

Abstract
Cancer is caused by dysregulation in cellular signaling systems that control cell proliferation, differentiation and cell death. Protein kinase C (PKC), a family of serine/threonine kinases, plays an important role in the growth factor signal transduction pathway. PKCε, however, is the only PKC isozyme that has been considered as an oncogene. It can contribute to malignancy by enhancing cell proliferation or by inhibiting cell death. This review focuses on how PKCε collaborates with other signaling pathways, such as Ras/Raf/ERK and Akt, to regulate cell survival and cell death. We have also discussed how PKCε mediates is antiapoptotic signal by altering the level or function of pro- and antiapoptotic Bcl-2 family members.
Keywords: Apoptosis, PKC, Akt, Ras, Raf, MAPK, Bcl-2, Bid
1. Introduction
An imbalance between life and death of cells is the hallmark of cancer. Cell growth is regulated by a complex cellular signaling network that coordinates extracellular signals with intracellular messages. Both genetic and epigenetic alterations lead to the genesis of cancer. Inappropriate expression or activation of cellular proto-oncogenes or inactivation of tumor suppressor genes can cause disordered cell growth.
Apoptosis is a highly orchestrated genetically determined cell suicidal program required to maintain tissue homeostasis and to eliminate unwanted cells, [1]. An aberration in apoptotic signaling may lead to many diseases. While too much apoptosis can cause neurological disorders, too little apoptosis can contribute to cancer. This process is also important for the treatment of cancer since most anticancer drugs kill cancer cells by inducing apoptosis.
Caspases, a family of cysteine proteases that cleave after aspartate residues, are central to the execution of apoptosis [2-4]. Caspases exist in inactive proenzyme form. In response to apoptotic stimuli, the local concentrations of initiator caspases are increased by protein-protein interaction leading to autoprocessing and activation [5]. Active initiator caspases then cleave effector procaspases to generate active caspases, thus initiating a cascade of events [6]. Activation of effector caspases in turn leads to cleavage of critical cellular proteins, resulting in cell death. While caspase-8, -9 and -10 participate in the initiation phase of apoptosis, caspase-3, -6 and -7 are involved in the execution phase of apoptosis. Caspase-2 can function as both initiator and effector caspase [7].
There are two major pathways of cell death. The extrinsic pathway is triggered by binding of ligand to members of tumor necrosis factor-α (TNF) receptor superfamily followed by oligomerization of the receptors and recruitment of procaspase-8 or -10 to the death receptors via adaptor proteins to form a death-inducing signaling complex (DISC) [8]. The intrinsic or mitochondrial pathway is triggered by cellular stress, such as DNA damage, which induces the release of mitochondrial cytochrome c and allows apoptosis promoting activating factor -1 (Apaf-1) to interact with the initiator procaspase-9 to form an active apoptosome complex [4]. Cross-talk between the receptor-mediated pathway and mitochondria occurs when active caspase-8 cleaves Bid and the truncated Bid translocates to the mitochondria causing activation of Bax/Bak, release of cytochrome c and activation of caspase-9 and downstream caspases [9-12].
Activation of caspases is regulated by various pro- and antiapoptotic signal transduction pathways. These include Bcl-2 family proteins [13], catalytically inactive versions of caspases (e.g., FLIP) [14] and inhibitors of apoptosis (IAP) [15]. Several protein kinases, including Akt/protein kinase B (PKB), protein kinase C (PKC) and mitogen-activated protein kinases (MAPKs) that play a central role in signal transduction also regulate activation of caspases. A deregulation in these kinases can contribute to human diseases.
Protein kinase C (PKC) is a family of phospholipid-dependent serine/threonine kinases that regulate a wide variety of cellular functions [16-21]. The PKC family consists of at least eleven members that have been categorized into three groups based on their structure and biochemical properties. Conventional or cPKCs (α,βI,βII and γ) require Ca2+ and diacylglycerol (DAG) for their activation. Novel or nPKCs (δ, ε, η and θ) are dependent on DAG but not Ca2+ whereas atypical or aPKCs (ζ and λ/ι) are independent of both Ca2+ and DAG [16-21]. The full-length PKCs contain an N-terminal regulatory domain and a C-terminal catalytic domain joined by a hinge region. The regulation of PKC is complex [19]. PKC resides in an inactive conformation due to interaction of the regulatory domain with the catalytic domain through pseudosubstrate sequences. The binding of cofactors, such as Ca2+ and DAG/phorbol esters to the regulatory domain induces a conformational change in the enzyme thereby exposing the substrate-binding site and catalysis takes place. Activation of PKC is usually associated with membrane translocation and prolonged cellular exposure to PKC activators can cause its degradation or down-regulation. Phosphorylation of PKCs at the activation loop, turn motif and hydrophobic site in the carboxyl terminal domain prime them for activation by cofactors [22]. The negative regulation on PKC activity can also be relieved by proteolytic dissociation of the regulatory domain from the catalytic domain by cleavage of PKC at the hinge region. The catalytic fragment thus generated does not require any activators or cofactors for activation.
Members of the PKC family have been shown to regulate cell death by apoptosis. Several PKC isozymes, including PKCδ, -ε, -θ and -ζ are substrates for caspases [23-27]. Cleavage of PKCs by caspases at the hinge region causes cofactor-independent activation [28]. Proteolytic activation of PKCδ has been directly linked with apoptosis [28]. PKCε, another member of novel PKCs, is believed to function as an antiapoptotic protein [23, 29-34]. It is the only PKC isozyme that has been associated with oncogenesis [35, 36]. There are several excellent review articles describing the role of PKCε in cardioprotection [37-39]. Although this review is primarily targeted to the cancer research aficionados, we have provided a comprehensive review of how PKCε interacts with various signaling pathways to regulate life and death of a cell and should benefit researchers in other fields as well.
2. PKCε, a unique PKC with oncogenic potential
PKCε is the only PKC isozyme that has been shown to behave as an oncoprotein [35, 36]. The first hint that PKCε may be involved in malignancy came from the study of Baxter et al. [40]. PKCε was constitutively active in a small cell lung cancer (SCLC) cell line. The majority of PKC ε was present in the particulate function and these cells expressed a cytosolic 40-kDa catalytic fragment of PKCε. The mitogenic effect of gastrin-releasing peptide was associated with an increase in the 40-kDa fragment [40]. Overexpression of PKCε in NIH 3T3 cells [36] and Rat 6 embryo fibroblasts [35] caused disordered cell growth, such as stimulation in growth rate, increased saturation density and growth in soft agar, and induced tumor formation in nude mice. Overexpression of PKCε was also oncogenic in colonic epithelial cells [41, 42]. Although a progressive increase in PKCε was noted in rat liver with increase in malignancy, overexpression of PKCε in rat liver MH1C1 cells did not result in disordered cell growth or tumor formation in nude mice [43]. Thus, whether or not PKCε will contribute to neoplastic transformation depends on the particular tissue or cell type.
Although the catalytic activity of PKCε is believed to be required for its oncogenic activity, PKCε regulatory domain can also exert effects independent of PKCε catalytic activity. Using reciprocal chimeras of PKCδ and PKCε regulatory and catalytic domains, it has been shown that both the catalytic and regulatory domains of PKCε could induce transformation in NIH 3T3 fibroblasts but only the catalytic domain was crucial for the tumorigenicity of PKCε in nude mice [44]. The N-terminal domain of PKCε, which contained part of the regulatory domain of PKCε, was associated with thyroid tumorigenicity [45]. PKCε gene was amplified and rearranged in WRO thyroid carcinoma cell line causing overexpression of a truncated PKCε (Tr-PKCε), (amino acids 1-116) that acted as a dominant-negative inhibitor of translocation of the wild-type PKCε to the membrane. Overexpression of Tr-PKCε caused an increase in saturation density and a decrease in apoptosis but had no effect on anchorage-independent growth or tumor formation. These results suggest that cell transformation and tumorigenicity could be uncoupled. However, no large-scale gene rearrangements in the PKCε gene were noted in thyroid papillary tumors compared to normal thyroid tissues although the levels of PKCε were substantially reduced in these tumors [46]. Thus, decreased abundance of PKCε was associated with thyroid carcinogenesis. Introduction of the PKCε regulatory domain was also sufficient to induce neuronal differentiation independent of PKCε catalytic activity [47].
PKCε was shown to be an important mediator of squamous cell carcinogenesis [48]. Targeted overexpression of PKCε in mouse epidermis caused development of squamous cell carcinoma (SCC) following application of 7,12-dimethylbenz(a)anthracene (DMBA) and 12-O-tetradecanoylphorbol-13-acetate (TPA) protocol or ultraviolet radiation (UVR). It was proposed that shedding of the proinflammatory cytokine TNF ectodomain was responsible for the development of SCC in PKCε transgenic mice [49]. Manganese superoxide dismutase (MnSOD) suppressed tumor promotion in a multi-stage skin carcinogenesis model by inhibiting PKCε [50]. The level of PKCε was increased in primary high-grade astroglial brain tumors [51] and in brain tumor-derived astroglial cell lines and overexpression of dominant-negative PKCε inhibited proliferation of U-373MG cells [52]. PKCε overexpression was sufficient to maintain prostate cancer growth and survival following androgen ablation and to convert androgen-dependent prostate cancer LNCaP cells to an androgen-independent variant [53].
PKCε has also been linked with invasion and metastasis. PKCε and -α membrane localization, which reflects the activation status of the enzymes, was associated with the invasiveness of human renal cell carcinomas (RCCs) [54]. NIH 3T3 fibroblasts overexpressing PKCε were invasive and displayed polarized morphology with extended long cellular membrane protrusions [55]. It has been reported that the regulatory domain of PKCε contains an actin binding site and direct interaction of PKCε with actin is essential for invasion and metastasis of tumors grown in vitro as well as in vivo [55]. PKCε transgenic mice developed highly metastatic SCCs [56, 57]. PKCε positively regulated integrin-mediated cell invasion and motility in glioma cells through interaction with the scaffolding protein Receptor for Activated C Kinase-1 (RACK1) which targeted PKCε to integrin β chains, leading to integrin clustering, focal adhesion formation, and increased adhesion and migration [58]. PKCε has been associated with aggressive, invasive and motile phenotype in breast cancer and human head and neck squamous cell carcinoma (HNSCC) [59, 60]. It has been postulated that PKCε mediates its action on invasion and motility via activation of Rho GTPases, which contain putative PKC phosphorylation sites [59]. Knockdown of PKCε decreased the level, activation status and phosphorylation of Rho GTPases. Furthermore, ectopic expression of constitutively-active RhoA and RhoC GTPase in PKCε-deficient cells completely restored invasion and motility defects [60]. Thus, Rho GTPases, which play an important role in invasion and motility, could be important downstream targets of PKCε.
3. PKCε, a killer or a savior?
PKCε can contribute to oncogenesis not only by inducing disordered cell growth but also by inhibiting cell death. Several studies have reported that the localization of PKCε is affected during apoptosis. Glucocorticoid-induced apoptosis was inhibited by PKC inhibitor GF 109203X and was associated with selective translocation of PKCε from the cytosol to the membrane in immature thymocytes, suggesting that activation of PKCε was linked to glucocorticoid-induced apoptosis [61]. The anti-proliferative activity of tamoxifen was also associated with membrane translocation of PKCε [62]. However, PKCε acted as an antiapoptotic protein in HT58 human lymphoblastic cells since TPA-induced downregulation of PKCε was associated with staurosporine-induced apoptosis [63]. Cytosolic translocation of PKCε and -δ correlated with ceramide-induced apoptosis in leukemic cells [64]. PKC activators, such as TPA that induced membrane translocation, inhibited ceramide-induced cytosolic translocation of PKCδ and -ε and prevented ceramide-induced apoptosis. It is not clear whether cytosolic translocation of PKCδ, -ε or both was associated with the apoptotic induction. In addition, since translocation of PKCs from the cytosol to the membrane is an indication of its activation, reverse translocation of PKCs from the plasma membrane to the cytosol may suggest that inactivation of PKC rather than its activation is responsible for ceramide-induced apoptosis. Furthermore, it remains to be established if cytosolic translocation of PKCδ and -ε simply correlates with apoptosis or is the cause of apoptosis. These earlier studies relied on pharmacological activators and inhibitors of PKCs that lacked specificity to PKCε. TPA also inhibited TNF- and calphostin-induced apoptosis in U937 histiocytic lymphoma cells [32]. A cell-permeable peptide that inhibited PKCε translocation blocked the protective effect of TPA on TNF- and calphostin C-induced apoptosis, suggesting that membrane translocation of PKCε by TPA was in fact necessary for its antiapoptotic effect. PKCε knockout mice exhibited significant decrease in survival, thus establishing a role for PKCε as an antiapoptotic protein [65].
PKCε has been shown to regulate both DNA damage- and receptor-mediated apoptosis. Ovarian cancer 2008 cells that acquired resistance to the DNA damaging anticancer drug cisplatin exhibited higher level of PKCε compared to drug-sensitive parental cells [66]. PKCε was also associated with induction of P-glycoprotein, which contributes to multiple drug resistance, in LNCaP cells [67]. We have shown that overexpression of PKCε in rat embryo fibroblasts inhibited apoptosis induced by cisplatin [29], thus providing first direct evidence that PKCε functions as an antiapoptotic protein during DNA damage-induced apoptosis. Increased level of PKCε was associated with chemoresistance of non-small cell lung cancer (NSCLC) cell lines and lack of PKCε was associated with chemosensitivity of SCLC cells [30]. Ectopic expression of PKCε in SCLC cells contributed to resistance to several anticancer drugs, including etoposide and doxorubicin [30]. Thus, PKCε inhibits DNA damage-induced apoptosis in a variety of cell lines and in response to several DNA damaging agents.
Several studies demonstrated that PKCε plays a protective role during receptor-mediated cell death. We have shown that the status of PKCε determined the ability of the PKC inhibitor bisindolylmaleimide (GF109203X) to sensitize breast cancer cells to TNF [68]. MCF-7, BT-20 and MDA-MB-231 cells that were most responsive to bisindolylmaleimide-mediated sensitization to TNF contained relatively high level of PKCε and proteolytic cleavage of PKCε correlated with TNF-induced cell death. Ectopic expression of PKCε protected breast cancer MCF-7 cells against TNF- [23] and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis [34]. It has been reported that cellular susceptibility to TRAIL cannot be explained by the status of TRAIL receptor expression but does correlate with PKCε level [34, 69]. Introduction of dominant-negative PKCε [69] or knockdown of PKCε [33] sensitized glioma cells to apoptosis. Variable responses of melanoma cells to TRAIL-induced apoptosis were also attributed to PKCε level. Introduction of wild-type PKCε decreased and dominant-negative PKCε enhanced TRAIL-induced apoptosis in melanoma cells [31].
4. PKCε, under attack by killer caspases
PKCε not only regulates apoptosis but it is also cleaved by caspases in response to several apoptotic stimuli, including chemotherapeutic agents [70], starvation [71] and TNF [23]. The cleavage of PKCε was inhibited by a cell permeable inhibitor of caspase-3, and PKCε was cleaved by purified human recombinant caspase-3, suggesting that PKCε is a substrate for caspase-3 [70, 71]. We have, however, found that PKCε was cleaved in response to TNF in MCF-7 cells lacking functional caspase-3 [23]. Recombinant caspases exhibit overlapping substrate specificities in in vitro cleavage assays. Caspase-3 and -7 have similar substrate specificities and they both recognize DXXD sequence. PKCε contains a DDVD↓C site that is recognized by caspase-3 and -7 [72]. Although both caspase-3 and -7 cleaved PKC at the DDVD↓C site, it was predominantly processed at the SSPD↓G site both in vitro and in intact cells [23, 70, 71]. Our results suggest that caspase-7 is the major caspase that cleaves PKCε at Asp383 site in intact cells [23].
The functional significance of proteolytic activation of PKCε remains to be established. Hoppe et al. have shown that the introduction of the catalytic domain or cleavage-resistant mutant of PKCε had no effect on cell death in AKR-2B fibroblasts [71]. We have, however, shown that blockage of caspase cleavage site at Asp383 prevented antiapoptotic effect of PKCε in MCF-7 cells [23], suggesting that proteolytic activation of PKCε may be necessary for its antiapoptotic function. In contrast, a caspase-resistant PKCε mutant (D383A) was more protective than PKCε against TRAIL-induced apoptosis in glioma cells [33], suggesting that the cleavage of PKCε contributed to the apoptotic effect of TRAIL. Since Asp383 site is located at the hinge region, cleavage of PKCε by caspases at SSPD↓G would generate a carboxy-terminal fragment containing the catalytic domain. In contrast, cleavage of PKCε at the DDVD↓C site would generate a carboxy-terminal fragment lacking the ATP binding site. We have shown that treatment of MCF-7 cells with TNF results in PKCε activation and mutation at the D383A site but not D451A site prevents this activation [23]. It is conceivable that in cells containing high levels of caspase-3, PKCε is processed further at DDVD↓C site following initial cleavage at SSPD↓G. This may result in downregulation of PKCε catalytic domain and a decrease in PKCε activity. Therefore, cleavage of PKCε may potentiate rather than protect against cell death. Thus, the cellular context may play an important role in deciding whether proteolytic cleavage of PKCε will induce, inhibit or have no effect on apoptosis.
5. PKCε and Ras/Raf/MAPK signaling
Mitogen-activated protein kinases (MAPK) are a family of structurally-related serine/threonine protein kinases that coordinate various extracellular signals to regulate cell proliferation, differentiation and cell survival [73-75]. Raf-1 interacts with the GTP-binding protein Ras, which recruits Raf-1 to the plasma membrane, where it can be activated by phosphorylation. Activation of Raf-1 initiates MAPK signaling cascade by activating mitogen-activated kinase kinase (MEK), which in turn activates extracellular signal-regulated kinase (ERK)-1 and -2.
Several studies suggested the involvement of Ras/Raf/MAPK pathway in mediating the oncogenic function of PKCε. First, overexpression of PKCε was associated with increase in Raf-1 kinase activity in Rat 6 fibroblasts [76], rat colonic epithelial cells [42], and COS cells [77]. Second, PKCε interacted with Raf-1 as evident by the presence of PKCε in Raf-1 immunoprecipitates [76]. Third, introduction of dominant-negative Raf-1 reversed the oncogenic effect of PKCε in Rat 6 fibroblasts and colonic epithelial cells [42, 76]. Fourth, overexpression of PKCε reversed growth inhibition caused by dominant-negative Ras in NIH 3T3 cells, and rat colonic epithelial cells [41, 77]. Expression of dominant-negative Ras had little effect on PKCε-mediated neoplastic transformation [41, 76]. These results suggest that PKCε interacts with the Ras signaling pathway acting downstream of Ras but upstream of Raf-1.
There are controversies whether or not PKCε directly phosphorylates and activates Raf-1. Cai et al have shown that human recombinant PKCε could activate Raf-1 in an in vitro kinase assay [77]. In contrast, Ueffing et al. have demonstrated that although PKCε interacted with Raf-1 at the kinase domain it activated Raf-1 indirectly by inducing secretion of autocrine growth factors that activated Raf-1 [78]. The production of TGFβ was considered to be one component contributing to Raf-1 activation since TGF-β was produced in PKCε overexpressing cells but not in parental cells and recombinant TGFβ resulted in activation of Raf-1. In addition, the production of active forms of TGFβ2 and β3 was associated with the oncogenic activity of PKCε [79].
The involvement of the MAPK pathway in mediating the growth promoting effect of PKCε is cell type-dependent. It has been reported that overexpression of PKCε enhanced nerve growth factor (NGF)-induced phosphorylation of ERK in PC12 pheochromocytoma cells and increased NGF-induced neurite outgrowth [80]. Ceramide inhibited growth of human embryonic kidney (HEK) 293 cells by attenuating ERK activity in a PKCε-dependent manner [81]. Ceramide directly inhibited PKCε activity in an in vitro kinase assay and interfered with the ability of PKCε to form a complex with Raf-1 and ERK in response to insulin-like growth factor [81]. It has been reported that transformation by oncogenic Ha-ras caused transcriptional activation of cyclin D1 via MEK/ERK in a PKCε-dependent manner. Expression of dominant-negative PKCε inhibited ERK activation by constitutively active Raf-1 [82]. However, overexpression of dominant-negative mutant of PKCε completely blocked proliferation of U-373MG human astrocytoma cells without affecting activation of MAPK [52]. Acquisition of androgen independence of LNCaP cells by PKCε overexpression was associated with constitutive activation of Raf-1 and ERK [53]. It has been suggested that PKCε acts upstream of Raf-1/ERK signaling and contributes to deregulation in cell cycle progression of LNCaP cells in the absence of testosterone through hyperphosphorylation of Rb [53]. Activated PKCε could stimulate the expression of parathyroid hormone-related protein, which promotes metastatic potential and proliferation of breast cancer cells in a MEK/ERK1-dependent manner [83]. Constitutive activation of ERK is frequently found in human melanoma [84]. ERK is a target of metabotropic glutamate receptor 1 (Grm1), a G protein coupled receptor, which has been associated with melanocytic neoplasia [85]. Introduction of kinase-dead mutant of PKCε inhibited Grm1-mediated ERK activation. Thus, PKCε is an important signaling molecule used by oncogenic Grm1 to activate ERK and to contribute to melanoma.
PKCε has also been implicated in ultraviolet (UV)-induced apoptosis and tumor promotion [86, 87]. It has been proposed that UVC irradiation could promote carcinogenesis by activating ERK via PKCε [87]. PKCε and PKCδ were involved in UVB-induced apoptosis via activation of ERK [86]. PKCε was also shown to contribute to hypoxia that promotes tumor progression [88]. The glucose-regulated protein 78 (GRP78) was overexpressed in primary gastric tumors and its expression was induced by chronic hypoxia in gastric cancer cells. It has been shown that PKCε directly participated in GRP78 induction by activating AP-1 via Raf-1/MEK/ERK pathway [88].
6. PKCε: AKTing in concert to promote survival
Akt, also known as PKB, a cellular homolog of the oncogene product v-Akt encoded by AKT8 retrovirus, belongs to a family of serine/threonine kinases [89-91]. It acts downstream of phosphoinositide 3-kinase (PI3-K) and plays a critical role in cell survival and oncogenesis [92]. Activation of Akt occurs when it is phosphorylated at both Thr308 in the activation loop and Ser473 in the C-terminal domain [93]. It is well established that phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates Akt at the Thr308 site [94, 95]. The identity of the kinase that phosphorylates the Ser473 site (designated PDK2), however, has been elusive. To date several candidates have been proposed, including integrin-linked kinase (ILK) [96, 97], mitogen-activated protein kinase-activated kinase-2 (MAPKAPK-2) [93, 98], DNA-dependent protein kinase (DNA-PK) [99], and rictor-mTOR complex [100]. Active Akt phosphorylates a large number of substrates, including GSK-3, caspase-9, Bad, MDM2 and Forkhead transcription factors, to mediate its antiapoptotic effects [101].
There is considerable evidence for PKCε-mediated increase in Akt phosphorylation and activity [102-104]. Matsumoto et al. reported that overexpression of a kinase-dead PKCε in CHO cells decreased Akt phosphorylation at Ser473 in response to insulin, hydrogen peroxide and heat shock, suggesting that PKCε activity was required for the phosphorylation of Akt [105]. Akt has been reported to be a downstream effector of PKCε for ethanol-induced cardioprotection because ethanol consumption caused an increase in expression and activity of PKCε and Akt, and inhibition of PKCε prevented the increase in Akt activity [104]. Cardiac-specific overexpression of PKCε in mice resulted in enhanced Akt phosphorylation at Ser473 [106] and protection against ischemia-reperfusion injury [107]. Li et al. found that overexpression of PKCε did not enhance Akt activity, nor did it protect 32D myeloid progenitor cells from interleukin-3 withdrawal [108]. This supports the notion that increased Akt activity may be required to mediate the antiapoptotic effects of PKCε. We have shown that overexpression of PKCε in MCF-7 cells increased Akt phosphorylation (Ser473) and depletion of Akt by siRNA inhibited the antiapoptotic effect of PKCε [102]. Therefore, it is reasonable to propose that these oncogene products collaborate to promote cell survival.
Several studies suggest that PKCε can directly interact with Akt although the functional significance of the association was not investigated in every case. A physical interaction between Akt and PKCε in cardiomyocytes was determined by functional proteomics [107]. Immunoprecipitation analyses revealed association of Akt and PKCε in several different cell lines, including recurrent prostate cancer CWR-R1 cells [103], breast cancer MCF-7 cells [102] and rat glomerular mesangial cells [109]. Generally, interaction of these two proteins was associated with an increase in Akt phosphorylation at Ser473 and consequently, resistance to stimulus-induced apoptosis. We have shown that PKCε activated Akt by enhancing interaction between Akt and DNA-PK, resulting in phosphorylation of Akt at Ser473 site [102]. In contrast, although PKCε and Akt were present in a constitutive complex in rat glomerular mesangial cells, neither Akt phosphorylation nor kinase activity was affected [109]. Interestingly, signaling via both proteins was required for efficient MAPK activation, suggesting that the PKCε-Akt complex can cross-talk with a third pathway to mediate its antiapoptotic effects.
PKCε may also enhance Akt activity indirectly. The most compelling example comes from reports of the reciprocal regulation of PKCε and β1 integrin signaling. PKCε overexpression increased β1 integrin protein levels, augmented the open (active) conformation of β1 integrin [103] and increased recycling of β1 integrin to the cell surface [110]. Engagement of integrins can trigger signaling through the PI3K pathway [111, 112]. In turn, integrins can activate PKCε [113]. Thus, PKCε, Akt and integrins are involved in a positive feed-back loop. PKCε overexpression in Rat 6 fibroblasts resulted in release of growth factors including TGFβ1 [78], which is a known activator of Akt Ser473 phosphorylation [114]. Therefore, PKCε may trigger Akt signaling via an autocrine loop. Thus, the indirect regulation of Akt by PKCε can involve integrins or secretion of growth factors that can elicit signaling through the pro-survival PI3K pathway.
Okhrimenko et al. showed that depletion of PKCε decreased Akt protein level and that overexpression of PKCε prevented TRAIL-induced degradation of Akt [33]. The authors suggested that the downregulation of Akt could be due to activation of caspase-3, since Akt is a substrate for caspase-3 [115]. Similarly, increased PKCε activity in transgenic mouse hearts resulted in a 6- to 10-fold induction in Akt protein levels [106]. PKCε overexpression also upregulated expression of stress-activated signaling pathways, though caspase activity was not studied [106]. Thus, it appears that the regulation of Akt by PKCε may not be restricted to phosphorylation, but also involves regulation of Akt protein levels. It is most likely that this effect is indirect, via changes in caspase-3 activation status. Whether PKCε can directly regulate Akt expression is questionable. For example, in MCF-7 cells that lack caspase-3, exposure to TNF did not affect Akt levels [102]. These observations, however, add a further layer of complexity to the interaction of PKCε with the PI3K pathway.
Contrary to the data cited above, there are some reports that suggest that PKC negatively regulates Akt function [116-121], and this was associated with increased apoptosis. Suppression of Akt phosphorylation in response to PKC activators or adenoviral expression of PKCε has been shown to occur via PP2A [117, 119]. Moreover, TPA co-treatment diminished the capacity of IGF to protect against UVC-induced cell death, while PKC inhibitors enhanced cell survival [119]. Similarly, in myeloid cell lines, Liu and colleagues showed that wild-type or constitutively-active PKCε attenuated cell proliferation stimulated by granulocyte colony-stimulating factor [120]. This inhibitory effect was associated with a decrease in Akt phosphorylation. Surprisingly, in the same model, IL3-stimulated cell growth was unaffected despite decreased Akt phosphorylation seen with PKCε overexpression. This suggests that the role of PKCε in cell survival depends on the stimulus and may be disconnected, in some cases, from its effect on Akt phosphorylation. Thus, there are contrasting reports as to whether PKCε can increase or suppress Akt activation and this appears to be determined in a cell-type and stimulus-specific manner. However, the evidence supporting cooperation between Akt and PKCε to promote survival is an overwhelming one.
7. PKCε and Bcl-2 family: many partners in the dance of death
The Bcl-2 family of proteins consists of at least 20 members that can be classified into anti- (e.g., Bcl-2, Bcl-xl, Mcl-1, and Bcl-w) and proapoptotic (e.g., Bax, Bad and Bid) family members based on structural and functional characteristics [122]. The function of these anti- and proapoptotic family members can be regulated by changes in expression (altering the ratio of anti- to proapoptotic proteins), post-translational modification (e.g., phosphorylation) or subcellular localization (e.g., mitochondrial translocation and association).
A number of studies have been performed in different systems using general PKC activators, including TPA and bryostatin-1 and determining effects on Bcl-2 family members. In general, PKC activator-mediated alterations in Bcl-2 were associated with protection/resistance from spontaneous or stimulus-induced apoptosis in a variety of cells, including B-CLL cells [123, 124], rat cardiomyocytes [125] and GT1-7 neuronal cells [126]. In contrast, there is some evidence that PKC can enhance drug-induced apoptosis by increasing the expression of BAD [127] or by inhibiting the expression of Bcl-2 [128]. These observations have been difficult to integrate because multiple PKC isoforms, both pro- and antiapoptotic, can be activated. Furthermore, prolonged exposure of cells to PKC activators could downregulate PKC protein levels. Therefore, the time of exposure to these agents is also a critical determinant of the final outcome of treatment.
Data supporting isoform-specific regulation of Bcl-2 family members are sparse, but quite consistent in that antiapoptotic PKCε enhances antiapoptotic Bcl-2 members [34, 129] while inhibiting the proapoptotic members of the family [31, 34, 130, 131]. Suzuki et al. first reported that intense expression of Bcl-2 during the development of pregnancy-dependent mammary tumors (PDMTs) to malignant tumors was associated with overexpression of PKCε but not of other isozymes, such as PKCα, -δ, -η, -ζ or –λ[132]. Gubina et al. provided first direct evidence that overexpression of PKCε could increase Bcl-2 protein levels and prevent both cell death and downregulation of Bcl-2 associated with IL-3 withdrawal in TF-1 hematopoietic cells [129]. Expression of truncated N-terminal domain of PKCε in thyroid cancer PCCL3 cells also caused an increase in Bcl-2, decrease in Bax and resistance to doxorubicin-induced apoptosis [45]. In MCF-7 cells, however, overexpression of the PKCε regulatory domain decreased Bcl-2 protein levels and increased susceptibility to apoptosis induced by multiple factors although the effects of PKCα regulatory domain was more pronounced [133]. It has been reported that EPO-driven upregulation of PKCε levels renders differentiating erythroid cells resistant to TRAIL, likely via Bcl-2 upregulation [134].
We recently showed that overexpression of PKCε in MCF-7 cells resulted in an increase in Bcl-2 at the mRNA and protein level with a concomitant decrease in proapoptotic Bid [34]. This dual regulation of pro- and antiapoptotic members of the Bcl-2 family contributed to TRAIL resistance. An increase in Bcl-2 phosphorylation was also associated with refraction to apoptosis [135]. A PKCε inhibitory peptide efficiently inhibited Bcl-2 phosphorylation and augmented hydrogen peroxide-induced apoptosis in a concentration-dependent manner in rat cardiomyocytes [135].
Overexpression of constitutively active PKCε increased phosphorylation of Bad at Ser112 [130, 136] but not Ser136 and inhibited CH11 (a FAS ligand mimic)-induced apoptosis in Jurkat T cells [130]. An interaction between PKCε and Bax was identified in CWR22 prostate tumor xenografts. In addition, PKCε-deficient LNCaP cells were sensitive to PMA-induced apoptosis and forced expression of PKCε in these cells conferred resistance to PMA-mediated apoptosis by preventing Bax activation and translocation to mitochondria [131]. In contrast, although ectopic expression of PKCε resulted in decreased Bax translocation to mitochondria in melanoma cell lines there was no association between PKCε and Bax [31]. Recent work by Pardo and colleagues showed that a higher expression level of PKCε in small cell lung cancer (SCLC) was associated with higher Bcl-XL and X-linked inhibitor of apoptosis (XIAP) protein levels [137]. Furthermore, adenoviral overexpression of PKCε in H69 cells protected them from VP-16-induced death and this was correlated with increased Bcl-XL and XIAP protein levels. In yeast cells, co-expression of PKCε with Bcl-XL was protective against acetic acid-induced apoptosis by preventing Bcl-XL phosphorylation [138]. Thus, the antiapoptotic effect of PKCε may be due, in part, to its ability to impinge upon one or more of the mechanisms that control the function of the Bcl-2 family of proteins.
8. Conclusions
Since the discovery of PKCε almost 20 years ago there have been numerous studies on the involvement of PKCε in cell survival and cell death. Although majority of the studies suggest that it favors life over death, a few studies showed that activation of PKCε could contribute to apoptosis. Since most of the cells express several members of the PKC family and some of the PKC isozymes have opposite effects on cell proliferation and cell death, the presence of these other PKC isozymes will also impact on the final outcome. In addition, PKCε interacts with several signaling pathways, including the Ras/Raf/MAPK and PI-3K/Akt pathway (Fig. 1). It appears that PKCε acts upstream of Akt as well as Raf-1. Thus, components of these signaling pathways and not just PKCε will determine the ultimate fate of a cell. Furthermore, since there is also cross-talk between the Ras/Raf/MAPK and Akt, there may be competition between these pathways.
Fig. 1.
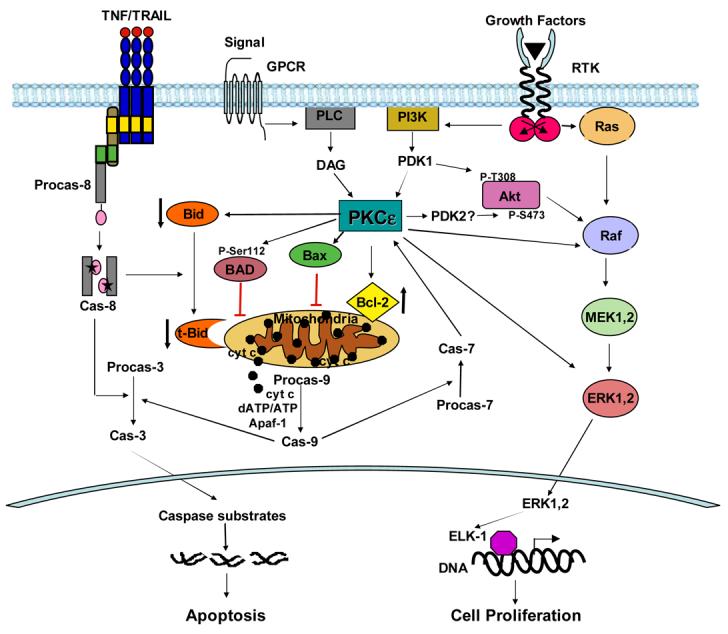
Regulation of cell proliferation and cell death by PKCε. PKCε can cause disordered cell growth by stimulating the Raf/MEK/ERK pathway downstream of Ras proto-oncogene. It can also send survival signal via the PI3K/Akt pathway. PKCε can increase phosphorylation of Akt at Ser473 by facilitating interaction of Akt with a putative PDK2, such as DNA-PK. PKCε has been shown to increase the levels of antiapoptotic Bcl-2. It can interact with Bax and inhibit its activation, dimerization and translocation to mitochondria. It can also phosphorylate Bad and prevents its translocation to the mitochondria. An increase in the ratio of anti- to proapoptotic Bcl-2 family members in the mitochondria inhibits release of cytochrome c from the mitochondria and prevents activation of procaspase-9. PKCε can also inhibit receptor-initiated cell death pathway by decreasing the level of Bid and thus decreasing the amount of truncated Bid in the mitochondria. PKCε is a substrate for caspase-7. In response to apoptotic stimuli, full-length PKCε is cleaved at the hinge region to generate the C-terminal catalytic domain, which is active in the absence of cofactors.
Based on the published reports, PKCε primarily exerts its antiapoptotic signaling by influencing the levels/activation status of Bcl-2 family proteins that regulate mitochondrial integrity (Fig. 1). It increases the ratio of anti- to proapoptotic Bcl-2 family members at the mitochondria by increasing the levels of antiapoptotic Bcl-2 family proteins, decreasing the levels of the proapoptotic Bcl-2 family members, or both. It remains to be established whether PKCε directly affects Bcl-2 family members or acts via Akt or MAPK pathway. PKCε is an important signaling molecule and an understanding of how it makes the life and death decision of a cell will help understand the process of carcinogenesis and facilitate identification of novel targets for cancer therapy.
Acknowledgements
We apologize if we inadvertently left out any major contribution in this field. This work was supported by the grant CA71727 from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kerr JFR, Winterford CM, Harmon BV. Cancer. 1994;73:2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 2.Salvesen GS, Dixit VM. Proc. Natl. Acd. Sci. USA. 1999;96:10964–10967. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen GM. Biochem. J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nunez G, Benedict MA, Hu Y, Inohara N. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- 5.Bao Q, Shi Y. Cell Death Differ. 2007;14(1):56–65. doi: 10.1038/sj.cdd.4402028. [DOI] [PubMed] [Google Scholar]
- 6.Timmer JC, Salvesen GS. Cell Death Differ. 2007;14(1):66–72. doi: 10.1038/sj.cdd.4402059. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S. Cell Death Differ. 2007;14(1):32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- 8.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Embo J. 1995;14(22):5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eskes R, Desagher S, Antonsson B, Martinou JC. Mol Cell Biol. 2000;20(3):929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. J Biol Chem. 1999;274(2):1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 11.Rudner J, Jendrossek V, Lauber K, Daniel PT, Wesselborg S, Belka C. Oncogene. 2005;24(1):130–140. doi: 10.1038/sj.onc.1208191. [DOI] [PubMed] [Google Scholar]
- 12.Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. Genes Dev. 1996;10(22):2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 13.Adams JM, Cory S. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 14.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer J-L, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 15.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 16.Nishizuka Y. Science. 1992;258:607–613. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 17.Newton AC. J. Biol. Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 18.Newton AC. Biochem. J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basu A. Pharmac. Ther. 1993;59:257–280. doi: 10.1016/0163-7258(93)90070-t. [DOI] [PubMed] [Google Scholar]
- 20.Parker PJ, Parkinson SJ. Biochem. Society Transact. 2001;29:860–863. doi: 10.1042/0300-5127:0290860. [DOI] [PubMed] [Google Scholar]
- 21.Stabel S, Parker PJ. Pharmac. Ther. 1991;51:71–95. doi: 10.1016/0163-7258(91)90042-k. [DOI] [PubMed] [Google Scholar]
- 22.Parekh DB, Ziegler W, Parker PJ. EMBO J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basu A, Lu D, Sun B, Moor AN, Akkaraju GR, Huang J. J Biol Chem. 2002;277(44):41850–41856. doi: 10.1074/jbc.M205997200. [DOI] [PubMed] [Google Scholar]
- 24.Datta R, Kojima H, Yoshida K, Kufe D. J. Biol. Chem. 1997;272:20317–20320. doi: 10.1074/jbc.272.33.20317. [DOI] [PubMed] [Google Scholar]
- 25.Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, Kufe D. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizuno K, Noda K, Araki T, Imaoka T, Kobayashi Y, Akita Y, Shimonaka M, Kishi S, Ohno S. Eur. J. Biochem. 1997;250:7–18. doi: 10.1111/j.1432-1033.1997.00007.x. [DOI] [PubMed] [Google Scholar]
- 27.Smith L, Chen L, Reyland ME, DeVries TA, Talanian RV, Omura S, Smith JB. J. Biol. Chem. 2000;275:40620–40627. doi: 10.1074/jbc.M908517199. [DOI] [PubMed] [Google Scholar]
- 28.Basu A. J. Cell Mol. Med. 2003;7:341–350. doi: 10.1111/j.1582-4934.2003.tb00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu A, Cline JS. Int J Cancer. 1995;63(4):597–603. doi: 10.1002/ijc.2910630422. [DOI] [PubMed] [Google Scholar]
- 30.Ding L, Wang H, Lang W, Xiao L. J Biol Chem. 2002;277(38):35305–35313. doi: 10.1074/jbc.M201460200. [DOI] [PubMed] [Google Scholar]
- 31.Gillespie S, Zhang XD, Hersey P. Mol Cancer Ther. 2005;4(4):668–676. doi: 10.1158/1535-7163.MCT-04-0332. [DOI] [PubMed] [Google Scholar]
- 32.Mayne GC, Murray AW. J Biol Chem. 1998;273(37):24115–24121. doi: 10.1074/jbc.273.37.24115. [DOI] [PubMed] [Google Scholar]
- 33.Okhrimenko H, Lu W, Xiang C, Hamburger N, Kazimirsky G, Brodie C. Cancer Res. 2005;65(16):7301–7309. doi: 10.1158/0008-5472.CAN-05-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sivaprasad U, Shankar E, Basu A. Cell Death Differ. 2007;14(4):851–860. doi: 10.1038/sj.cdd.4402077. [DOI] [PubMed] [Google Scholar]
- 35.Cacace AM, Guadagno SN, Krauss RS, Fabbro D, Weinstein IB. Oncogene. 1993;8(8):2095–2104. [PubMed] [Google Scholar]
- 36.Mischak H, Goodnight JA, Kolch W, Martiny-Baron G, Schaechtle C, Kazanietz MG, Blumberg PM, Pierce JH, Mushinski JF. J Biol Chem. 1993;268(9):6090–6096. [PubMed] [Google Scholar]
- 37.Chou WH, Messing RO. Trends Cardiovasc Med. 2005;15(2):47–51. doi: 10.1016/j.tcm.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Inagaki K, Churchill E, Mochly-Rosen D. Cardiovasc Res. 2006;70(2):222–230. doi: 10.1016/j.cardiores.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Vondriska TM, Ping P. J Mol Cell Cardiol. 2003;35(9):1027–1033. doi: 10.1016/s0022-2828(03)00175-5. [DOI] [PubMed] [Google Scholar]
- 40.Baxter G, Oto E, Daniel-Issakani S, Strulovici B. J Biol Chem. 1992;267(3):1910–1917. [PubMed] [Google Scholar]
- 41.Perletti GP, Concari P, Brusaferri S, Marras E, Piccinini F, Tashjian AH., Jr. Oncogene. 1998;16(25):3345–3348. doi: 10.1038/sj.onc.1201871. [DOI] [PubMed] [Google Scholar]
- 42.Perletti GP, Folini M, Lin HC, Mischak H, Piccinini F, Tashjian AH., Jr. Oncogene. 1996;12(4):847–854. [PubMed] [Google Scholar]
- 43.Perletti G, Tessitore L, Sesca E, Pani P, Dianzani MU, Piccinini F. Biochem Biophys Res Commun. 1996;221(3):688–691. doi: 10.1006/bbrc.1996.0657. [DOI] [PubMed] [Google Scholar]
- 44.Wang QJ, Acs P, Goodnight J, Blumberg PM, Mischak H, Mushinski JF. Oncogene. 1998;16(1):53–60. doi: 10.1038/sj.onc.1201507. [DOI] [PubMed] [Google Scholar]
- 45.Knauf JA, Elisei R, Mochly-Rosen D, Liron T, Chen XN, Gonsky R, Korenberg JR, Fagin JA. J Biol Chem. 1999;274(33):23414–23425. doi: 10.1074/jbc.274.33.23414. [DOI] [PubMed] [Google Scholar]
- 46.Knauf JA, Ward LS, Nikiforov YE, Nikiforova M, Puxeddu E, Medvedovic M, Liron T, Mochly-Rosen D, Fagin JA. J Clin Endocrinol Metab. 2002;87(5):2150–2159. doi: 10.1210/jcem.87.5.8441. [DOI] [PubMed] [Google Scholar]
- 47.Zeidman R, Lofgren B, Pahlman S, Larsson C. J Cell Biol. 1999;145(4):713–726. doi: 10.1083/jcb.145.4.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verma AK, Wheeler DL, Aziz MH, Manoharan H. Mol Carcinog. 2006;45(6):381–388. doi: 10.1002/mc.20230. [DOI] [PubMed] [Google Scholar]
- 49.Wheeler DL, Ness KJ, Oberley TD, Verma AK. Cancer Res. 2003;63(19):6547–6555. [PubMed] [Google Scholar]
- 50.Zhao Y, Xue Y, Oberley TD, Kiningham KK, Lin SM, Yen HC, Majima H, Hines J, Clair D. Cancer Res. 2001;61(16):6082–6088. [PubMed] [Google Scholar]
- 51.Sharif TR, Sharif M. Int J Oncol. 1999;15(2):237–243. [PubMed] [Google Scholar]
- 52.Sharif TR, Sasakawa N, Sharif M. Int J Mol Med. 2001;7(4):373–380. doi: 10.3892/ijmm.7.4.373. [DOI] [PubMed] [Google Scholar]
- 53.Wu D, Foreman TL, Gregory CW, McJilton MA, Wescott GG, Ford OH, Alvey RF, Mohler JL, Terrian DM. Cancer Res. 2002;62(8):2423–2429. [PubMed] [Google Scholar]
- 54.Engers R, Mrzyk S, Springer E, Fabbro D, Weissgerber G, Gernharz CD, Gabbert HE. Br J Cancer. 2000;82(5):1063–1069. doi: 10.1054/bjoc.1999.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tachado SD, Mayhew MW, Wescott GG, Foreman TL, Goodwin CD, McJilton MA, Terrian DM. J Cell Biochem. 2002;85(4):785–797. doi: 10.1002/jcb.10164. [DOI] [PubMed] [Google Scholar]
- 56.Jansen AP, Verwiebe EG, Dreckschmidt NE, Wheeler DL, Oberley TD, Verma AK. Cancer Res. 2001;61(3):808–812. [PubMed] [Google Scholar]
- 57.Reddig PJ, Dreckschmidt NE, Zou J, Bourguignon SE, Oberley TD, Verma AK. Cancer Res. 2000;60(3):595–602. [PubMed] [Google Scholar]
- 58.Besson A, Wilson TL, Yong VW. J Biol Chem. 2002;277(24):22073–22084. doi: 10.1074/jbc.M111644200. [DOI] [PubMed] [Google Scholar]
- 59.Pan Q, Bao LW, Kleer CG, Sabel MS, Griffith KA, Teknos TN, Merajver SD. Cancer Res. 2005;65(18):8366–8371. doi: 10.1158/0008-5472.CAN-05-0553. [DOI] [PubMed] [Google Scholar]
- 60.Pan Q, Bao LW, Teknos TN, Merajver SD. Cancer Res. 2006;66(19):9379–9384. doi: 10.1158/0008-5472.CAN-06-2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iwata M, Iseki R, Sato K, Tozawa Y, Ohaka Y. International Immunology. 1994;6:431–438. doi: 10.1093/intimm/6.3.431. [DOI] [PubMed] [Google Scholar]
- 62.Lavie Y, Zhang Z-c., Cao H-t., Han T-Y, Jones RC, Liu Y-Y, Jarman M, Hardcastle IR, Giuliano AE, Cabot MC. Int. J. Cancer. 1998;77:928–932. doi: 10.1002/(sici)1097-0215(19980911)77:6<928::aid-ijc22>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 63.Mihalik R, Farkas G, Kopper L, Benczur M, Farago A. Int J Biochem Cell Biol. 1996;28(8):925–933. doi: 10.1016/1357-2725(96)00020-9. [DOI] [PubMed] [Google Scholar]
- 64.Sawai H, Okazaki T, Takeda Y, Tashima M, Sawada H, Okuma M, Kishi S, Umehara H, Domae N. J Biol Chem. 1997;272(4):2452–2458. doi: 10.1074/jbc.272.4.2452. [DOI] [PubMed] [Google Scholar]
- 65.Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Bosca L. J. Exp. Med. 2001;194:1231–1242. doi: 10.1084/jem.194.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Basu A, Weixel KM. Int. J. Cancer. 1995;62:457–460. doi: 10.1002/ijc.2910620416. [DOI] [PubMed] [Google Scholar]
- 67.Flescher E, Rotem R. Cell Signal. 2002;14(1):37–43. doi: 10.1016/s0898-6568(01)00215-7. [DOI] [PubMed] [Google Scholar]
- 68.Basu A, Mohanty S, Sun B. Biochem. Biophys. Res. Commun. 2001;280:883–891. doi: 10.1006/bbrc.2000.4209. [DOI] [PubMed] [Google Scholar]
- 69.Shinohara H, Kayagaki N, Yagita H, Oyaizu N, Ohba M, Kuroki T, Ikawa Y. Biochem Biophys Res Commun. 2001;284(5):1162–1167. doi: 10.1006/bbrc.2001.5104. [DOI] [PubMed] [Google Scholar]
- 70.Koriyama H, Kouchi Z, Umeda T, Saido TC, Momoi T, Ishiura S, Suzuki K. Cell. Signal. 1999;11:831–838. doi: 10.1016/s0898-6568(99)00055-8. [DOI] [PubMed] [Google Scholar]
- 71.Hoppe J, Hoppe V, Schafer R. Exp Cell Res. 2001;266(1):64–73. doi: 10.1006/excr.2001.5211. [DOI] [PubMed] [Google Scholar]
- 72.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 73.Johnson GL, Lapadat R. Science. 2002;298(5600):1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 74.Marshall CJ. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 75.Chang L, Karin M. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 76.Cacace AM, Ueffing M, Philipp A, Han EK, Kolch W, Weinstein IB. Oncogene. 1996;13(12):2517–2526. [PubMed] [Google Scholar]
- 77.Cai H, Smola U, Wixler V, Eisenmann-Tappe I, Diaz-Meco MT, Moscat J, Rapp U, Copper GM. Mol. Cell. Biol. 1997;17:732–741. doi: 10.1128/mcb.17.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ueffing M, Lovric J, Philipp A, Mischak H, Kolch W. Oncogene. 1997;15(24):2921–2927. doi: 10.1038/sj.onc.1201477. [DOI] [PubMed] [Google Scholar]
- 79.Cacace AM, Ueffing M, Han EK, Marme D, Weinstein IB. J Cell Physiol. 1998;175(3):314–322. doi: 10.1002/(SICI)1097-4652(199806)175:3<314::AID-JCP9>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 80.Hundle B, McMahon T, Dadgar J, Messing RO. J Biol Chem. 1995;270(50):30134–30140. doi: 10.1074/jbc.270.50.30134. [DOI] [PubMed] [Google Scholar]
- 81.Bourbon NA, Yun J, Berkey D, Wang Y, Kester M. Am J Physiol Cell Physiol. 2001;280(6):C1403–1411. doi: 10.1152/ajpcell.2001.280.6.C1403. [DOI] [PubMed] [Google Scholar]
- 82.Kampfer S, Windegger M, Hochholdinger F, Schwaiger W, Pestell RG, Baier G, Grunicke HH, Uberall F. J Biol Chem. 2001;276(46):42834–42842. doi: 10.1074/jbc.M102047200. [DOI] [PubMed] [Google Scholar]
- 83.Lindemann RK, Braig M, Hauser CA, Nordheim A, Dittmer J. Biochem J. 2003;372(Pt 3):787–797. doi: 10.1042/BJ20030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, Van Belle P, Elder DE, Herlyn M. Cancer Res. 2003;63(4):756–759. [PubMed] [Google Scholar]
- 85.Marin YE, Namkoong J, Cohen-Solal K, Shin SS, Martino JJ, Oka M, Chen S. Cell Signal. 2006;18(8):1279–1286. doi: 10.1016/j.cellsig.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 86.Chen N, Ma W, Huang C, Dong Z. J. Biol. Chem. 1999;274:15389–15394. doi: 10.1074/jbc.274.22.15389. [DOI] [PubMed] [Google Scholar]
- 87.Zhuang S, Hirai S, Mizuno K, Suzuki A, Akimoto K, Izumi Y, Yamashita A, Ohno S. Biochem Biophys Res Commun. 1997;240(2):273–278. doi: 10.1006/bbrc.1997.7474. [DOI] [PubMed] [Google Scholar]
- 88.Song MS, Park YK, Lee JH, Park K. Cancer Res. 2001;61(22):8322–8330. [PubMed] [Google Scholar]
- 89.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJC, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. J. Biol. Chem. 1997;272(50):31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 90.Toker A. Mol Pharmacol. 2000;57(4):652–658. [PubMed] [Google Scholar]
- 91.Bellacosa A, Franke TF, Gonzalez-Portal ME, Datta K, Taguchi T, Gardner J, Cheng JQ, Testa JR, Tsichlis PN. Oncogene. 1993;8(3):745–754. [PubMed] [Google Scholar]
- 92.Brazil DP, Hemmings BA. Trends in Biochemical Sciences. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 93.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Embo J. 1996;15(23):6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 94.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Curr Biol. 1997;7(4):261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 95.Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR. Curr Biol. 2000;10(8):439–448. doi: 10.1016/s0960-9822(00)00441-3. [DOI] [PubMed] [Google Scholar]
- 96.Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Proc Natl Acad Sci U S A. 1998;95(19):11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Troussard AA, Mawji NM, Ong C, Mui A, Arnaud R, Dedhar S. J Biol Chem. 2003;278(25):22374–22378. doi: 10.1074/jbc.M303083200. [DOI] [PubMed] [Google Scholar]
- 98.Rane MJ, Coxon PY, Powell DW, Webster R, Klein JB, Pierce W, Ping P, McLeish KR. J. Bio. Chem. 2001;276:3517–3523. doi: 10.1074/jbc.M005953200. [DOI] [PubMed] [Google Scholar]
- 99.Feng J, Park J, Cron P, Hess D, Hemmings BA. J. Biol. Chem. 2004;279(39):41189–41196. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 100.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 101.Woodgett JR. Curr Opin Cell Biol. 2005;17(2):150–157. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 102.Lu D, Huang J, Basu A. J Biol Chem. 2006;281(32):22799–22807. doi: 10.1074/jbc.M603390200. [DOI] [PubMed] [Google Scholar]
- 103.Wu D, Thakore CU, Wescott GG, McCubrey JA, Terrian DM. Oncogene. 2004;23(53):8659–8672. doi: 10.1038/sj.onc.1207900. [DOI] [PubMed] [Google Scholar]
- 104.Zhou H, Karliner JS, Gray MO. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H165–H174. doi: 10.1152/ajpheart.00408.2001. [DOI] [PubMed] [Google Scholar]
- 105.Matsumoto M, Ogawa W, Hino Y, Furukawa K, Ono Y, Takahashi M, Ohba M, Kuroki T, Kasuga M. J. Biol. Chem. 2001;276:14400–14406. doi: 10.1074/jbc.M011093200. [DOI] [PubMed] [Google Scholar]
- 106.Zhang J, Baines CP, Zong C, Cardwell EM, Wang G, Vondriska TM, Ping P. Am J Physiol Heart Circ Physiol. 2005;288(2):H954–961. doi: 10.1152/ajpheart.00756.2004. [DOI] [PubMed] [Google Scholar]
- 107.Ping P, Zhang J, Pierce WM, Jr., Bolli R. Circ. Res. 2001;88:59–62. doi: 10.1161/01.res.88.1.59. [DOI] [PubMed] [Google Scholar]
- 108.Li W, Zhang J, Flechner L, Hyun T, Yam A, Franke TF, Pierce JH. Oncogene. 1999;18(47):6564–6572. doi: 10.1038/sj.onc.1203065. [DOI] [PubMed] [Google Scholar]
- 109.Venkatesan BA, Mahimainathan L, Ghosh-Choudhury N, Gorin Y, Bhandari B, Valente AJ, Abboud HE, Choudhury GG. Cell Signal. 2006;18(4):508–518. doi: 10.1016/j.cellsig.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 110.Ivaska J, Whelan DH, Watson R, Parker PJ. EMBO J. 2002;21:3608–3619. doi: 10.1093/emboj/cdf371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cordes N, Seidler J, Durzok R, Geinitz H, Brakebusch C. Oncogene. 2006;25(9):1378–1390. doi: 10.1038/sj.onc.1209164. [DOI] [PubMed] [Google Scholar]
- 112.Folgiero V, Bachelder RE, Bon G, Sacchi A, Falcioni R, Mercurio AM. Cancer Res. 2007;67(4):1645–1652. doi: 10.1158/0008-5472.CAN-06-2980. [DOI] [PubMed] [Google Scholar]
- 113.Parekh DB, Katso RMT, Leslie NR, Downes CP, Procyk KJ, Waterfield MD, Parker PJ. Biochem. J. 2000;352:425–433. [PMC free article] [PubMed] [Google Scholar]
- 114.Kuang PP, Zhang XH, Rich C, Foster JA, Subramanian M, Goldstein RH. Am J Physiol Lung Cell Mol Physiol. 2007 doi: 10.1152/ajplung.00184.2006. [DOI] [PubMed] [Google Scholar]
- 115.Bachelder RE, Wendt MA, Fujita N, Tsuruo T, Mercurio AM. J. Bio. Chem. 2001;276:34702–34707. doi: 10.1074/jbc.M102806200. [DOI] [PubMed] [Google Scholar]
- 116.Barthel A, Nakatani K, Dandekar AA, Roth RA. Biochem Biophys Res Commun. 1998;243(2):509–513. doi: 10.1006/bbrc.1998.8134. [DOI] [PubMed] [Google Scholar]
- 117.Guan L, Song K, Pysz MA, Curry KJ, Hizli AA, Danielpour D, Black AR, Black JD. J Biol Chem. 2007 doi: 10.1074/jbc.M610513200. [DOI] [PubMed] [Google Scholar]
- 118.Ikeda Y, Olsen GS, Ziv E, Hansen LL, Busch AK, Hansen BF, Shafrir E, Mosthaf-Seedorf L. Diabetes. 2001;50:584–592. doi: 10.2337/diabetes.50.3.584. [DOI] [PubMed] [Google Scholar]
- 119.Li L, Sampat K, Hu N, Zakari J, Yuspa SH. J Biol Chem. 2006;281(6):3237–3243. doi: 10.1074/jbc.M512167200. [DOI] [PubMed] [Google Scholar]
- 120.Liu H, Qiu Y, Xiao L, Dong F. J Immunol. 2006;176(4):2407–2413. doi: 10.4049/jimmunol.176.4.2407. [DOI] [PubMed] [Google Scholar]
- 121.Wen HC, Huang WC, Ali A, Woodgett JR, Lin WW. Cell. Signal. 2003;15:37–45. doi: 10.1016/s0898-6568(02)00047-5. [DOI] [PubMed] [Google Scholar]
- 122.Cory S, Huang DC, Adams JM. Oncogene. 2003;22(53):8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 123.Perez-Chacon G, Vargas JA, Jorda J, Morado M, Rosado S, Martin-Donaire T, Losada-Fernandez I, Rebolleda N, Perez-Aciego P. Leuk Res. 2007;31(2):183–193. doi: 10.1016/j.leukres.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 124.Thomas A, Pepper C, Hoy T, Bentley P. Leuk Lymphoma. 2004;45(5):997–1008. doi: 10.1080/10428190310001639470. [DOI] [PubMed] [Google Scholar]
- 125.Choi H, Kim SH, Chun YS, Cho YS, Park JW, Kim MS. Exp Biol Med (Maywood) 2006;231(4):463–472. doi: 10.1177/153537020623100412. [DOI] [PubMed] [Google Scholar]
- 126.Neithardt A, Farshori MP, Shah FB, Catt KJ, Shah BH. J Cell Physiol. 2006;208(3):586–593. doi: 10.1002/jcp.20697. [DOI] [PubMed] [Google Scholar]
- 127.Farrow B, Thomas RP, Wang XF, Evers BM. Int J Gastrointest Cancer. 2002;32(23):63–72. doi: 10.1385/IJGC:32:2-3:63. [DOI] [PubMed] [Google Scholar]
- 128.Zong ZP, Matsui S, Katsuda S, Han JF, Fujikawa-Yamamoto K. Eur J Pharmacol. 2004;489(12):3–11. doi: 10.1016/j.ejphar.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 129.Gubina E, Rinaudo MS, Szallasi Z, Blumberg PM, Mufson RA. Blood. 1998;91(3):823–829. [PubMed] [Google Scholar]
- 130.Bertolotto C, Maulon L, Filippa N, Baier G, Auberger P. J. Biol. Chem. 2000;275:37246–37250. doi: 10.1074/jbc.M007732200. [DOI] [PubMed] [Google Scholar]
- 131.McJilton MA, Van Sikes C, Wescott GG, Wu D, Foreman TL, Gregory CW, Weidner DA, Harris Ford O, Morgan Lasater A, Mohler JL, Terrian DM. Oncogene. 2003;22(39):7958–7968. doi: 10.1038/sj.onc.1206795. [DOI] [PubMed] [Google Scholar]
- 132.Suzuki A, Goto Y, Iguchi T. Carcinogenesis. 1997;18(5):883–887. doi: 10.1093/carcin/18.5.883. [DOI] [PubMed] [Google Scholar]
- 133.Soh JW, Lee YS, Weinstein IB. J Exp Ther Oncol. 2003;3(3):115–126. doi: 10.1046/j.1359-4117.2003.01087.x. [DOI] [PubMed] [Google Scholar]
- 134.Mirandola P, Gobbi G, Ponti C, Sponzilli I, Cocco L, Vitale M. Blood. 2006;107(2):508–513. doi: 10.1182/blood-2005-07-2676. [DOI] [PubMed] [Google Scholar]
- 135.Mangat R, Singal T, Dhalla NS, Tappia PS. Am J Physiol Heart Circ Physiol. 2006;291(2):H854–860. doi: 10.1152/ajpheart.01205.2005. [DOI] [PubMed] [Google Scholar]
- 136.Baines CP, Zhang J, Wang G-W, Zheng Y-T, Xiu JX, Cardwell EM, Bolli R, Ping P. Mol. Med. 2002:390–397. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 137.Pardo OE, Wellbrock C, Khanzada UK, Aubert M, Arozarena I, Davidson S, Bowen F, Parker PJ, Filonenko VV, Gout IT, Sebire N, Marais R, Downward J, Seckl MJ. Embo J. 2006;25(13):3078–3088. doi: 10.1038/sj.emboj.7601198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Saraiva L, Silva RD, Pereira G, Goncalves J, Corte-Real M. J Cell Sci. 2006;119(Pt 15):3171–3181. doi: 10.1242/jcs.03033. [DOI] [PubMed] [Google Scholar]