

Abstract
Platelet thrombi are central to the development of most vascular ischemic events. There is marked inter-individual variation in platelet responsiveness, with some subjects displaying platelet hyperreactivity. An increasing number of reports indicate there are laboratory measures of platelet function that predict clinical thrombotic events. Some, but not all measures of platelet function are reproducible. Platelet hyperreactivity can be assessed with multiple stimuli in multiple assays, and is more likely to be present in females and in subjects with elevated fibrinogen levels.
Keywords: platelets, platelet reactivity, platelet aggregation, coronary heart disease
Introduction
Occlusive vascular diseases, such as myocardial infarction (MI), are the major causes of mortality and morbidity in the U.S. MI and other acute coronary syndromes develop when a platelet thrombus forms at the site of a ruptured or eroded unstable coronary atherosclerotic plaque. Pathologic platelet thrombi also cause ischemia in the brain (stroke) and extremities (peripheral arterial disease). There is increasing evidence that enhanced platelet reactivity can prospectively identify subjects at risk for these conditions. Increased platelet reactivity may also mitigate hemorrhage in inherited or acquired bleed conditions. There is a well-known inter-individual variation in platelet reactivity, which may contribute to the risk of thrombosis in one individual, but protection in another. On the other hand, reduced platelet function may also contribute to hemorrhagic disorders, usually in very different clinical settings. This review will consider inherent differences in platelet function and the clinical relevance of platelet hyperreactivity. The first section will examine patient studies linking ex vivo platelet functional assays to clinical events. The second section will consider those variables and subject characteristics associated with platelet hyperreactivity.
Platelets in arterial thrombosis
The pathophysiology of arterial thrombosis is complex, with many genes dictating physiologic and biochemical traits that modify one another to increase or decrease the risk for developing adverse clinical outcomes. Environmental factors also impinge on the risk, as do age and gender. Figure 1 emphasizes the central role of platelets in the pathogenesis of clinical thrombosis in arterial disease. The position of platelets in acute coronary syndromes is well established and is grounded in physiologic, pathologic, and clinical studies [1-3]. Platelet deposition onto the subendothelium is proportional to the shear rate [4], such that platelets play a particularly important role in arterial thrombosis. Upon arterial plaque rupture, von Willebrand factor (VWF) molecules are rapidly localized to the subendothelium and the initial platelet contact with the wound is a tethering of platelets to this insoluble form of VWF. The glycoprotein (GP) GPIbα subunit of the GPIb-IX-V complex [5,6] mediates this tethering to VWF, resulting in a slower platelet velocity, which in turn permits platelet GPVI to bind to collagen [7]. Signaling between and through GPIbα and GPVI causes platelet activation, resulting in secretion, firm platelet adhesion through activated integrins: α2β1 binding to exposed collagen and GPIIb-IIIa (integrin αIIbβ3) binding to VWF and fibrinogen [8]. An expanding thrombus ensues when platelets aggregate via the intercellular bridging of fibrinogen and VWF binding to the activated conformation of GPIIb-IIIa. Blood flow ceases when an occlusive platelet plug forms. The importance of GPIIb-IIIa, GPIbα and α2β1 is underscored by the moderate to severe bleeding seen in patients with inherited defects in these genes.
Figure 1. Schema indicating the multifactorial nature of thrombosis.
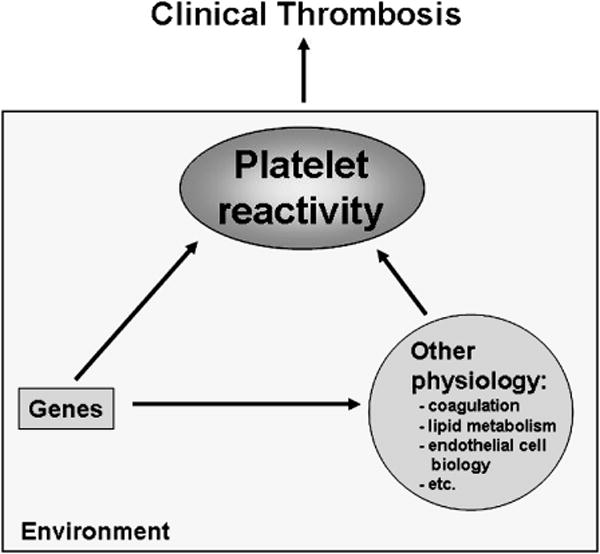
The central role of platelets is emphasized.
Platelet assays as predictors of CHD events
Substantial evidence has accumulated linking platelet hyperreactivity with vascular thrombotic diseases. Table 1 lists conditions in which enhanced platelet reactivity have been demonstrated. Other than heparin-induce thrombocytopenia, the mechanisms responsible for the hyperreactivity are generally poorly understood. Most studies in this area have included patients with acute coronary events, and have measured ex vivo markers of platelet activation. For example, patients admitted to the coronary care unit have increased levels of circulating P-selectin [9]. However, cross-sectional studies cannot distinguish the cart from the horse; i.e., did underlying platelet hyperreactivity contribute to the thrombotic event? Or did the stress of the acute event induce platelet activation? The plethora of cross-sectional studies will not address these questions. Prospective studies are required to appropriately test the hypothesis that ex vivo platelet reactivity predicts coronary heart disease (CHD) outcomes. In fact, a modest body of literature exists supporting this hypothesis. The design of most prospective studies is as follows: 1) high-risk patients are identified, 2) any of a variety of platelet functional assays are performed, and 3) patients are followed for variable periods of time for a variety of different outcomes. Table 2 lists the different platelet assays that have been utilized in these types of clinical studies.
Table 1.
Conditions associated with enhanced platelet reactivity.
| Coronary artery disease |
| Cerebrovascular disease |
| Peripheral artery disease |
| Acute coronary syndromes |
| Percutaneous coronary interventions |
| Coronary artery bypasses graft surgery |
| Antiphospholipid syndrome |
| Myeloproliferative disorders |
| Heparin-induced thrombocytopenia |
Table 2.
Platelet measures reported to prospectively predict CHD outcomes.
| Assay | Reference |
|---|---|
| Platelet aggregation | [10-17] |
| PFA-100® | [18-22] |
| Mean Platelet Volume | [12,23-25] |
| Urinary thromboxane | [26] |
| Platelet count | [11] |
| Platelet secretion | [27] |
| Platelet-leukocyte aggregates | [28] |
| Platelet microaggregation (PA-200®) | [29,30] |
| Platelet fibrinogen binding | [31,32] |
| Soluble CD40L | [33] |
| Platelet nitric oxide responsiveness | [34] |
| Ultegra RPFA® | [35] |
The vast majority of research linking platelet reactivity to CHD events has utilized three routine clinical measures of platelet reactivity: standard lumiaggregometry with platelet rich plasma (PRP), the PFA-100® and the mean platelet volume (MPV). As summarized in Table 3A, spontaneous aggregation, and aggregation induced by arachidonic acid, epinephrine, ADP and collagen have all been found to predict clinical CHD outcomes. On the other hand, most studies have not found platelet aggregation predicts re-stenosis at angiography (Table 3B), the exception being one of the two studies by Torres et al. [12]. Both the PFA-100® and the MPV have been found to predict CHD outcomes (Table 3C). Of note, the study by Frossard et al. [19] utilized an intermediate outcome of troponin T levels. The largest study to test whether a platelet parameter predicted a CHD outcome was that of Martin et al. [23]. These investigators identified 2,033 male MI patients, and obtained hematologic studies on 1,716 patients six months post-event. After a follow-up of approximately 24 months 126 patients had experienced a CHD death or recurrent MI. The average MPV was significantly higher in those patients who experienced a recurrent ischemic event compared to those who did not (P<0.001). Only one or two prospective studies have been performed that tested the value of the last nine measures of platelet function listed in Table 2, and these will not be reviewed in detail. However, the report by Eikelboom et al. is noteworthy for its sample size [26]. These investigators found that high levels of urinary thromboxane in patients on aspirin predicted recurrent vascular events.
Table 3A.
Enhanced platelet aggregation may predict CHD outcomes.
| n | Population | n | Outcome | Follow-up (mo) | Predictive | Ref |
|---|---|---|---|---|---|---|
| 149 | MI survivors | 12 | Recurrent CHD events | 60 | Spontaneous | Trip 1990 |
| 150 | Healthy subjects | 11 | Fatal events | 60 | ADP | Thaulow 1991 |
| 47 | PTCA patients | 10 | Re-occlusion by angiography | 6 | epi, ADP, collagen | Terres 1995 |
| 326 | CAD | 34 | Recurrent CHD events | 3 | AA and ADP | Gum 2003* |
| 106 | NSTEMI → PCI | 12 | Recurrent CHD events | 24 | ADP | Cuisset 2006 |
stratified by aspirin responsiveness
MI, myocardial infarction; CHD, coronary heart disease; PTCA, Percutaneous transluminal coronary angioplasty; NSTEMI, non-ST elevation MI; PCI, Percutaneous coronary intervention; ADP, adenosine diphosphate; epi, epinephrine; AA, arachidonic acid
Table 3B.
Enhanced platelet aggregation may not predict re-stenosis.
| n | Population | n | Outcome | Follow-up (mo) | Not predictive | Ref |
|---|---|---|---|---|---|---|
| 98 | PTCA patients | - | Angiography | 3 | ADP, collagen | Terres 1992 |
| 53 | CAD | - | Angiography | 24 | epi, ADP, collagen, PF4; thrombin was predictive | Lam 1994 |
| 155 | PCTA | - | Angiography | 20 | Spontaneous | Capanni 1999 |
CAD, coronary artery disease; PTCA, Percutaneous transluminal coronary angioplasty; ADP, adenosine diphosphate; epi, epinephrine; PF4, platelet factor 4
Table 3C.
PFA-100 and MPV may predict CHD outcomes.
| n | Population | n | Outcome | Follow-up (mo) | Predictive | Ref. | |
|---|---|---|---|---|---|---|---|
| PFA-100® | |||||||
| 98 | PAD → angioplasty | 25 | Restenosis by Doppler | 12 | ADP | Ziegler 2002 | |
| 212 | ACS | - | troponin T | - | ADP, epi associated w levels | Frossard 2004 | |
| 100 | ACS | 5 | Deaths | 6 | epi<138 sec | Sambola 2004 | |
| 175 | ACS → PCI | 12? | Recurrent ACSs | 6 | ADP, epi | Gianetti 2006 | |
| 208 | ACS | 58 | Recurrent ACSs | 28 | ADP, epi | Fuchs 2006 | |
| MPV | |||||||
| 1716 | Male MI survivors | 126 | Recurrent MI, CHD death | 24 | MPV | Martin 1991 | |
| 47 | PTCA | - | Restenosis by angiography | 4-8 | MPV | Smyth 1993 | |
| 47 | PCI | 10 | Re-occlusion by angiography | 6 | MPV | Terres 1995 | |
| 398 | STMI → PCI | 91/29 | Restenosis/mortality | 6 | MPV | Huczek 2005 |
PAD, peripheral arterial disease; ACS, acute coronary syndrome; MI, myocardial infarction; CHD, coronary heart disease; PTCA, Percutaneous transluminal coronary angioplasty; STMI, ST elevation MI; PCI, Percutaneous coronary intervention; ADP, adenosine diphosphate; epi, epinephrine; AA, arachidonic acid; MPV, mean platelet volume
It must be emphasized that there are limitations in drawing conclusions from these prospective studies (Table 4). The patients comprising the data summarized in Tables 3A-3C are quite heterogeneous. Differences include acute versus chronic disease, coronary versus peripheral vessels, different lengths of follow-up, different endpoints, etc. Most of these studies were rather small – many with fewer than 30 CHD events. There were technical differences as well – the timing of phlebotomy from the acute event, as well as different assays and different agonist concentrations. This type of research also lends itself to publication bias, since a study finding no predictive value for a give platelet assay has less impact. Finally, since most study populations included patients with vascular ischemia, nearly all subjects were on anti-platelet medications. Many of these agents are known to affect the results of in vitro assays. In and of itself, this does not undermine the conclusions from these studies because patients biologically resistant to the platelet inhibitory effects of these agents might be expected to have both worse clinical outcomes and enhanced ex vivo measures of platelet reactivity. However, only a small number of these studies were designed specifically to consider the effect of anti-platelet agents, with the vast majority not considering this variable in the analysis. Having acknowledged these limitations, it is nevertheless fair to conclude that there is a substantial body of evidence – both cross sectional and prospective – supporting the hypothesis that ex vivo platelet reactivity predicts CHD outcomes.
Table 4.
Study limitations
| 1. | Different disease groups (healthy, acute MI, PCI, etc.) |
| 2. | Generally small sample sizes (many with 10-30 CHD events) with little or no consideration of confounders |
| 3. | Nearly all on anti-platelet medications and other medications |
| 4. | Different platelet assays, different stimuli |
| 5. | Different lengths of follow-up |
| 6. | Different endpoints (angiographic stenosis, clinical events, myocardial necrosis) |
| 7. | Timing of phlebotomy |
| 8. | Publication bias |
Are these platelet assays clinically useful? This is quite a separate issue, and the answer is largely related to the failure of anti-platelet therapy and whether there are alternative approaches to “rescue” an inadequate ex vivo response to anti-platelet therapy. For example, if a compliant patient taking aspirin for secondary prevention of MI is found to have enhanced epinephrine-induced platelet aggregation and an MPV in the upper quartile of the normal distribution, will they benefit from the addition of clopidogrel? These are important and unresolved questions that need additional clinical research.
Intrinsic platelet function variation
There is a common belief that substantial inter- and intra-individual variation exists in platelet responsiveness. This variation persistently challenges the interpretation of clinical research involving platelet assays. To more rigorously characterize this variation and to determine the factors contributing to this variation, we recruited a large number of healthy subjects to donate blood samples. The design of this study has been previously detailed [36,37] and Table 5 lists the assays that were performed. Figure 2 illustrates the variability observed amongst the first 280 subjects studied, using collagen as the agonist in platelet lumiaggregometry. Similar variability was observed with all other agonists (not shown). Note the striking variation in a healthy population on the “steep” portion of the dose-response curve. At the two extremes (very little stimulation and maximal stimulation) there is considerably less variability, as indicated by the smaller standard deviations. From the point of view of platelet risk factors for arterial thrombosis, the subjects with maximal aggregation at low concentrations of agonist might be considered to have a hyperreactive platelet phenotype. It is this population that is especially interesting. Knowing the characteristics of this group could provide valuable clinical information and provide insights into the molecular basis of platelet reactivity.
Table 5.
Platelet functional assays.
| Assay | Condition (concentration or shear rate)* |
|---|---|
| PRP aggregation† | |
| Epinephrine | 0.1, 0.2, 0.4, 1.5, 3, 10 μM |
| CRP | 0.005, 0.01, 0.02, 0.05 μg/mL |
| ADP | 0.05, 0.5, 1, 4, 20 μM |
| Collagen | 2.5, 5. 10, 20, 50, 200 μg/mL |
| Arachidonic acid | 0.5 mg/mL |
| Ristocetin | 0.1, 0.5, 0.75, 1 mg/mL |
| Spontaneous | |
| Whole Blood flow cytometry | |
| P-selectin expression | 0, 1, 10 μM ADP |
| PAC-1 binding | 0, 1, 10 μM ADP |
| Shear assays† | |
| PFA-100 (ADP) | |
| PFA-100 (epinephrine) | |
| SIPE (cone and plate viscometer) | 0, 500, 1000, 2000, 5000, 10000 sec−1 |
| SIPA (cone and plate viscometer) | 0, 500, 1000, 2000, 5000, 10000 sec−1 |
| Receptor density | |
| αIIbβ3 (CD41) | resting |
| GPIbα (CD42b) | resting |
| α2β1 (CD49) | resting |
| FcγRIIA (CD32) | resting |
| Clot retraction† |
Blank means only a single condition was used.
Assays also performed in PPACK anticoagulant
CRP, collagen-related peptide; SIPE, shear-induced P-selectin expression; SIPA, shear-induced platelet aggregation
Figure 2. Variation in platelet reactivity.
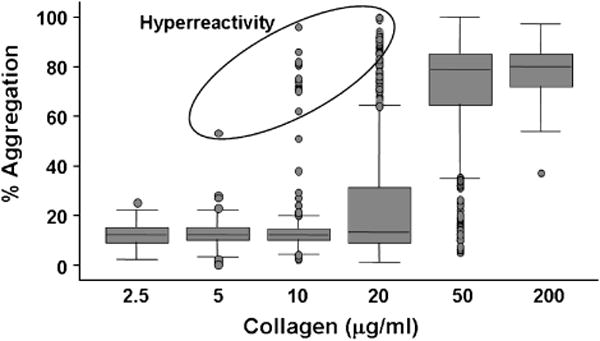
The lumiaggregation response in plateletrich plasma in response to increasing concentrations of type I acid soluble collagen is shown. The box indicates the 25th-75th percentiles, the line in the box indicates the median, the lines outside the box indicate the 10th and 90th percentiles, and the dots are the outliers below the 10th percentile and above the 90th percentile. An arbitrary group of hyperreactive outliers is circled.
Reproducibility of ex vivo platelet assays
Table 6 lists some of the potential sources of variation in platelet functional assays. If one wanted to characterize the contribution of any one of the factors listed in Table 6, then the extent of variation contributed by the other factors must either be minimal or must be known and considered in the analyses. In practical terms, knowing the reproducibility of the platelet assays becomes critical. Surprisingly little information exists on the reproducibility of platelet function. This is especially important for studies with small numbers of subjects, typical of many clinical studies and virtually all basic research studies. Because of our interest in identifying genes that modify platelet function, we performed a formal reproducibility study of the assays listed in Table 5. Twenty seven subjects were studied on a weekly basis for four weeks. Subjects were studied weekly because of concerns that 1) a longer time interval might introduce new variables, such as age, and 2) a shorter time interval would not account for the effects of new platelet production. For agonist concentrations selected to identify a hyperreactive phenotype, poor reproducibility was observed for collagen and ristocetin, but there was excellent reproducibility for epinephrine and a collagen-related-peptide (CRP) [36]. Reproducibility analyses with submaximal concentrations ADP were inconclusive. Fifteen of the 27 subjects had had platelet function assays 2-25 months prior to the reproducibility study. Fourteen of these 15 showed remarkably consistent results over this time period in response to 0.4 μM epinephrine [36]. This study demonstrated that submaximal concentrations of epinephrine and CRP can be used to assess in vitro platelet hyperreactivity. We recommend that such a classification be limited to subjects whose platelets demonstrate more than 60% aggregation to epinephrine or more than 50% aggregation to CRP on at least 2 occasions.
Table 6.
Sources of variation in platelet functional assays
| Genetic/intrinsic | Environmental | Technical |
|---|---|---|
| Genes | Cigarette smoking | Instruments |
| Alcohol | Different lots of reagents | |
| Sex hormones | Phlebotomy techniques | |
| Heparin | Time of day | |
| Plasma | Changes in personnel | |
| Unknown | Processing (e.g., time, temp., transportation) | |
| Unknown |
Platelet hyperreactivity is global
Subjects with hyperreactivity to epinephrine are more likely to exhibit hyperfunction in each major aspect of platelet activity, including adhesion (response to ristocetin), activation (surface P-selectin expression and PAC-1 binding after stimulation) and aggregation to applied shear stress (PFA-100 and cone-and-plate viscometer) [37]. Figure 3 shows that hyperreactivity to epinephrine was strongly associated with hyperreactivity to submaximal concentrations of other agonists. The consistent results using different platelet function assays show that platelet hyperreactivity is not specific to epinephrine mediated aggregation, but generalizes to multiple forms of platelet stimulation. This indicates underlying mechanisms involved in the early aspects of platelet activation and that are shared by multiple pathways affecting distinct aspects of platelet function.
Figure 3. Association between platelet hyperreactivity to epinephrine and hyperreactivity to other agonists.
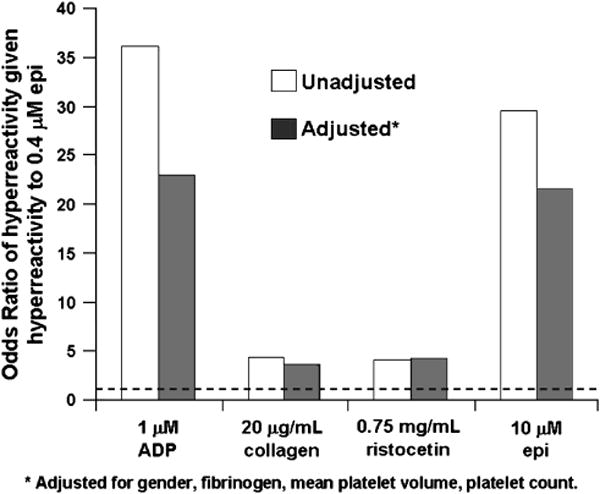
The unadjusted and adjusted Odds Ratios (probability of hyperreactivity to the specified agonist given hyperreactivity to epinephrine/probability of hyperreactivity to the specified agonist given no hyperreactivity to epinephrine) are shown for three different agonists. As expected, hyperreactivity to 0.4 μM epinephrine predicts hyperreactivity to 10 μM epinephrine. The dashed line indicates the position of an OR=1.0. Every bar represents an Odds Ratio that is significant at the <0.001 level.
Factors associated with platelet hyperreactivity
Factors associated with platelet hyperreactivity are listed in Table 7. Healthy subjects demonstrating platelet hyperreactivity did not differ significantly from subjects without hyperreactive platelets for age, race, body mass index, smoking status, or the presence of hypertension. The predilection of platelet hyperreactivity for female gender has been noted in other studies in both humans [38-40] and mice [41]. The lack of association between platelet hyperreactivity and oral contraceptive use, menopausal status, or phase of the menstrual cycle at the time of testing among female subjects suggests an underlying genetic basis (XX vs. XY) for this gender difference. Feng et al also found that higher fibrinogen levels were associated with increased aggregation to epinephrine, but other factors must also contribute [37]. It seems likely that no single factor will induce platelet hyperreactivity in most subjects, but it remains to be determined whether combinations of the factors listed in Table 7 increase the risk of this platelet phenotype.
Table 7.
Characteristics of subjects with hyperreactive platelets
| • | Female (p=0.016) |
| • | Higher fibrinogen levels (p=0.009) |
| • | Higher MPV (p<0.001) |
| • | Hyperreactive to other agonists |
| • | Increased platelet secretion |
| • | Increased reactivity to shear |
| • | Increased levels of integrin αIIbβ3 (GP IIb-IIIa) |
| • | 825 T allele of the GNB3 exon 10 polymorphism |
Platelet genes regulating platelet variation
The genetic contributions to coronary artery disease (CAD) and arterial thrombosis are well established [42]. We have observed strong heritability (h2) in platelet function studies on extended family structures in whites and African Americans [43]. There are well-known examples of inherited gene variations affecting platelet function, the best understood being mutations that lead to a clinical hemorrhagic phenotype (e.g., Glanzmann thrombasthenia and storage pool diseases).
In 1990 we reported the first genetic variation in a platelet-mediated inherited bleeding disorder [44], and since that time many other platelet gene defects have been described that produce a clinical hemorrhagic phenotype due to platelet hypofunction. Only in the last decade have investigators begun to consider inherited variations that enhance platelet reactivity. In 1996 we reported the first platelet genetic variation associated with acute myocardial infarction (MI) [45]. Since then many platelet genetic epidemiology studies have been performed, but the results have often been inconsistent (for review, see reference [46]). These inconsistencies should not be interpreted to mean platelet genes are not involved in the pathogenesis of these thrombotic disorders. Since thrombosis is one of several components involved in the pathophysiology of pathologic thrombi (such as MI), the effect of a prothrombotic variations are “diluted” by the effects of genes affecting other components of the pathogenesis of the clinical phenotype. Ten of 11 polymorphisms in 8 platelet genes were not associated with platelet hyperreactivity, including functional polymorphisms in genes encoding the α2 adrenergic receptor, integrins β3 and α2, and GPIbα and GP VI [37]. However, platelet hyperreactivity was associated with the C825T polymorphism in the gene encoding the β3 subunit of G-proteins.
Summary
Platelets play a central role in ischemic arterial occlusions and enhanced platelet reactivity is a risk factor for clinical CHD events. The use of state-of-the-art genomic, proteomic and RNA expression studies for gene discovery will require rigorously defined platelet phenotypes. Fortunately, platelet hyperreactivity can be easily and reproducibly detected in healthy subjects in the absence of anti-platelet therapy. Formal reproducibility studies are needed in patients taking aspirin and clopidogrel. Platelet hyperreactivity appears to be a global phenomenon, observed with multiple stimuli, suggesting mechanisms that involve the early pathways of platelet activation. There is marked inter-individual variation in platelet reactivity, and this variation is associated with female gender, high plasma fibrinogen and genetic variation. The future identification of novel genetic variants associated with platelet reactivity may be useful predictors of clinical thrombotic events, as well as modifiers of bleeding disorders or pharmacologic effects.
Acknowledgments
Supported by grants HL65229 and HL65967 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fuster V, Badimon L, Badimon JJ, Chesebro JH. The pathogenesis of coronary artery disease and the acute coronary syndromes (1) N Engl J Med. 1992;326:242–250. doi: 10.1056/NEJM199201233260406. [DOI] [PubMed] [Google Scholar]
- 2.Fitzgerald DJ, Roy L, Catella F, FitzGerald GA. Platelet activation in unstable coronary disease. N Engl J Med. 1986;315:983–989. doi: 10.1056/NEJM198610163151602. [DOI] [PubMed] [Google Scholar]
- 3.DeWood MA, Spores J, Notske R, et al. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N Engl J Med. 1980;303:897–902. doi: 10.1056/NEJM198010163031601. [DOI] [PubMed] [Google Scholar]
- 4.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996;88:1525–1541. [PubMed] [Google Scholar]
- 5.Bolhuis PA, Sakariassen KS, Sander HJ, Bouma BN, Sixma JJ. Binding of factor VIII-von Willebrand factor to human arterial subendothelium precedes increased platelet adhesion and enhances platelet spreading. J Lab Clin Med. 1981;97:568–576. [PubMed] [Google Scholar]
- 6.Fredrickson BJ, Dong JF, McIntire LV, López JA. Shear-dependent rolling on von Willebrand factor of mammalian cells expressing the platelet glycoprotein Ib-IX-V complex. Blood. 1998;92:3684–3693. [PubMed] [Google Scholar]
- 7.Nieswandt B, Brakebusch C, Bergmeier W, et al. Glycoprotein VI but not alpha2beta1 is essential for platelet interaction with collagen. EMBO J. 2001;20:2120–2130. doi: 10.1093/emboj/20.9.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 9.Ikeda H, Nakayama H, Oda T, et al. Soluble form of P-selectin in patients with acute myocardial infarction. Coron Artery Dis. 1994;5:515–518. [PubMed] [Google Scholar]
- 10.Trip MD, Cats VM, van Capelle FJ, Vreeken J. Platelet hyperreactivity and prognosis in survivors of myocardial infarction. N Engl J Med. 1990;322:1549–1554. doi: 10.1056/NEJM199005313222201. [DOI] [PubMed] [Google Scholar]
- 11.Thaulow E, Erikssen J, Sandvik L, Stormorken H, Cohn PF. Blood platelet count and function are related to total and cardiovascular death in apparently healthy men. Circulation. 1991;84:613–617. doi: 10.1161/01.cir.84.2.613. [DOI] [PubMed] [Google Scholar]
- 12.Terres W, Lund GK, Hubner A, Ehlert A, Reuter H, Hamm CW. Endogenous tissue plasminogen activator and platelet reactivity as risk factors for reocclusion after recanalization of chronic total coronary occlusions. Am Heart J. 1995;130:711–716. doi: 10.1016/0002-8703(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 13.Terres W, Hamm CW, Ruchelka A, Weilepp A, Kupper W. Residual platelet function under acetylsalicylic acid and the risk of restenosis after coronary angioplasty. J Cardiovasc Pharmacol. 1992;19:190–193. doi: 10.1097/00005344-199202000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Lam JY, Latour JG, Lesperance J, Waters D. Platelet aggregation, coronary artery disease progression and future coronary events. Am J Cardiol. 1994;73:333–338. doi: 10.1016/0002-9149(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 15.Capanni M, Prisco D, Antonucci E, et al. The pre-procedural platelet state predicts clinical recurrence after coronary angioplasty. Int J Clin Lab Res. 1999;29:145–149. doi: 10.1007/s005990050081. [DOI] [PubMed] [Google Scholar]
- 16.Gum PA, Kottke-Marchant K, Welsh PA, White J, Topol EJ. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol. 2003;41:961–965. doi: 10.1016/s0735-1097(02)03014-0. [DOI] [PubMed] [Google Scholar]
- 17.Cuisset T, Frere C, Quilici J, et al. High post-treatment platelet reactivity identified low-responders to dual antiplatelet therapy at increased risk of recurrent cardiovascular events after stenting for acute coronary syndrome. J Thromb Haemost. 2006;4:542–549. doi: 10.1111/j.1538-7836.2005.01751.x. [DOI] [PubMed] [Google Scholar]
- 18.Ziegler S, Maca T, Alt E, Speiser W, Schneider B, Minar E. Monitoring of antiplatelet therapy with the PFA-100(R) in peripheral angioplasty patients. Platelets. 2002;13:493–497. doi: 10.1080/0953710021000057866. [DOI] [PubMed] [Google Scholar]
- 19.Frossard M, Fuchs I, Leitner JM, et al. Platelet function predicts myocardial damage in patients with acute myocardial infarction. Circulation. 2004;110:1392–1397. doi: 10.1161/01.CIR.0000141575.92958.9C. [DOI] [PubMed] [Google Scholar]
- 20.Sambola A, Heras M, Escolar G, et al. The PFA-100 detects sub-optimal antiplatelet responses in patients on aspirin. Platelets. 2004;15:439–446. doi: 10.1080/69537100412351272550. [DOI] [PubMed] [Google Scholar]
- 21.Gianetti J, Parri MS, Sbrana S, et al. Platelet activation predicts recurrent ischemic events after percutaneous coronary angioplasty: a 6 months prospective study. Thromb Res. 2006;118:487–493. doi: 10.1016/j.thromres.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs I, Frossard M, Spiel A, Riedmuller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost. 2006;4:2547–2552. doi: 10.1111/j.1538-7836.2006.02239.x. [DOI] [PubMed] [Google Scholar]
- 23.Martin JF, Bath PM, Burr ML. Influence of platelet size on outcome after myocardial infarction. Lancet. 1991;338:1409–1411. doi: 10.1016/0140-6736(91)92719-i. [DOI] [PubMed] [Google Scholar]
- 24.Smyth DW, Martin JF, Michalis L, Bucknall CA, Jewitt DE. Influence of platelet size before coronary angioplasty on subsequent restenosis. Eur J Clin Invest. 1993;23:361–367. doi: 10.1111/j.1365-2362.1993.tb02037.x. [DOI] [PubMed] [Google Scholar]
- 25.Huczek Z, Kochman J, Filipiak KJ, et al. Mean platelet volume on admission predicts impaired reperfusion and long-term mortality in acute myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol. 2005;46:284–290. doi: 10.1016/j.jacc.2005.03.065. [DOI] [PubMed] [Google Scholar]
- 26.Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–1655. doi: 10.1161/01.cir.0000013777.21160.07. [DOI] [PubMed] [Google Scholar]
- 27.Heptinstall S, Mulley GP, Taylor PM, Mitchell JR. Platelet-release reaction in myocardial infarction. Br Med J. 1980;280:80–81. doi: 10.1136/bmj.280.6207.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faraday N, Braunstein JB, Heldman AW, et al. Prospective evaluation of the relationship between platelet-leukocyte conjugate formation and recurrent myocardial ischemia in patients with acute coronary syndromes. Platelets. 2004;15:9–14. doi: 10.1080/09537100310001644006. [DOI] [PubMed] [Google Scholar]
- 29.Miyamoto S, Kawano H, Kudoh T, et al. Usefulness of preprocedural platelet aggregation to predict restenosis after percutaneous coronary intervention. Am J Cardiol. 2005;96:71–73. doi: 10.1016/j.amjcard.2005.02.048. [DOI] [PubMed] [Google Scholar]
- 30.Miyamoto S, Kawano H, Sakamoto T, et al. Formation of platelet microaggregates correlates with adverse clinical outcome in patients with coronary artery disease. Thromb Haemost. 2003;89:681–686. [PubMed] [Google Scholar]
- 31.Kabbani SS, Watkins MW, Ashikaga T, et al. Platelet reactivity characterized prospectively: a determinant of outcome 90 days after percutaneous coronary intervention. Circulation. 2001;104:181–186. doi: 10.1161/01.cir.104.2.181. [DOI] [PubMed] [Google Scholar]
- 32.Kabbani SS, Watkins MW, Ashikaga T, Terrien EF, Sobel BE, Schneider DJ. Usefulness of platelet reactivity before percutaneous coronary intervention in determining cardiac risk one year later. Am J Cardiol. 2003;91:876–878. doi: 10.1016/s0002-9149(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 33.Turker S, Guneri S, Akdeniz B, et al. Usefulness of preprocedural soluble CD40 ligand for predicting restenosis after percutaneous coronary intervention in patients with stable coronary artery disease. Am J Cardiol. 2006;97:198–202. doi: 10.1016/j.amjcard.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 34.Willoughby SR, Stewart S, Holmes AS, Chirkov YY, Horowitz JD. Platelet nitric oxide responsiveness: a novel prognostic marker in acute coronary syndromes. Arterioscler Thromb Vasc Biol. 2005;25:2661–2666. doi: 10.1161/01.ATV.0000193622.77294.57. [DOI] [PubMed] [Google Scholar]
- 35.Steinhubl SR, Talley JD, Braden GA, et al. Point-of-care measured platelet inhibition correlates with a reduced risk of an adverse cardiac event after percutaneous coronary intervention: results of the GOLD (AU-Assessing Ultegra) multicenter study. Circulation. 2001;103:2572–2578. doi: 10.1161/01.cir.103.21.2572. [DOI] [PubMed] [Google Scholar]
- 36.Yee DL, Sun CW, Bergeron AL, Dong JF, Bray PF. Aggregometry detects platelet hyperreactivity in healthy individuals. Blood. 2005;106:2723–2729. doi: 10.1182/blood-2005-03-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yee DL, Bergeron AL, Sun CW, Dong JF, Bray PF. Platelet hyperreactivity generalizes to multiple forms of stimulation. J Thromb Haemost. 2006;4:2043–2050. doi: 10.1111/j.1538-7836.2006.02089.x. [DOI] [PubMed] [Google Scholar]
- 38.Johnson M, Ramey E, Ramwell PW. Sex and age differences in human platelet aggregation. Nature. 1975;253:355–357. doi: 10.1038/253355a0. [DOI] [PubMed] [Google Scholar]
- 39.Faraday N, Goldschmidt-Clermont PJ, Bray PF. Gender differences in platelet GPIIb-IIIa activation. Thromb Haemost. 1997;77:748–754. [PubMed] [Google Scholar]
- 40.Kurrelmeyer K, Becker L, Becker D, Yanek L, Goldschmidt-Clermont P, Bray PF. Platelet hyperreactivity in women from families with premature atherosclerosis. j am med woman assos. 2003;85:272–277. [PubMed] [Google Scholar]
- 41.Leng XH, Hong SY, Larrucea S, et al. Platelets of female mice are intrinsically more sensitive to agonists than are platelets of males. Arterioscler Thromb Vasc Biol. 2004;24:376–381. doi: 10.1161/01.ATV.0000110445.95304.91. [DOI] [PubMed] [Google Scholar]
- 42.Williams MS, Bray PF. Genetics of arterial prothrombotic risk states. Exp Biol Med (Maywood) 2001;226:409–419. doi: 10.1177/153537020122600504. [DOI] [PubMed] [Google Scholar]
- 43.Bray PF, Mathias RA, Herrera-Galeano JE, et al. Heritability of platelet reactivity in White and African American subjects at moderately high risk of coronary artery disease. Circulation. 2005;112:443a. [Google Scholar]
- 44.Bray PF, Shuman MA. Identification of an abnormal gene for the GPIIIa subunit of the platelet fibrinogen receptor resulting in Glanzmann's thrombasthenia. Blood. 1990;75:881–888. [PubMed] [Google Scholar]
- 45.Weiss EJ, Bray PF, Tayback M, et al. A polymorphism of a platelet glycoprotein receptor as an inherited risk factor for coronary thrombosis. N Engl J Med. 1996;334:1090–1094. doi: 10.1056/NEJM199604253341703. [DOI] [PubMed] [Google Scholar]
- 46.Yee DL, Bray PF. Clinical and functional consequences of platelet membrane glycoprotein polymorphisms. Semin Thromb Hemost. 2004;30:591–600. doi: 10.1055/s-2004-835679. [DOI] [PubMed] [Google Scholar]