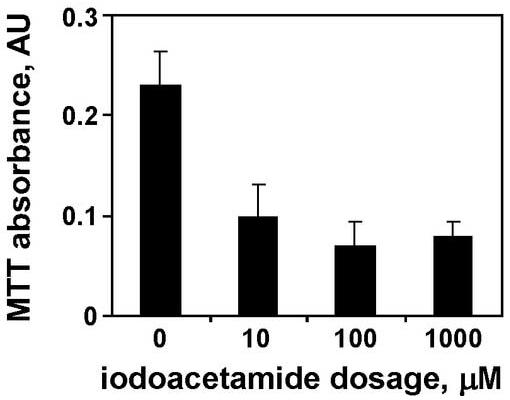
Absorbance from MTT assay for mitochondrial reductases in tissue chips. Data are averages from six tissue chips after 6 h at each dosage, with standard deviations as error bars. All treatments were statistically different from control at P < 0.05 (Student’s t-test with Welch’s correction).
3.3 Fluorescent Indicators for Tissue Chip Viability
Having established the viability and the response of the model toxicant in tissue chips with traditional viability indicators, fluorescent-probe assays were investigated, beginning with assays for intracellular sulfhydryls. Two methods were compared: the absorbance-based Ellman’s reagent (Haugaard et al. 1969; Rootwelt 1967; Sedlak and Lindsay 1968), and the fluorescence-based MBB assay (Kosower et al. 1979; Kosower et al. 1980). Following IAM challenge to the tissue chip, GSH levels were reduced in a dose-dependent manner, as shown both for absorbance of Ellman’s reagent and fluorescence from MBB assays (Figure 5).
Figure 5.
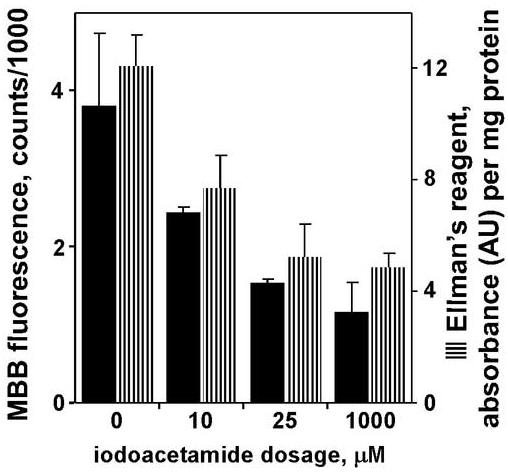
Sulfhydryl assays for viability of tissue chips after 6-h incubations in varying concentrations of iodoacetamide. Data are averages of N = 4 each for monobromobimane or N = 26 each for Ellman’s reagent, with error bars representing standard deviation. In both assays, the lowest treatment level (10 μM) was statistically different from control, and the two higher treatment levels (25 μm, 1 mM) were statistically different from the 10-μM treatment (P < 0.05, Tukey’s).
Given the success of the MBB fluorescence assay in tissue chip lysate, two other fluorescent probes for general viability indicators were investigated: FURA-2 as a calcium indicator, and 6-carboxyfluorescein to measure damage to membrane integrity. Tissue chips incubated with IAM displayed an increase in FURA-2 fluorescence, indicative of damage to normal cellular homeostasis and an increase in intracellular calcium levels, as shown in Figure 6. To further studies of the cellular membrane, tissue chips were directly incubated with 6-CF. Iodoacetamide increased the uptake of 6-CF (Figure 6), suggesting damage to the cellular membrane integrity that allowed 6-CF to enter the cell.
Figure 6.
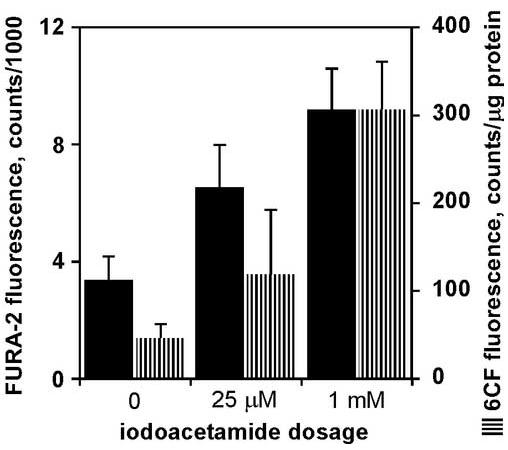
Fluorescent probes to measure membrane integrity of tissue chips after 6-h incubations in varying concentrations of iodoacetamide. Each bar represents average of data from 7 − 12 tissue chips, with error bars as standard deviation. In both assays, all three dosages were statistically different from each other (P < 0.05, Tukey’s).
4. DISCUSSION
Using a 4-mm biopsy punch on 8-mm tissue slices, smaller-diameter tissue chips were successfully generated, which greatly increased the number of samples generated from the same organ of a single animal. The extra time required to punch the tissue slice is minimal, usually only a few more seconds per tissue slice. Usually three 4-mm tissue chips could be produced from a single tissue slice, and 15 − 20 slices were produced per liver – with tissue chips, the number of samples per liver were increased to between 30 and 60. Therefore, a significant savings in the number of animals used per experiment has been achieved, increasing the total number of samples per animal. Moreover, this procedure allowed selectivity for optimal tissue appearance; blood vessels in the slices could be avoided when selecting punch targets for chips. With the use of this procedure, the means to generate larger, more similar sample sets was realized, allowing, the total number of analyses per experiment to be greatly augmented. Though previous studies have shown that shaker platform incubators can yield viability controls comparable to roller culture incubators (Olinga et al. 1997), the most consistent method for the incubation of tissue chips was the dynamic roller culture incubator.
Standard tissue-slice viability indicators—such as K+/DNA, MTT, and Ellman’s assays—were successfully applied to these tissue chips. However, the fluorescent probes 6-CF (for viability), FURA-2 (for calcium), and MBB (for sulfhdryls) were also useful, which made toxicity screening much quicker. FURA-2 was chosen because previous studies demonstrated this compound’s applicability with calcium analysis in simple buffer systems (McCall and Fierke 2000). Additionally, no enzymatic activation step is required, unlike with the acetyl methoxy (AM ester) derivative of FURA-2. As demonstrated with 6-CF, the application of these probes directly to the incubation media is possible, with subsequent analysis of the lysate in a relatively rapid manner. Compared to the standard indicator of tissue slice perturbation, intracellular potassium ion content, the amount of sample preparation time saved per experiment is conservatively approximated at 12 hours. Analyses which typically required overnight incubations or snap-freezing samples can now be done when the experimental time course is complete, thus allowing same-day results.
This reduction in required amount of tissue makes possible the use of an even smaller biomass. This is important because it allows the use of readily-available inbred mouse strains (with liver sizes less than 2 g) instead of the more commonly used, wild-type-only rats (liver ∼9 g). These fluorescent probes may also enable assays of smaller organs of the mouse. Applications with organs such as the spleen (100 mg), ovary (5 − 10 mg), and adrenal glands (2 − 4 mg) have not yet been demonstrated with tissue slices from mice. The fluorescent indicators described in these analyses could easily be applied to these other smaller tissues, since the weight of a single tissue chip was between 5- and 7-mg. Therefore, the examination of a potential toxicant in a large variety of organs from a single animal could now be realized, while maximizing the biomass of each organ, and decreasing the total analysis time.
In conclusion, a method is now in place to further the use of tissue slice technology as a high-volume method for toxicant evaluation. These studies provide the necessary groundwork to create a system which can accurately reflect target organ toxicity, and can also be used to quickly screen large numbers of compounds from the same organ of a single mouse. The multiple fluorescent probes allow for rapid detection, while providing multiple processes to be explored. Additionally, this decreased biomass allows many samples to be generated from one organ, with the potential of using the smaller organs of the mouse. This approach offers a number of benefits: less animal use, increased similarity of sample set, increased sample numbers, multiple endpoint analyses from the same animal, and a significant decrease in the time involved.
Acknowledgments
The authors would like to thank Dana D. Wise for reviewing the manuscript. This work was supported by NIH R33 CA97449 and NIEHS STTR R41 ES10668.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST STATEMENT The authors indicate no conflict of interest.
REFERENCES
- Azri S, Mata HP, Gandolfi AJ, Brendel K. CCl4-induced cytochrome P-450 loss and lipid peroxidation in rat liver slices. Adv Exp Med Biol. 1991;283:669–74. doi: 10.1007/978-1-4684-5877-0_84. [DOI] [PubMed] [Google Scholar]
- Brandon EF, Raap CD, Meijerman I, Beijnen JH, Schellens JH. An update on in vitro test methods in human hepatic drug biotransformation research: pros and cons. Toxicol Appl Pharmacol. 2003;189:233–46. doi: 10.1016/s0041-008x(03)00128-5. [DOI] [PubMed] [Google Scholar]
- Drobner C, Glockner R, Muller D. Optimal oxygen tension conditions for viability and functioning of precision-cut liver slices. Exp Toxicol Pathol. 2000;52:335–8. doi: 10.1016/S0940-2993(00)80059-7. [DOI] [PubMed] [Google Scholar]
- Fisher R, Nau H, Gandolfi AJ, Putnam CW, Brendel K. Valproic acid hepatotoxicity in human liver slices. Drug Chem Toxicol. 1991;14:375–94. doi: 10.3109/01480549109011640. [DOI] [PubMed] [Google Scholar]
- Gahwiler BH, Capogna M, Debanne D, McKinney RA, Thompson SM. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 1997;20:471–7. doi: 10.1016/s0166-2236(97)01122-3. [DOI] [PubMed] [Google Scholar]
- Gandolfi AJ, Wijeweera J, Brendel K. Use of precision-cut liver slices as an in vitro tool for evaluating liver function. Toxicol Pathol. 1996;24:58–61. doi: 10.1177/019262339602400108. [DOI] [PubMed] [Google Scholar]
- Groneberg DA, Grosse-Siestrup C, Fischer A. In vitro models to study hepatotoxicity. Toxicol Pathol. 2002;30:394–9. doi: 10.1080/01926230252929972. [DOI] [PubMed] [Google Scholar]
- Haugaard N, Lee NH, Kostrzewa R, Haugaard ES. Effects of a disulfide (Ellman’s reagent) and thiols on oxidative phosphorylation and ion transport by rat liver mitochondria. Biochem Pharmacol. 1969;18:2385–91. doi: 10.1016/0006-2952(69)90353-0. [DOI] [PubMed] [Google Scholar]
- Kosower NS, Kosower EM, Newton GL, Ranney HM. Bimane fluorescent labels: labeling of normal human red cells under physiological conditions. Proc Natl Acad Sci U S A. 1979;76:3382–6. doi: 10.1073/pnas.76.7.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosower NS, Newton GL, Kosower EM, Ranney HM. Bimane fluorescent labels. Characterization of the bimane labeling of human hemoglobin. Biochim Biophys Acta. 1980;622:201–9. [PubMed] [Google Scholar]
- Lerche-Langrand C, Toutain HJ. Precision-cut liver slices: characteristics and use for in vitro pharmaco-toxicology. Toxicology. 2000;153:221–53. doi: 10.1016/s0300-483x(00)00316-4. [DOI] [PubMed] [Google Scholar]
- McCall KA, Fierke CA. Colorimetric and fluorimetric assays to quantitate micromolar concentrations of transition metals. Anal Biochem. 2000;284:307–15. doi: 10.1006/abio.2000.4706. [DOI] [PubMed] [Google Scholar]
- Olinga P, Groen K, Hof IH, De Kanter R, Koster HJ, Leeman WR, Rutten AA, Van Twillert K, Groothuis GM. Comparison of five incubation systems for rat liver slices using functional and viability parameters. J Pharmacol Toxicol Methods. 1997;38:59–69. doi: 10.1016/s1056-8719(97)00060-9. [DOI] [PubMed] [Google Scholar]
- Rootwelt K. Quantitative determination of thiols and disulphides in urine by means of Ellman’s reagent and thiolated Sephadex and its application in cystinuria. Scand J Clin Lab Invest. 1967;19:325–30. doi: 10.3109/00365516709090646. [DOI] [PubMed] [Google Scholar]
- Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem. 1968;25:192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- van De Water B, Wang Y, Asmellash S, Liu H, Zhan Y, Miller E, Stevens JL. Distinct endoplasmic reticulum signaling pathways regulate apoptotic and necrotic cell death following iodoacetamide treatment. Chem Res Toxicol. 1999;12:943–51. doi: 10.1021/tx990054q. [DOI] [PubMed] [Google Scholar]
- Vickers AE, Fisher RL. Organ slices for the evaluation of human drug toxicity. Chem Biol Interact. 2004;150:87–96. doi: 10.1016/j.cbi.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Wijeweera J, Gandolfi J, Zheng XH. Interactive toxicity and stress protein expression by vinylidene chloride and monochloroacetate in precision-cut rat liver slices. Environ Health Perspect. 1998;106(Suppl 6):1319–23. doi: 10.1289/ehp.98106s61319. [DOI] [PMC free article] [PubMed] [Google Scholar]