

Abstract
Either the absence or dysfunction of a number of critical pathways, such as those that involve the nuclear retinoblastoma protein (Rb) and the transcription factor E2F1, may account for the aberrant induction of the cell cycle in post-mitotic neurons that can be responsible for oxidative stress-induced apoptotic cellular destruction. Yet, it is unclear whether early programs of apoptotic injury that involve membrane phosphatidylserine (PS) exposure and calreticulin expression as well as later phases of apoptotic injury with nuclear DNA injury require the critical modulation of Rb and E2F1. We demonstrate that both the post-translational of phosphorylation of Rb to prevent E2F1 transcription as well as the protein integrity of Rb are closely aligned with the modulation of cell cycle induction in post mitotic neurons during oxidative stress. More importantly, we illustrate that both the initial onset of apoptosis with either membrane PS exposure or calreticulin analysis as well as the more terminal phases of apoptosis that involve nuclear DNA degradation proceed concurrently in the same neuronal cells with cell cycle induction. Progression of attempted cell cycle induction is closely associated with the phosphorylation of Rb, its inability to bind to E2F1, and the degradation of the Rb protein. Inhibition of Rb phosphorylation using cyclin dependent kinase inhibitors maintains the integrity of the E2F1/Rb complex and is neuroprotective during free radical exposure. Furthermore, maintenance of the integrity of the Rb protein is specifically dependent upon caspase 3-like activity, since caspase 3 can cleave Rb during free radical activity and this degradation of Rb can be blocked during the inhibition of caspase 3 activity. Our studies not only highlight the critical role of attempted cell cycle induction during oxidative stress-induced neuronal apoptotic injury, but also bring to light the significant impact of the Rb and E2F1 pathways upon early apoptotic programs that can directly influence both intrinsic cell survival as well as extrinsic inflammatory cell activation.
Keywords: Apoptosis, bromodeoxyuridine, calreticulin, nitric oxide, E2F1, oxidative stress, phosphatidylserine, proliferating cell nuclear antigen, retinoblastoma protein
INTRODUCTION
Oxidative stress through the production of reactive oxygen species, such as nitric oxide (NO), can precipitate significant damage in the nervous system. In particular, generation of reactive oxygen species can lead to a number of neuronal cell injuries that involve cell membrane lipid destruction, the cleavage of nuclear DNA (Vincent, AM and Maiese, K, 1999, Wang, JY et al., 2003), the peroxidation of cellular membrane lipids (Siu, AW and To, CH, 2002), and the oxidation of proteins that yield protein carbonyl derivatives and nitrotyrosine (Adams, S et al., 2001). In addition to the destruction of cellular integrity, reactive oxygen species also can block mitochondrial respiration (Smeitink, JAM et al., 2004, Yamamoto, T et al., 2002).
Neuronal injury as a result of oxidative stress is believed to function through apoptotic pathways. For example, in clinical disorders such as Alzheimer’s disease, studies in human and in vitro models of Alzheimer’s disease demonstrate an association between neuronal DNA damage and plaque density (Colurso, GJ et al., 2003). Other lines of evidence link apoptotic cellular injury with mutations in the amyloid precursor protein (McPhie, DL et al., 2003). Interestingly, in patients with either mild cognitive impairment or with Alzheimer’s disease, cell cycle proteins, such as cyclin D, cyclin B, and proliferating cell nuclear antigen (PCNA), are significantly increased in the hippocampus and basal nucleus (McShea, A et al., 1997, Yang, Y et al., 2003), suggesting that attempted cell cycle induction in post-mitotic neurons may be responsible for neuronal apoptotic injury (Becker, EB and Bonni, A, 2004, Lin, SH et al., 2001).
The deficiency or dysfunction of several vital components for the complete execution of the cell cycle in post-mitotic neurons is believed to lead to apoptotic injury in neurons (Maiese, K and Chong, ZZ, 2004a). For example, during a cellular insult, deregulation of cell cycle proteins, such as cyclin, cyclin-dependent kinase, and the retinoblastoma protein (Rb), can ensue (Padmanabhan, J et al., 1999). In regards to the nuclear phosphoprotein Rb, it can prevent the induction of apoptosis through cell cycle inhibition (Lin, SH et al., 2001) and the inactivation of the transcription factor E2F1 (Kortylewski, M et al., 1999). Yet, it is unclear whether early programs of apoptotic injury that involve membrane phosphatidylserine (PS) exposure (Chong, ZZ et al., 2005b) and calreticulin expression (Gardai, SJ et al., 2005) as well as later phases of apoptotic injury with DNA injury (Maiese, K et al., 2004b) are reliant upon Rb regulation. Furthermore, modulation of Rb activity can occur at several levels. These may involve the phosphorylation state of Rb, since only hypophosphorylated Rb can bind to its transcription factor E2F1 to prevent apoptosis (Qin, XQ et al., 1995). In addition, Rb contains a caspase 3 - like recognition sequence, a CED-3/ICE cleavage site (DEADG), present at the C-terminus of all reported homologues except in related Rb proteins of p107 and p130 (Chen, WD et al., 1997, Tan, X et al., 1997) that can result in the internal cleavage of Rb to promote apoptotic injury (Chen, WD et al., 1997).
In our present studies, we demonstrate that apoptotic cellular injury during free radical exposure is correlated with aberrant cell cycle induction in post-mitotic neurons during both the initial onset of apoptosis with membrane PS exposure and the presence of calreticulin as well as with the more terminal phases of apoptosis that involve nuclear DNA degradation. Progression of attempted cell cycle induction relies upon the phosphorylation of Rb, its inability to bind to E2F1, and the degradation of the Rb protein. Inhibition of Rb phosphorylation using cyclin dependent kinase inhibitors maintains the integrity of the E2F1/Rb complex and is neuroprotective during free radical exposure. Furthermore, maintenance of the integrity of the Rb protein is crucial for this neuroprotective process since the degradation of Rb is tightly aligned with caspase 3-like activity during free radical activity and can be blocked during specific inhibition of caspase 3-like activity.
MATERIALS AND METHODS
Primary Hippocampal Neuronal Cultures
The hippocampi were obtained from E-19 Sprague-Dawley rat pups and incubated in dissociation medium (90 mM Na2SO4, 30 mM K2SO4, 5.8 mM MgCl2, 0.25 mM CaCl2, 10 mM kynurenic acid, and 1 mM HEPES with the pH adjusted to 7.4) containing papain (10 U ml-1) and cysteine (3 mM) for two 20 min periods. The hippocampi were then rinsed in dissociation medium and incubated in dissociation medium containing trypsin inhibitor (10-20 U ml-1) for three 5-minute periods. The cells were washed in growth medium (Leibovitz’s L-15 medium, Gibco BRL, Gaithersburg, MD) containing 6% sterile rat serum (ICN Biomedicals, Aurora, OH), 150 mM NaHCO3, 2.25 mg ml-1 of transferrin, 2.5 μg ml-1 of insulin, 10 nM progesterone, 90 μM putrescine, 15 nM selenium, 35 mM glucose, 1 mM L-glutamine, penicillin and streptomycin (50 μg ml-1), and vitamins. The dissociated cells were plated at a density of ~1.5 ×103 cells/mm2 in 35 mm polylysine/laminin-coated plates (Falcon Labware, Lincoln Park, NJ). Neurons were maintained in growth medium at 37 °C in a humidified atmosphere of 5% CO2 and 95% room air for 2 weeks.
Experimental Treatments
Nitric oxide (NO) administration was performed by replacing the culture media with media containing sodium nitroprusside (SNP) (300 μM) (Sigma, St. Louis, MO) or 6-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-hexan-amine (NOC-9) (300 μM) (Calbiochem, San Diego, CA) per the experimental paradigm (Maiese, K and Vincent, AM, 2000). More than one NO generator was used as a control to demonstrate that the neurons were responding to NO rather than to other by-products of these agents. During the experimental paradigms, inhibitors of Rb phosphorylation (butyrolactone (50 μM) or olomoucine (50 μM)) application was continuous.
Assessment of Neuronal Survival
Hippocampal neuronal injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 h following NO exposure per our previous protocols (Chong, ZZ et al., 2003, Lin, SH et al., 2000). Neurons were identified by morphology and the mean survival was determined by counting eight randomly selected non-overlapping fields with each containing approximately 10-20 neurons (viable + non-viable) in each 35 mm2 Petri dish.
Assessment of DNA Fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay (Lin, SH et al., 2000, Maiese, K et al., 2000). Briefly, neurons were fixed in 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde and the 3’-hydroxy ends of cut DNA were labeled with biotinylated dUTP using the enzyme terminal deoxytransferase (Promega, Madison, WI) followed by streptavidin-peroxidase and visualized with 3,3’-diaminobenzidine (Vector Laboratories, Burlingame, CA). The mean number of cells positive for TUNEL was determined by counting eight randomly selected non-overlapping fields with each containing approximately 20 cells (TUNEL (+) + TUNEL (-)).
Assessment of Membrane Phosphatidylserine (PS) Residue Externalization
Per our prior protocols (Lin, SH et al., 2000, Maiese, K et al., 2000), a 30 μg ml-1 stock solution of annexin V conjugated to phycoerythrin (PE) (R&D Systems, Minneapolis, MN) was diluted to 3 μg ml-1 in warmed calcium containing binding buffer (10 mM Hepes, pH 7.5, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2). Plates were incubated with 500 μl of diluted annexin V for 10 min. Images were acquired with “blinded” assessment with a Leitz DMIRB microscope (Leica, McHenry, IL) and a Fuji/Nikon Super CCD (6.1 megapixels) using transmitted light and fluorescent single excitation light at 490 nm and detected emission at 585 nm. The mean number of cells positive for membrane PS exposure was determined by counting eight randomly selected non-overlapping fields with each containing approximately 20 cells (PS (+) + PS (-)).
Assessment of Proliferating Nuclear Antigen (PCNA) Expression, Phospho-Rb Expression, and Bromodeoxyuridine (BrdU) Uptake
Per our prior protocols (Kang, Kang), proliferating cell nuclear antigen (PCNA) staining for microglial activation and bromodeoxyuridine (BrdU) staining for microglial proliferation were performed with anti-mouse monoclonal antibody PCNA (1:200) or BrdU (1:100) conjugated with biotinylated anti-mouse IgG (1:50) (Calbiochem, San Diego, CA) and visualized through fluorescein avidin (1:50) for PCNA and Texas Red streptavidin (Vector laboratories, Burlingame, CA) for BrdU. BrdU (10 μM) and fluorodexyuridine (1μM) (Sigma, St. Louis, MO) were applied 1 h prior to the time of fixation. For phospho (p)-Rb cell expression, neurons were incubated with a rabbit antibody against p-Rb (1:100, Cell Signaling, Beverly, MA) and then visualized by incubation with anti-rabbit IgG conjugated with Texas Red (Vector Laboratories, Burlingame, CA) for 2 hr.
Western Blot Analysis for Rb, Rb Phosphorylation, E2F1, and Calreticulin Phosphorylation
Cells were homogenized and following protein determination, each sample (50 μg/lane) was then subjected to 7.5% (Rb, phosphorylated (phospho-) Rb (p-Rb)) or 12.5% (E2F1, calreticulin) SDS-polyacrylamide gel electrophoresis. The membranes were incubated with a primary rabbit polyclonal antibody against Rb (1:200, Santa Cruz Biotechnologies, Santa Cruz, CA), a goat polyclonal antibody against phos-pho-Rb (p-Rb, 1:200) (Santa Cruz Biotechnologies, Santa Cruz, CA), a primary rabbit polyclonal antibody against E2F1 (1:200, Santa Cruz Biotechnologies, Santa Cruz, CA), and a rabbit antibody against calreticulin (1:1000, ABR Affinity Bioreagents, Golden, CO). After washing, the membranes were incubated with a horseradish peroxidase conjugated secondary antibody (goat anti-mouse IgG, 1:2000) (Pierce, Rockford, IL) or rabbit anti-goat IgG (1:5000) (Santa Cruz Biotechnologies, Santa Cruz, CA). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, NJ).
Immunoprecipitation of the Rb/E2F1 Complex
Cells were lysed, the crude homogenates were centrifuged (5 min, at 10,000 rpm, 4 °C), and the pellets were discarded. Supernatants were subsequently precleared by incubation with a mixture of protein A/ G-agarose conjugates (Santa Cruz Biotechnologies, Santa Cruz, CA) for 30 min at 4 °C and further centrifugation (2,500 rpm, 30 second). Total protein (200μg) was incubated in the presence of 2 μl of antibodies against Rb (Santa Cruz Biotechnologies, Santa Cruz, CA). The complexes were collected by incubation with 20μl of a mixture of protein A/G-agarose beads (Santa Cruz Biotechnologies, Santa Cruz, CA) and centrifugation. Pellets were then washed three times with cold PBS and underwent Western analysis for E2F1.
Statistical Analysis
For each experiment involving assessment of neuronal cell survival, DNA degradation, membrane PS exposure, PCNA expression, and BrdU uptake, the mean and standard error were determined from 4 to 6 replicate experiments. Statistical differences between groups were assessed by means of analysis of variance (ANOVA) with the post-hoc Student’s t-test. Results are expressed as the mean ± the standard error. Statistical significance was considered at P<0.05.
RESULTS
Cellular DNA Fragmentation Occurs in Conjunction with Attempted Cell Cycle Induction in Post-Mitotic Neurons During Free Radical Injury
To investigate whether the onset of DNA fragmentation following NO exposure was associated with the attempted induction of the cell cycle in post-mitotic neurons, we initially assessed the expression of PCNA that can become evident during the S and early G2 cell cycle phases (Hall, PA et al., 1990) with DNA fragmentation at 6 and 24 hr following NO exposure (NOC-9 or SNP, 300 μM) by double staining of immunocytochemistry. In Fig 1A, representative images demonstrate that 24 hr following NO (NOC-9, 300 μM) exposure significant expression of PCNA and TUNEL occurs in the same neuronal cells. Merged images illustrated the co-localization of DNA fragmentation with PCNA.
Fig. (1). Expression of proliferating cell nuclear antigen (PCNA) occurs in conjunction with DNA fragmentation following NO exposure.
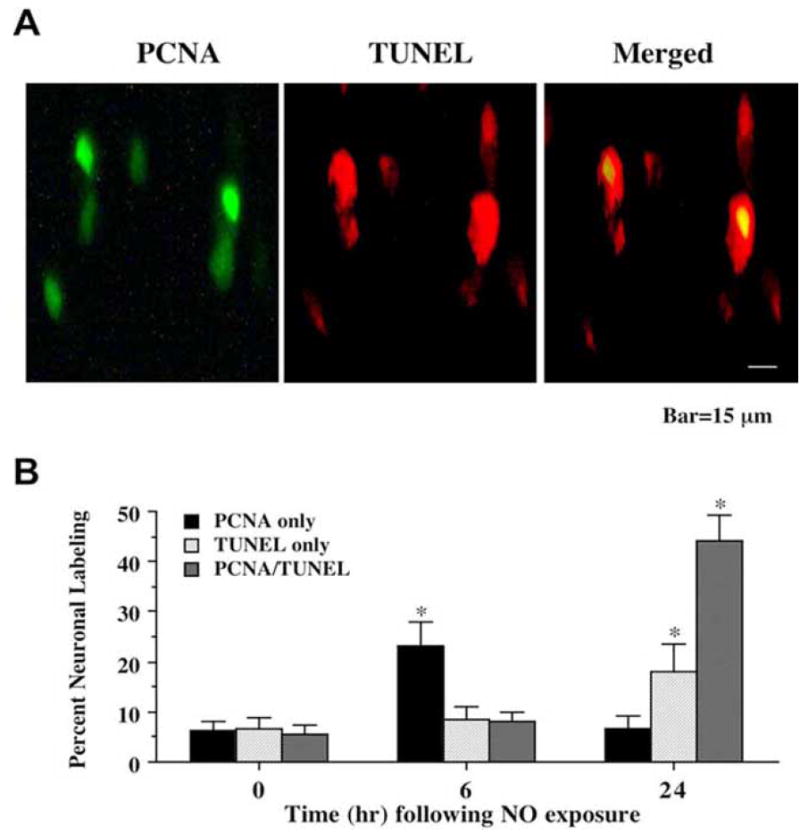
(A) Representative fields illustrate the double staining of neurons with TUNEL and PCNA expression. Dual labeling for TUNEL and PCNA expression in the same neuronal cultures was performed 24 hr following NO exposure (NOC-9, 300 μM). PCNA labeling (green) was evident in neuronal cultures exposed to NO. DNA fragmentation was determined by TUNEL staining (red). PCNA expression and DNA fragmentation were co-localized in hippocampal neurons 24 hr following NO exposure. (B) Quantitative results for either PCNA alone, TUNEL alone, or combined PCNA with TUNEL were determined 6 and 24 hr following NO exposure (SNP or NOC-9, 300 μM). PCNA positive neurons progressively became positive for TUNEL staining over a 24 hr period during NO exposure (*p<0.01 vs. control untreated neurons). To simplify the figures, results for the two NO donors were combined and data were represented as mean ± SEM.
We further quantitated our results and to simplify the analysis, data for the NO generators NOC-9 (300 μM) and SNP (300 μM) were combined. At time 0 hr (untreated control not exposed to NO), approximately 6-7% of neurons labeled for PCNA only or TUNEL only and another 4-5% of the neuronal population labeled for both PCNA and TUNEL (Fig 1B). However, following the application of NO, expression of combined PCNA and TUNEL in the same neurons significantly increases. Initially, PCNA at 6 hr increases significantly to approximately 25% while TUNEL expression or combined PCNA and TUNEL expression remains at approximately 8%, suggesting that early attempted cell cycle induction precedes DNA cell injury (Fig 1B). Over the course of the next 18 hr following NO application, the majority of neurons expressed either TUNEL only (approximately 20%) or combined PCNA and TUNEL (approximately 43%) (Fig 1B) with at least 63% of the neuronal population entering the initial stages of apoptosis and more than half of these cells have attempted to enter the cell cycle.
To further investigate the role of a cell cycle induction in post-mitotic neurons during the initial phases of apoptosis, we next extended our analysis with the DNA precursor BrdU to assess whether neurons attempt to re-enter the cell cycle at the G1/S phase (Lau, YF and Arrighi, FE, 1980) following exposure to NO. In Fig 2A, representative images demonstrate that 24 hr following NO (NOC-9, 300 μM) exposure significant uptake of BrdU and TUNEL occurs in the same neuronal cells. Merged images illustrated the co-localization of DNA fragmentation with BrdU.
Fig. (2). Uptake of bromodeoxyuridine (BrdU) occurs in conjunction with DNA fragmentation following NO exposure.
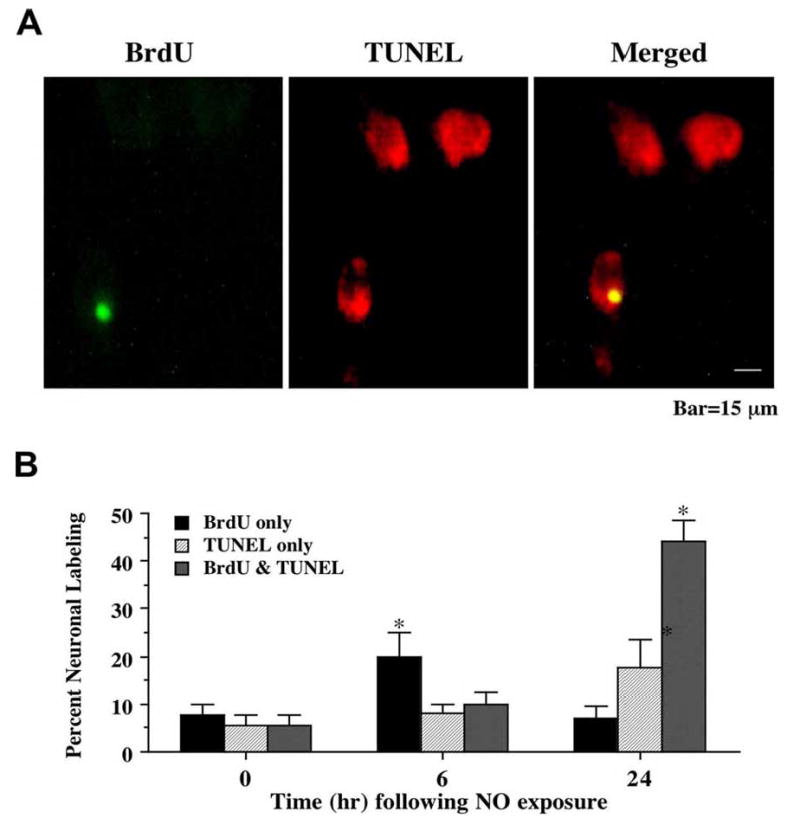
(A) Representative fields illustrate the double staining of neurons with TUNEL and BrdU incorporation. Dual labeling for BrdU and TUNEL in the same neuronal cultures was performed 24 hr following NO exposure (NOC-9, 300 μM). BrdU labeling (green) was evident in neuronal cultures exposed to NO. DNA fragmentation was determined by TUNEL staining (red). BrdU expression and DNA fragmentation were co-localized in hippocampal neurons 24 hr following NO exposure. (B) Quantitative results for either BrdU alone, TUNEL alone, or combined BrdU with TUNEL were determined 6 and 24 hr following NO exposure (SNP or NOC-9, 300 μM). BrdU positive neurons progressively became positive for TUNEL staining over a 24 hr period during NO exposure (*p<0.01 vs. control untreated neurons). To simplify the figures, results for the two NO donors were combined and data were represented as mean ± SEM.
Similar to our studies with PCNA, results for the NO generators NOC-9 (300 μM) and SNP (300 μM) were combined. Prior to NO administration (untreated control not exposed to NO), less than 10% of the neurons were positive for BrdU, TUNEL, or combined BrdU and TUNEL (Fig 2B). In contrast, within 6 hr following exposure to NO, a rapid and significant increase in neurons that expressed BrdU was present. Within 24 h following NO exposure, approximately 20% of neurons were positive for only TUNEL, but 45% of neurons labeled for combined BrdU and TUNEL (Fig 2B), illustrating that the majority of post-mitotic neurons undergoing the initial stages of apoptosis were also attempting to enter the cell cycle. Similar to our analysis with PCNA, a small group of neurons that was positive for BrdU only prior to NO exposure continued to label for BrdU only without significant change (Fig 2B).
Presence of Phosphorylated Retinoblastoma Protein (p-Rb) Parallels PCNA Expression and BrdU Uptake in Post-Mitotic Neurons During Free Radical Injury
Given the regulatory role the retinoblastoma (Rb) protein exerts over the induction of the cell cycle, we next examined the association of phosphorylated Rb (p-Rb) with the onset of an attempted cell cycle induction in post-mitotic neurons through the expression of PCNA and the uptake of BrdU. A NO donor (NOC-9 or SNP, 300 μM) was applied to neuronal cultures directly and double staining for p-Rb with PCNA or BrdU was performed 6 and 24 hr following NO exposure. As shown in Fig. 3A, representative images demonstrate that 24 hr following NO (NOC-9, 300 μM) exposure significant expression of p-Rb and PCNA occurs in the same neuronal cells. Merged images illustrated the co-localization of p-Rb with PCNA. In addition, representative images in Fig. 4A show that 24 hr following NO (NOC-9, 300 μM) exposure significant uptake of BrdU and p-Rb is present in the same neuronal cells. Merged images illustrated the co-localization of p-Rb with BrdU. Quantative analysis with results for the NO generators NOC-9 (300 μM) and SNP (300 μM) combined illustrates early p-Rb expression alone at approximately 30-35% in neurons at 6 hr post NO exposure with a significant rise in PCNA expression (approximately 20%) and BrdU uptake (approximately 23%) during this same time interval). Yet, by 24 hr post NO exposure, approximately 45-48% of neurons double label for p-Rb and PCNA or for p-Rb and BrdU with a total count of approximately 65-70% of post-mitotic neurons positive for p-Rb expression with or without PCNA or BrdU labeling, suggesting that a large proportion of neurons following free radical injury have attempted cell cycle induction with the phosphorylation of Rb (Figs. 3B and 4B).
Fig. (3). Expression of proliferating cell nuclear antigen (PCNA) occurs in conjunction with phosphorylation of the retinoblastoma protein (Rb) following NO exposure.
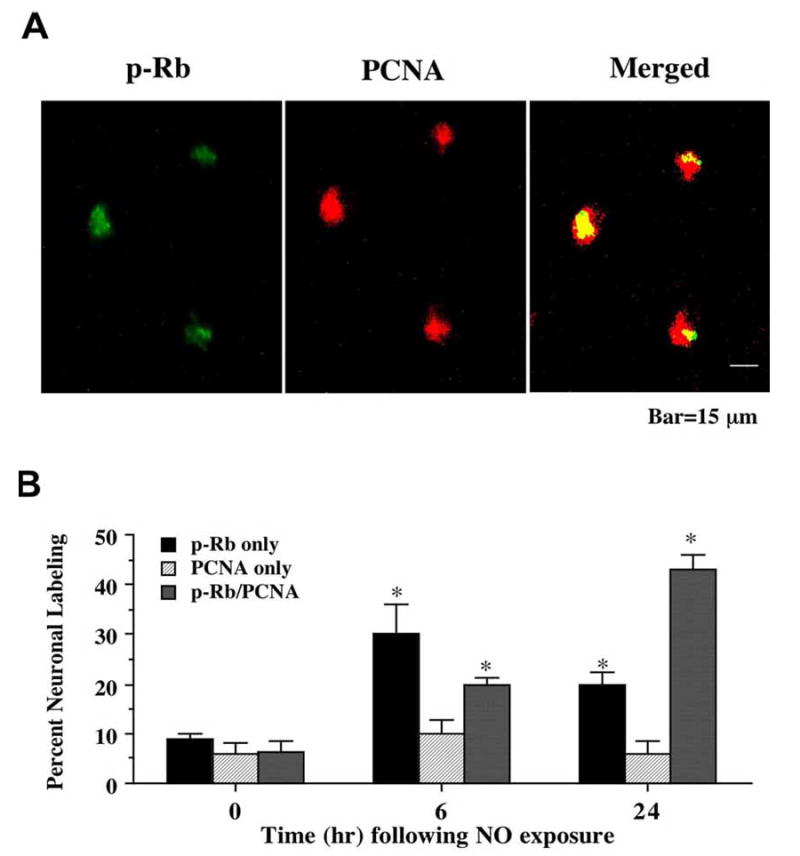
(A) Representative fields illustrate the double staining of neurons with phospho-(p-) Rb and PCNA expression. Dual labeling for p-Rb and PCNA expression in the same neuronal cultures was performed 24 hr following NO exposure (NOC-9, 300 μM). NO exposure resulted in significant p-Rb staining (green). PCNA labeling (red) also was evident in the neuronal cultures exposed to NO. Merged images reveal staining for both proteins in the same neuronal cells. (B) Quantitative results for either p-Rb alone, PCNA alone, or combined PCNA and p-Rb were determined 6 and 24 hr following NO exposure (SNP or NOC-9, 300 μM). p-Rb positive neurons progressively became positive for PCNA staining over a 24 hr period during NO exposure (*p<0.01 vs. control untreated neurons). To simplify the figures, results for the two NO donors were combined and data were represented as mean ± SEM.
Fig. (4). Uptake of bromodeoxyuridine (BrdU) occurs in conjunction with phosphorylation of the retinoblastoma protein (Rb) following NO exposure.
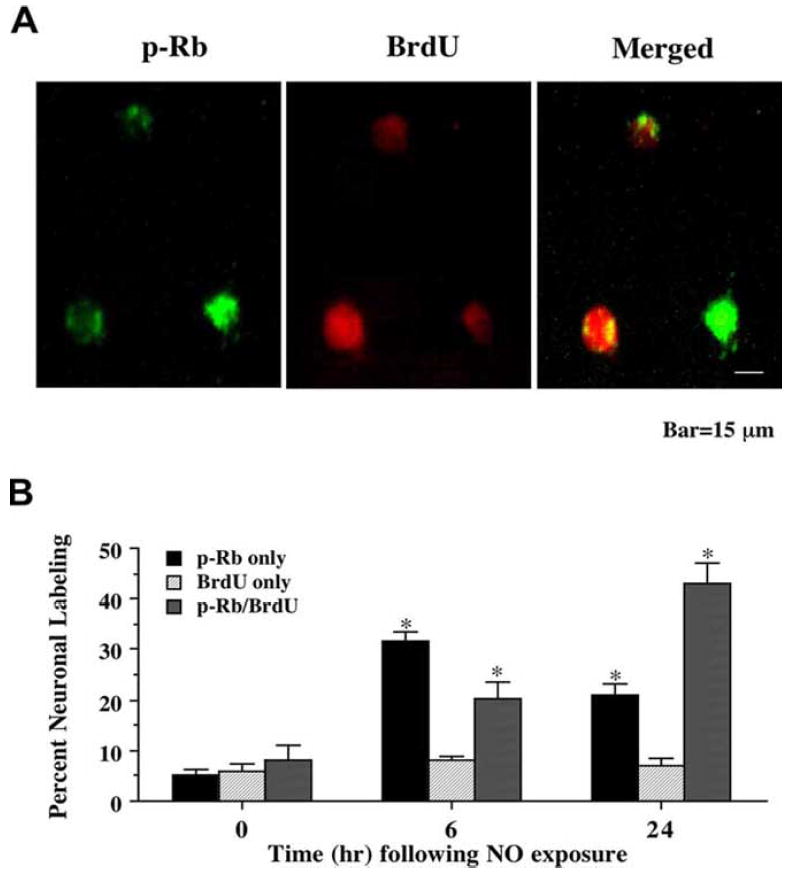
(A) Representative fields illustrate the double staining of neurons with p-Rb and BrdU incorporation. Dual labeling for BrdU and p-Rb in the same neuronal cultures was performed 24 hr following NO exposure (NOC-9, 300 μM). NO exposure resulted in significant p-Rb staining (green). BrdU labeling (red) also was evident in the neuronal cultures exposed to NO. Merged images reveal staining for both proteins in the same neuronal cells. (B) Quantitative results for either p-Rb alone, BrdU alone, or combined BrdU with p-Rb were determined 6 and 24 hr following NO exposure (SNP or NOC-9, 300 μM). p-Rb positive neurons progressively became positive for BrdU staining over a 24 hr period during NO exposure (*p<0.01 vs. control untreated neurons). To simplify the figures, results for the two NO donors were combined and data were represented as mean ± SEM.
Expression of p-Rb Occurs During Early and Late Indicators of Apoptosis in Post-Mitotic Neurons During Free Radical Injury
Since phosphorylation of p-Rb was found to be closely tied to attempted cell cycle induction in post-mitotic neurons with our studies examining PCNA expression and BrdU uptake, we assessed the role of p-Rb expression with the induction of both early apoptotic programs tied to phosphatidylserine (PS) membrane exposure and late apoptotic injury linked to nuclear DNA fragmentation. In Fig. 5A, representative images demonstrate that 24 hr following NO (NOC-9, 300 μM) administration p-Rb and PS externalization are evident in the same post-mitotic neurons. Merged images confirm the co-localization of p-Rb with PS. In a similar manner, representative images in Fig. 6A demonstrate that 24 hr following NO (NOC-9, 300 μM) exposure both p-Rb and DNA fragmentation per TUNEL labeling are present in the same neuronal cells. Merged images show the co-localization of p-Rb with TUNEL. Quantative analysis with results for the NO generators NOC-9 (300 μM) and SNP (300 μM) combined demonstrates early p-Rb expression alone at approximately 35-40% in neurons at 6 hr post NO exposure with an increase in PS expression (approximately 25%). Interestingly, by 24 hr post NO exposure, approximately 53-55% of neurons double label for p-Rb and PS or for p-Rb and TUNEL with a total count of approximately 70% of post-mitotic neurons positive for p-Rb expression with or without PS or TUNEL labeling, illustrating that a majority of neurons following free radical injury have attempted cell cycle induction with the phosphorylation of Rb as they enter both early and late stages of apoptosis (Figs. 5B and 6B).
Fig. (5). Cellular membrane phosphatidylserine (PS) exposure occurs in conjunction with phosphorylation of the retinoblastoma protein (Rb) following NO exposure.
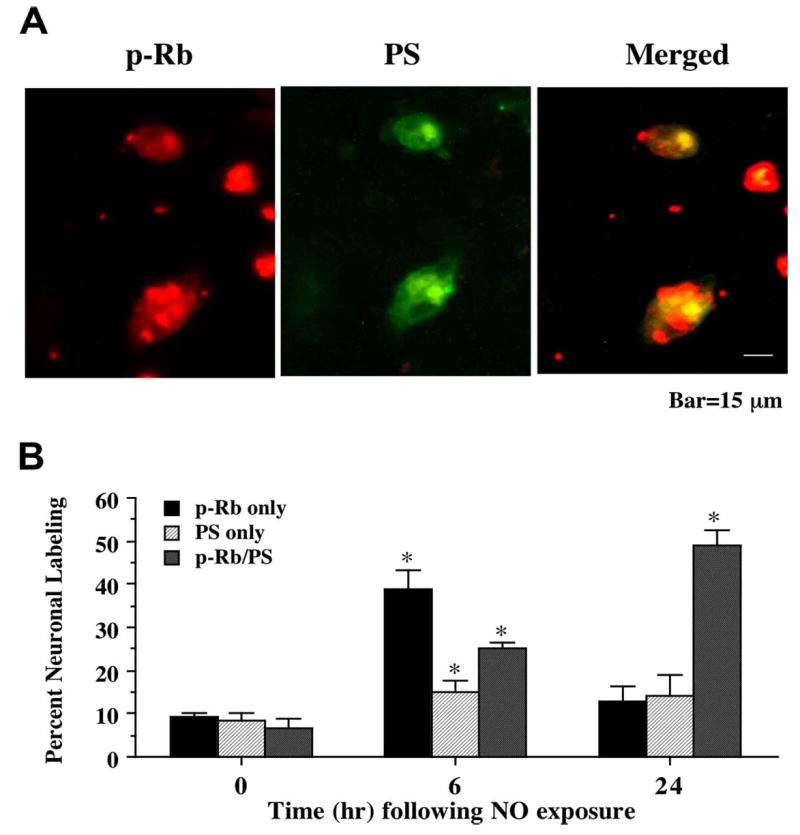
(A) Representative fields illustrate the double staining of neurons with p-Rb and annexin V labeling (for PS) performed 24 hr following NO exposure (NOC-9, 300 μM). NO exposure resulted in significant p-Rb staining (red). PS exposure (green) also was evident in neuronal cultures exposed to NO. Merged images reveal staining for both proteins in the same neuronal cells. (B) Quantitative results for either p-Rb alone, annexin V alone, or combined annexin V with p-Rb were determined 6 and 24 hr following NO exposure (SNP or NOC-9, 300 μM). p-Rb positive neurons progressively became positive for annexin V staining over a 24 hr period during NO exposure (*p<0.01 vs. control untreated neurons). To simplify the figures, results for the two NO donors were combined and data were represented as mean ± SEM.
Fig. (6). Nuclear DNA fragmentation occurs in conjunction with phosphorylation of the retinoblastoma protein (Rb) following NO exposure.
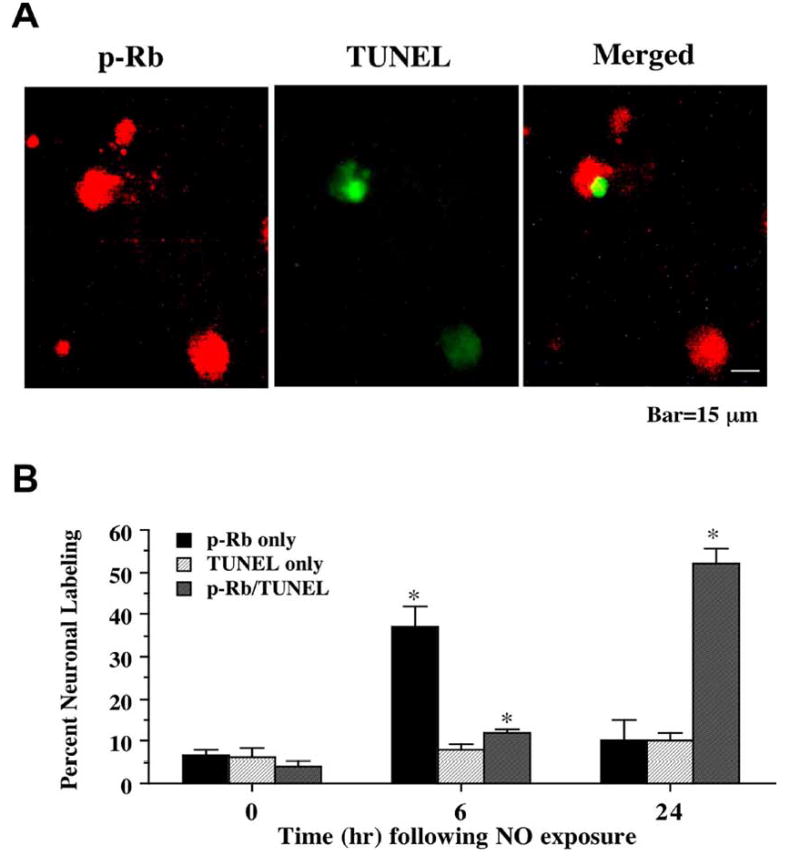
(A) Representative fields illustrate the double staining of neurons with p-Rb and TUNEL performed 24 hr following NO exposure (NOC-9, 300 μM). NO exposure resulted in significant p-Rb staining (red). p-Rb labeling (green) also was evident in neuronal cultures exposed to NO. Merged images reveal staining for both proteins in the same neuronal cells. (B) Quantitative results fro either p-Rb alone, TUNEL alone, or combined TUNEL with p-Rb were determined 6 and 24 hr following NO exposure (SNP or NOC-9, 300 μM). p-Rb positive neurons progressively became positive for TUNEL staining over a 24 hr period during NO exposure (*p<0.01 vs. control untreated neurons). To simplify the figures, results for the two NO donors were combined and data were represented as mean ± SEM.
Free radical Exposure Leads to the Phosphorylation of Rb and the Increased Expression of the Transcription Factor E2F1
Vital to the ability of Rb to block apoptotic injury with attempted cell cycle induction in post-mitotic neurons is the transcription factor E2F1. Although their exists five cloned E2Fs (E2F1 through 5), E2F1 in particular, preferentially binds to Rb. Deregulation of E2F during the absence of bound Rb can result in apoptosis in several cellular systems (Qin, XQ et al., 1995). We therefore examined the effect of free radical exposure on the phosphorylation of Rb and the expression of E2F1 in neurons. Increased phosphorylation of Rb allows E2F1 to proceed with attempted cell cycle induction.
Hippocampal neurons were exposed to a NO donor (NOC-9 or SNP, 300 μM) and Western analysis was performed at 1, 2, 4, 6, and 24 hr post NO exposure (Fig. 7A). Minimal expression of p-Rb was present in control untreated hippocampal neurons. Yet, following NO exposure (representative Western analysis shown with NOC-9, 300 μM), p-Rb expression significantly increased at 2, 4, 6 hr, and was maintained through the 24 hr period (Fig. 7A). In conjunction with the phosphorylation of Rb, E2F1 expression was significantly increased at 4, 6, and 24 hr suggesting that the loss of regulatory binding of Rb during its phosphorylation was associated with enhanced E2F1 expression (Fig. 7A).
Fig. (7). Maintenance of an intact Rb/E2F1 complex during blockade of Rb phosphorylation is neuroprotective during NO exposure.
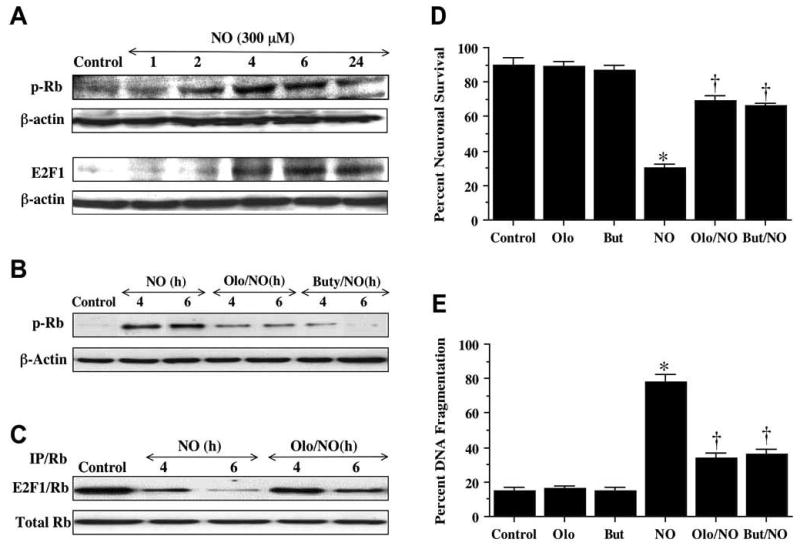
(A) A representative Western blot for Rb phosphorylation and E2F1 expression is illustrated for analysis at specific time periods following application of the NO donor NOC-9 (300 μM) using 50 μg/lane for neuronal lysates. The expression of p-Rb was significantly increased at 2 hr and continued over a 24 hr period following NO. E2F1 was significantly increased at 4 hr and continued over a 24 hr period following NO. (B) Primary hippocampal neurons were pretreated with the Rb phosphorylation inhibitors olomoucine (Olo, 50 μM) or butyrolactone (But, 50 μM) 24 hr prior to exposure to a NO generator (SNP, 300 μM or NOC-9, 300 μM)). Subsequently, the expression of p-Rb was determined at 4 and 6 hr following NO exposure. Representative Western analysis shown using 50 μg/lane for neuronal lysates. Olo and But significantly reduced p-Rb expression following NO exposure. (C) Representative results for E2F1/Rb immunoprecipitation are illustrated. Total protein extract was immunoprecitated by an antibody against Rb. E2F1/Rb complex expression by Western analysis reveals significant decreased expression of E2F1/Rb at 4 and 6 hr post NO exposure, but olomoucine (Olo, 50 μM) pretreatment significantly maintained E2F1/Rb expression at 4 and 6 hr following NO exposure. (D) Olomoucine (Olo, 50 μM) or butyrolactone (But, 50 μM) was applied to neuronal cultures 24 hr prior to exposure to a NO generator (SNP, 300 μM or NOC-9, 300 μM) and neuronal survival was determined by trypan blue exclusion 24 hr following NO exposure. Olo and But significantly increased neuronal survival from approximately 30% with NO only to approximately 70% (*p<0.01 vs. control untreated neurons; +p<0.01 vs. NO only treated neurons). (E) Olomoucine (Olo, 50 μM) or buty-rolactone (But, 50 μM) was applied to neuronal cultures 24 hr prior to exposure to a NO generator (SNP, 300 μM or NOC-9, 300 μM) and DNA fragmentation was determined by TUNEL assay 24 hr following NO exposure. Olo and But significantly decreased DNA fragmentation 24 hr following NO exposure (*p<0.01 vs. control untreated neurons; +p<0.01 vs. NO only treated neurons). In D and E, each data point represents the mean and SEM of n=4 determinations from four separate experiments. Control= untreated neurons not exposed to NO.
Prevention of Rb Phosphorylation Maintains Rb/E2F1 Binding and Prevents Neuronal Injury, DNA Fragmentation, and Early Apoptotic Signaling During Free Radical Exposure
We employed the inhibitors of cyclin-dependent kinases olomoucine and butyrolactone (Gray, N et al., 1999) to block the phosphorylation of Rb during free radical exposure and examine the effects upon E2F1 binding, neuronal injury, nuclear DNA fragmentation, and early cellular membrane apoptotic signaling. On Western analysis, application of the cyclin dependent kinase inhibitors olomoucine (Olo, 50 μM) and butyrolactone (But, 50 μM) significantly prevented the phosphorylation of Rb and the expression of p-Rb at 4 hr and 6 hr following NO exposure (Fig. 7B). A representative Western is shown with NOC-9, 300 μM.
In Fig. 7C, the binding of E2F1 to Rb was determined by immunoprecipitation at 4 and 6 hr following NO exposure with a representative Western shown with NOC-9, 300 μM. Binding of Rb to E2F1 blocks transcriptional activity of E2F1 and subsequent cell cycle induction. Immunoprecipitation of the E2F1/Rb complex was reduced at 4 hr following NO exposure and almost absent at 6 hr following NO exposure. Yet, the E2F1/Rb complex remained intact at 4 hr and 6 hr following NO exposure during application of the cyclin dependent kinase inhibitor olomoucine (Olo, 50 μM), suggesting that the cyclin dependent kinase inhibitors can prevent phosphorylation of Rb and maintain the integrity of the E2F1/Rb complex to block cell cycle induction (Figs. 7B and 7C)
We next assessed whether prevention of Rb phosphorylation which would maintain the E2F1/Rb complex was relevant during free radical exposure in regards to neuronal survival, nuclear DNA fragmentation, and early apoptotic membrane changes. In Figs. 7D and 7E, cyclin dependent kinase inhibitors of Rb phosphorylation were applied to neuronal cultures 24 hr prior to NO exposure (NOC-9 or SNP, 300 μM) and neuronal survival and DNA fragmentation were determined 24 hr following NO exposure. Results for the NO generators NOC-9 (300 μM) and SNP (300 μM) were combined to simplify the analysis. No toxicity was observed when the Rb phosphorylation inhibitors olomoucine (Olo, 50 μM) and butyrolactone (But, 50 μM) were applied to untreated control cultures of hippocampal neurons. Yet, inhibition of Rb phosphorylation by Olo and But protected neurons against NO toxicity and significantly increased neuronal survival following NO exposure, illustrating that Rb and its regulation of the E2F1 transcription factor was necessary to protect neurons against free radical injury (Fig. 7D). In a similar manner, administration of Olo and But also prevented late apoptotic injury as indicated by the significant reduction of DNA fragmentation during free radical exposure (Fig. 7E).
Calreticulin functions as a principal calcium binding and buffering protein in the endoplasmic reticulum (Groenendyk, J et al., 2004). As a result, calreticulin is involved in several cellular processes that modulate calcium homeostasis, protein folding, and cellular development. Interestingly, cell surface calreticulin has recently been suggested to function in early apoptotic signaling similar to PS exposure that can lead to inflammatory cell activation (Gardai, SJ et al., 2005). Therefore, we examined whether prevention of Rb phosphorylation could alter calreticulin expression similar to PS expression. Administration of the cyclin dependent kinase inhibitors olomoucine (Olo, 50 μM) and butyrolactone (But, 50 μM) prevented the upregulation of calreticulin expression during NO exposure, illustrating that regulation of post-translational activity of Rb impacts both cellular PS expression and calreticulin expression which together may affect inflammatory cell activation (Fig. 8).
Fig. (8). Inhibition of Rb phosphorylation down-regulates the expression of calreticulin during NO exposure.

A representative Western blot analysis for calreticulin expression following NO exposure is illustrated using 50 μg/lane for neuronal lysates. Rb phosphorylation inhibitors olomoucine (Olo, 50 μM) or butyrolactone (But, 50 μM) were applied to neuronal cultures 24 hr prior to a NO generator (SNP, 300 μM or NOC-9, 300 μM) and total protein extracts were prepared at 24 hr following NO exposure. Olo and But significantly reduced the expression of calreticulin following NO exposure.
Inhibition of Caspase 3-like Activity Prevents Degradation for Rb During Free Radical Exposure
Modulation of the cell cycle to prevent apoptosis may occur at different checkpoints to maintain the integrity and hypophosphorylated state of Rb. One mechanism may involve may involve the regulation of caspase 3 - like activity, since Rb is a substrate for caspase 3 and contains a consensus DEADG site at the carboxyl terminus (Chen, WD et al., 1997). Cleavage of Rb by caspase 3 appears to disable its ability to inhibit E2F activity and to subsequently avert apoptosis (An, B and Dou, QP, 1996, Tan, X et al., 1997). We therefore examined whether total Rb expression during free radical exposure could be altered by modulation of caspase 3-like activity. Modulation of caspase 1-like activity was used as an experimental control.
In Fig. 9A, total Rb expression was maintained within 6 hr following NO exposure, but, the expression of total Rb was significantly decreased 24 hr following NO exposure (representative Western shown with NOC-9, 300 μM). Next, the caspase-1 inhibitor (YVAD, 50 μM) and the caspase-3 inhibitor (DEVD, 50 μM) were added into neuronal cultures 1 hr prior to NO exposure (NOC-9, 300 μM used as illustration) and a Western blot for C-terminal Rb was carried out 24 hr later (Fig. 9B). The C-terminal Rb was cleaved yielding a significant decrease in Rb expression (Fig. 9B) following NO administration. Yet, administration of DEVD during NO exposure significantly prevented Rb degradation, but minimal protection against Rb degradation was seen with YVAD, a caspase 1 inhibitor, suggesting that maintenance of Rb integrity is closely aligned with modulation of caspase 3 -like activity.
Fig. (9). Rb degradation during NO expsoure is dependent on caspase 3 activation.
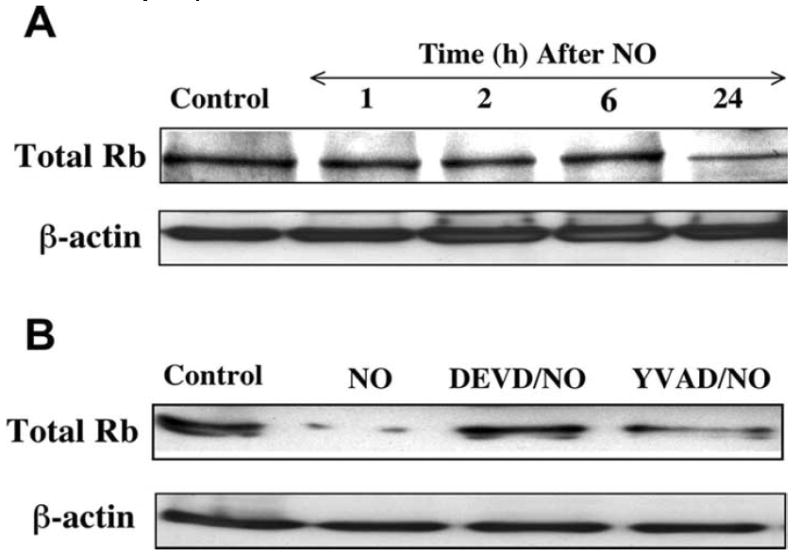
(A) A representative Western blot for Rb expression is illustrated for analysis at specific time periods following application of the NO donor NOC-9 (300 μM) using 50 μg/lane for neuronal lysates. The expression of Rb was significantly decreased 24 hr following NO exposure. (B) The caspase 1 inhibitor (YVAD, 50 μM) and the caspase 3 inhibitor (DEVD, 50 μM) were applied to cultures 1 hr prior to NO exposure and Rb expression was determined 24 hr following NO exposure. Degradation of Rb by NO was significantly prevented by the caspase 3 inhibitor DEVD.
DISCUSSION
Oxidative stress associated with free radical injury may lead to attempted cell cycle induction in neurons (Chong, ZZ et al., 2005d, Chong, ZZ et al., 2005f). Furthermore, a number of studies have provided direct evidence that cell cycle induction in post-mitotic neurons can activate cellular mechanisms that lead to neuronal apoptosis (El-Khodor, BF et al., 2003, Ino, H and Chiba, T, 2001, Lin, SH et al., 2001, Rideout, HJ et al., 2003). If one also examines clinical neurodegenerative diseases, such as Alzheimer’s disease, attempted cell cycle induction also can yield neuronal injury (Arendt, T, 2000, Becker, EB and Bonni, A, 2005, Chong, ZZ et al., 2005e). Expression of components of the cell cycle, such as cyclin D, CDK4, PCNA, and cyclin B1 have been shown to be present in patients with Alzheimer’s disease and in animal models in regions that include the hippocampus, subiculum, locus coeruleus, and dorsal raphe nuclei. A close association appears to exist between injured cells and cell cycle protein expression, since staining for cell cycle proteins have been shown to be absent in brain regions without neuronal injury in Alzheimer’s disease patients and in age-matched brains (Chong, ZZ et al., 2005e, Stoothoff, WH and Johnson, GV, 2005, Wen, Y et al., 2004).
Given the prior work that links attempted cell cycle induction in post-mitotic neurons to neuronal injury, our present work sheds further insight into the potential mechanisms that regulate attempted cell cycle neuronal injury during both early and late programs of apoptosis. Apoptotic injury is believed to contribute significantly to a variety of diseases that involve the nervous system such as cerebral ischemic disease, Alzheimer’s disease, and trauma (Doonan, F and Cotter, TG, 2004, Ferretti, P, 2004, Koyama, R and Ikegaya, Y, 2004, Li, F et al., 2004). Apoptosis consists of membrane PS exposure and DNA fragmentation (Maiese, K et al., 2005). As an early event in the dynamics of cellular apoptosis, membrane PS externalization can become a signal for the phagocytosis of cells (Chong, ZZ et al., 2002, Chong, ZZ et al., 2005c). In contrast to the early externalization of membrane PS residues, the cleavage of genomic DNA into fragments is considered to be a delayed event that occurs late during apoptosis (Dombroski, D et al., 2000, Jessel, R et al., 2002, Kang, JQ et al., 2003b, Maiese, K et al., 2000).
In regards to DNA fragmentation, we show that PCNA expression, which can become evident during the S and early G2 cell cycle phases (Hall, PA et al., 1990), is closely associated with the majority of neurons with nuclear DNA fragmentation. In addition, our analysis with the DNA precursor BrdU to assess whether neurons attempt to re-enter the cell cycle at the G1/S phase (Lau, YF et al., 1980) further complemented the studies with PCNA and illustrated that a significant proportion of neurons with nuclear DNA fragmentation also had uptake of BrdU, suggesting that the late phase of apoptotic injury was closely tied to attempted cell cycle induction.
Interestingly, post-translational modification of Rb by phosphorylation during free radical oxidative stress appears to be associated with cell cycle induction in post-mitotic neurons. The Rb protein plays an important regulatory role in the cell cycle and apoptotic cell injury injury. Rb can control the cell cycle at the G1/S checkpoint (Goodrich, DW et al., 1991, Lee, EY et al., 1994). Unphosphorylated Rb can block cell cycle progression by binding to the transcription factor E2F1 in the G1 phase and repress E2F1-responsive genes (Dick, FA and Dyson, N, 2003, Harbour, JW and Dean, DC, 2000). The phosphorylation of Rb results in the dissociation of the E2F1/Rb complex and the release of E2F1 that contributes to S phase entry and apoptosis (Denchi, EL and Helin, K, 2005). We illustrate that a large proportion of neurons following free radical injury attempt to enter the cell cycle as indicated by PCNA expression or BrdU uptake during the phosphorylation of Rb. Furthermore, these same neurons that are exposed to the detrimental effects of NO also label for both TUNEL and PS with p-Rb expression, illustrating that a majority of neurons following free radical injury have attempted cell cycle induction with the phosphorylation of Rb as they enter both the early and late stages of apoptosis. Possibly more important is the observation that Rb may exert control over membrane PS expression which can lead to inflammatory cell activation and subsequent microglial activation (Chong, ZZ et al., 2005a, Kang, JQ et al., 2003a, Kang, JQ et al., 2003b).
As previously described, phosphorylation of Rb can determine its ability to bind to its transcription factor E2F1 and prevent transcription. Passage through the G1 restriction point and entry into the S phase is controlled by cyclin-dependent protein kinases involved in the phosphorylation process of Rb that are sequentially regulated by the cyclins D, E, and A (Beijersbergen, RL et al., 1995, Quelle, DE et al., 1993, Resnitzky, D et al., 1995). In our present study, we show that following NO exposure, p-Rb expression is significantly increased at 2, 4, 6 hr and is maintained through a 24 hr period. In conjunction with the phosphorylation of Rb, E2F1 expression also is significantly increased at 4, 6, and 24 hr, suggesting that the loss of regulatory binding of Rb during its phosphorylation is associated with enhanced E2F1 expression. We subsequently employed the inhibitors of cyclin-dependent kinases olomoucine and butyrolactone (Gray, N et al., 1999) to block the phosphorylation of Rb during free radical exposure. We show that that the cyclin dependent kinase inhibitors can prevent phosphorylation of Rb and maintain the integrity of the E2F1/Rb complex to block cell cycle induction. Similar to prior models with potassium chloride withdrawal cell injury and cyclin dependent kinase inhibition (Padmanabhan, J et al., 1999), we demonstrate that inhibition of Rb phosphorylation by Olo and But significantly increased neuronal survival following NO exposure. Our work suggests that Rb and its regulation of the E2F1 transcription factor is necessary to protect neurons against free radical neuronal injury as well as late apoptotic injury with DNA fragmentation.
Our observations that phosphorylation of Rb becomes a critical modulator of neuronal DNA fragmentation also extends to early apoptotic mechanisms through analysis with calreticulin. Calreticulin is a principal calcium binding and buffering protein in the endoplasmic reticulum (Groenendyk, J et al., 2004). Yet, cell surface calreticulin has recently been suggested to function in early apoptotic signaling similar to PS exposure that can lead to inflammatory cell activation (Gardai, SJ et al., 2005). We illustrate that administration of Olo or But prevented the upregulation of calreticulin expression during NO exposure, illustrating that regulation of post-translational activity of Rb impacts upon both cellular PS externalization and calreticulin expression, providing further evidence that modulation of Rb pathways may directly control neuronal inflammatory cell injury (Chong, ZZ et al., 2005a, Kang, JQ et al., 2003a, Kang, JQ et al., 2003b).
In addition to the post-translational modification of Rb through phosphorylation, Rb also may be regulated through cleavage by enhanced caspase activity. Rb is a substrate for caspase 3 and contains a consensus DEADG site at the carboxyl terminus (Chen, WD et al., 1997). Cleavage of Rb by caspase 3 can disable the ability of Rb to inhibit E2F1 activity and prevent apoptosis (An, B et al., 1996, Tan, X et al., 1997). We therefore elected to investigate whether total Rb expression during free radical exposure could be altered by modulation of caspase 3-like activity. NO exposure can directly lead to caspase 3 activity (Li, F et al., 2006, Maiese, K and Chong, ZZ, 2003). We show that the integrity of Rb is aligned with caspase 3-like activity during free radical activity and that blockade of caspase 3-like activity prevents Rb degradation.
In summary, we show that attempted cell cycle induction during NO-induced oxidative stress requires the phosphorylation of Rb, the inability of Rb to bind to E2F1, and the loss of integrity of the Rb protein. Yet, potentially more important is the observation that aberrant cell cycle induction in post-mitotic neurons occurs during both the later committed phases of apoptosis with nuclear DNA degradation as well as during the initial onset of apoptosis that involves membrane PS externalization and calreticulin expression which can significantly influence inflammatory cell activation. In addition, both modulation of Rb phosphorylation and the maintenance of Rb integrity during caspase 3 activity are necessary to block aberrant cell cycle induction and protect against apoptotic neuronal injury during free radical exposure. Future investigations should continue to elucidate the expanding role of cell cycle regulation in post-mitotic neurons that impacts upon both early and late apoptotic programs.
Acknowledgments
This research was supported by the following grants (KM): American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
References
- Adams S, Green P, Claxton R, Simcox S, Williams MV, Walsh K, Leeuwenburgh C. Reactive carbonyl formation by oxidative and non-oxidative pathways. Front Biosci. 2001;6:A17–24. doi: 10.2741/adams. [DOI] [PubMed] [Google Scholar]
- An B, Dou QP. Cleavage of retinoblastoma protein during apoptosis: an interleukin 1 beta-converting enzyme-like protease as candidate. Cancer Res. 1996;56(3):438–42. [PubMed] [Google Scholar]
- Arendt T. Alzheimer’s disease as a loss of differentiation control in a subset of neurons that retain immature features in the adult brain. Neurobiol Aging. 2000;21(6):783–96. doi: 10.1016/s0197-4580(00)00216-5. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol. 2004;72(1):1–25. doi: 10.1016/j.pneurobio.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A. Beyond proliferation--cell cycle control of neuronal survival and differentiation in the developing mammalian brain. Semin Cell Dev Biol. 2005;16(3):439–48. doi: 10.1016/j.semcdb.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Beijersbergen RL, Carlee L, Kerkhoven RM, Bernards R. Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev. 1995;9(11):1340–53. doi: 10.1101/gad.9.11.1340. [DOI] [PubMed] [Google Scholar]
- Chen WD, Otterson GA, Lipkowitz S, Khleif SN, Coxon AB, Kaye FJ. Apoptosis is associated with cleavage of a 5 kDa fragment from RB which mimics dephosphorylation and modulates E2F binding. Oncogene. 1997;14(10):1243–8. doi: 10.1038/sj.onc.1201096. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang J, Li F, Maiese K. mGluRI Targets Microglial Activation and Selectively Prevents Neuronal Cell Engulfment Through Akt and Caspase Dependent Pathways. Curr Neurovasc Res. 2005a;2(3):197–211. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106(23):2973–9. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol. 2003;138(6):1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005b;20(1):299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal Injury during beta-amyloid toxicity. Curr Neurovasc Res. 2005c;2(4):387–399. doi: 10.2174/156720205774962683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005d;75(3):207–46. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Stress in the brain: novel cellular mechanisms of injury linked to Alzheimer’s disease. Brain Res Brain Res Rev. 2005e;49(1):1–21. doi: 10.1016/j.brainresrev.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li FQ, Maiese K. Employing new cellular therapeutic targets for Alzheimer’s disease: A change for the better? Curr Neurovasc Res. 2005f;2(1):55–72. doi: 10.2174/1567202052773508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colurso GJ, Nilson JE, Vervoort LG. Quantitative assessment of DNA fragmentation and beta-amyloid deposition in insular cortex and midfrontal gyrus from patients with Alzheimer’s disease. Life Sci. 2003;73(14):1795–803. doi: 10.1016/s0024-3205(03)00512-5. [DOI] [PubMed] [Google Scholar]
- Denchi EL, Helin K. E2FZ1 is crucial for E2F-dependent apoptosis. EMBO Rep. 2005;6(7):661–8. doi: 10.1038/sj.embor.7400452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, Dyson N. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol Cell. 2003;12(3):639–49. doi: 10.1016/s1097-2765(03)00344-7. [DOI] [PubMed] [Google Scholar]
- Dombroski D, Balasubramanian K, Schroit AJ. Phosphatidylserine expression on cell surfaces promotes antibody- dependent aggregation and thrombosis in beta2-glycoprotein I-immune mice. J Autoimmun. 2000;14(3):221–9. doi: 10.1006/jaut.2000.0365. [DOI] [PubMed] [Google Scholar]
- Doonan F, Cotter TG. Apoptosis: A potential therapeutic target for retinal degenerations. Curr Neurovasc Res. 2004;1(1):41–53. doi: 10.2174/1567202043480215. [DOI] [PubMed] [Google Scholar]
- El-Khodor BF, Oo TF, Kholodilov N, Burke RE. Ectopic expression of cell cycle markers in models of induced programmed cell death in dopamine neurons of the rat substantia nigra pars compacta. Exp Neurol. 2003;179(1):17–27. doi: 10.1006/exnr.2002.8047. [DOI] [PubMed] [Google Scholar]
- Ferretti P. Neural stem cell plasticity: Recruitment of endogenous populations for regeneration. Curr Neurovasc Res. 2004;1(3):215–229. doi: 10.2174/1567202043362397. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321–34. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Goodrich DW, Wang NP, Qian YW, Lee EY, Lee WH. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell. 1991;67(2):293–302. doi: 10.1016/0092-8674(91)90181-w. [DOI] [PubMed] [Google Scholar]
- Gray N, Detivaud L, Doerig C, Meijer L. ATP-site directed inhibitors of cyclin-dependent kinases. Curr Med Chem. 1999;6(9):859–75. [PubMed] [Google Scholar]
- Groenendyk J, Lynch J, Michalak M. Calreticulin, Ca2+, and calcineurin - signaling from the endoplasmic reticulum. Mol Cells. 2004;17(3):383–9. [PubMed] [Google Scholar]
- Hall PA, Levison DA, Woods AL, Yu CC, Kellock DB, Watkins JA, Barnes DM, Gillett CE, Camplejohn R, Dover R, et al. Proliferating cell nuclear antigen (PCNA) immunolocalization in paraffin sections: an index of cell proliferation with evidence of deregulated expression in some neoplasms. J Pathol. 1990;162(4):285–94. doi: 10.1002/path.1711620403. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14(19):2393–409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- Ino H, Chiba T. Cyclin-dependent kinase 4 and cyclin D1 are required for excitotoxin-induced neuronal cell death in vivo. J Neurosci. 2001;21(6):6086–94. doi: 10.1523/JNEUROSCI.21-16-06086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessel R, Haertel S, Socaciu C, Tykhonova S, Diehl HA. Kinetics of apoptotic markers in exogeneously induced apoptosis of EL4 cells. J Cell Mol Med. 2002;6(1):82–92. doi: 10.1111/j.1582-4934.2002.tb00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003a;74(1):37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003b;64(3):557–69. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- Kortylewski M, Heinrich PC, Mackiewicz A, Schniertshauer U, Kling-muller U, Nakajima K, Hirano T, Horn F, Behrmann I. Interleukin-6 and oncostatin M-induced growth inhibition of human A375 melanoma cells is STAT-dependent and involves upregulation of the cyclin-dependent kinase inhibitor p27/Kip1. Oncogene. 1999;18(25):3742–53. doi: 10.1038/sj.onc.1202708. [DOI] [PubMed] [Google Scholar]
- Koyama R, Ikegaya Y. Mossy fiber sprouting as a potential therapeutic target for epilepsy. Curr Neurovasc Res. 2004;1(1):3–10. doi: 10.2174/1567202043480242. [DOI] [PubMed] [Google Scholar]
- Lau YF, Arrighi FE. Studies of mammalian chromosome replication. I. BrdU-induced differential staining patterns in interphase and metaphase chromosomes. Cytogenet Cell Genet. 1980;27(23):176–83. doi: 10.1159/000131479. [DOI] [PubMed] [Google Scholar]
- Lee EY, Hu N, Yuan SS, Cox LA, Bradley A, Lee WH, Herrup K. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 1994;8(17):2008–21. doi: 10.1101/gad.8.17.2008. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Erythropoietin on a Tightrope: Balancing Neuronal and Vascular Protection between Intrinsic and Extrinsic Pathways. Neurosignals. 2004;13(6):265–89. doi: 10.1159/000081963. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Winding through the Wnt pathway during cellular development and demise. Histol and Histopath. 2006;21:103–124. doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SH, Chong ZZ, Maiese K. Cell cycle induction in post-mitotic neurons proceeds in concert with the initial phase of programmed cell death in rat. Neurosci Lett. 2001;310(23):173–7. doi: 10.1016/s0304-3940(01)02118-8. [DOI] [PubMed] [Google Scholar]
- Lin SH, Vincent A, Shaw T, Maynard KI, Maiese K. Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J Cereb Blood Flow Metab. 2000;20(9):1380–91. doi: 10.1097/00004647-200009000-00013. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol Sci. 2003;24(5):228–32. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Insights into oxidative stress and potential novel therapeutic targets for Alzheimer disease. Restor Neurol Neurosci. 2004a;22(2):87–104. [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. Erythropoietin in the brain: can the promise to protect be fulfilled? Trends Pharmacol Sci. 2004b;25(11):577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005;293(1):90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Vincent AM. Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J Neurosci Res. 2000;59(4):568–80. doi: 10.1002/(SICI)1097-4547(20000215)59:4<568::AID-JNR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- McPhie DL, Coopersmith R, Hines-Peralta A, Chen Y, Ivins KJ, Manly SP, Kozlowski MR, Neve KA, Neve RL. DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J Neurosci. 2003;23(17):6914–27. doi: 10.1523/JNEUROSCI.23-17-06914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol. 1997;150(6):1933–9. [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan J, Park DS, Greene LA, Shelanski ML. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci. 1999;19(20):8747–56. doi: 10.1523/JNEUROSCI.19-20-08747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin XQ, Livingston DM, Ewen M, Sellers WR, Arany Z, Kaelin W., Jr The transcription factor E2F-1 is a downstream target of RB action. Mol Cell Biol. 1995;15(2):742–55. doi: 10.1128/mcb.15.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle DE, Ashmun RA, Shurtleff SA, Kato JY, Bar-Sagi D, Roussel MF, Sherr CJ. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993;7(8):1559–71. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- Resnitzky D, Hengst L, Reed SI. Cyclin A-associated kinase activity is rate limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol Cell Biol. 1995;15(8):4347–52. doi: 10.1128/mcb.15.8.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout HJ, Wang Q, Park DS, Stefanis L. Cyclin-dependent kinase activity is required for apoptotic death but not inclusion formation in cortical neurons after proteasomal inhibition. J Neurosci. 2003;23(4):1237–45. doi: 10.1523/JNEUROSCI.23-04-01237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu AW, To CH. Nitric oxide and hydroxyl radical-induced retinal lipid peroxidation in vitro. Clin Exp Optom. 2002;85(6):378–82. [PubMed] [Google Scholar]
- Smeitink JAM, van den Heuvel L, Koopman WJH, Nijtmans LGJ, Ugalde C, Willems P. Cell biological consequences of mitochondrial NADH: Ubiquinone oxidoreductase deficiency. Curr Neurovasc Res. 2004;1(1):29–40. doi: 10.2174/1567202043480224. [DOI] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739(23):280–97. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Tan X, Martin SJ, Green DR, Wang J. Degradation of retino-blastoma protein in tumor necrosis factor- and CD95-induced cell death. J Biol Chem. 1997;272(15):9613–6. doi: 10.1074/jbc.272.15.9613. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. Nitric oxide induction of neuronal endonuclease activity in programmed cell death. Exp Cell Res. 1999;246(2):290–300. doi: 10.1006/excr.1998.4282. [DOI] [PubMed] [Google Scholar]
- Wang JY, Shum AY, Ho YJ. Oxidative neurotoxicity in rat cerebral cortex neurons: synergistic effects of H2O2 and NO on apoptosis involving activation of p38 mitogen-activated protein kinase and caspase-3. J Neurosci Res. 2003;72(4):508–19. doi: 10.1002/jnr.10597. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer’s disease-like tauopathy in female rats. J Biol Chem. 2004;279(21):22684–92. doi: 10.1074/jbc.M311768200. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Maruyama W, Kato Y, Yi H, Shamoto-Nagai M, Tanaka M, Sato Y, Naoi M. Selective nitration of mitochondrial complex I by peroxynitrite: involvement in mitochondria dysfunction and cell death of dopaminergic SH-SY5Y cells. J Neural Transm. 2002;109(1):1–13. doi: 10.1007/s702-002-8232-1. [DOI] [PubMed] [Google Scholar]
- Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci. 2003;23(7):2557–63. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]