

Abstract
Saturated free fatty acid (FFA) is a major source of metabolic stress that activates the cJun NH2-terminal kinase (JNK). This FFA-stimulated JNK pathway is relevant to hallmarks of metabolic syndrome, including insulin resistance. Here we used gene ablation studies in mice to demonstrate a central role for mixed-lineage protein kinases (MLK) in this signaling pathway. Saturated FFA causes protein kinase C (PKC)-dependent activation of MLK3 that subsequently causes increased JNK activity by a mechanism that requires the MAP kinase kinases MKK4 and MKK7. Loss of PKC, MLK3, MKK4, or MKK7 expression prevents FFA-stimulated JNK activation. Together, these data establish a signaling pathway that mediates effects of metabolic stress on insulin resistance.
Introduction
Obesity is a world-wide health problem that is associated with metabolic syndrome, including insulin resistance and the development of type 2 diabetes (Boden, 2003). Obesity is associated with increased blood levels of free fatty acids (FFA). This increase in FFA is considered to be a causative link between obesity and insulin resistance (Arner, 2002; Boden, 2006; Kahn et al., 2006; Kovacs and Stumvoll, 2005). The mechanism that accounts for FFA-induced insulin resistance is incompletely understood. However, activation of the cJun NH2-terminal kinase (JNK) stress signaling pathway appears to play a major role in the development of obesity-induced insulin resistance (Hirosumi et al., 2002). One molecular mechanism that contributes to JNK-induced insulin resistance is the phosphorylation of the insulin receptor adapter protein IRS1 on the inhibitory site Ser-307 (Aguirre et al., 2000; Aguirre et al., 2002; Lee et al., 2003). FFA-stimulated JNK signaling is therefore an important physiological mechanism of insulin resistance.
Although progress has been achieved towards understanding the function of JNK in the development of insulin resistance (Weston and Davis, 2007), the mechanism that accounts for FFA-stimulated JNK activation remains unclear. Two MAP kinase kinases (MAP2K; MKK4 and MKK7) can phosphorylate and activate JNK (Tournier et al., 2001). These kinases, in turn, are phosphorylated and activated by MAP kinase kinase kinases (MAP3K). Several different groups of MAP3K are implicated in JNK regulation, including MEKK, MLK, TAK1, ASK1, and TPL2 (Weston and Davis, 2007). Knowledge of specific roles of individual MAP3K in the response to particular stimuli is incomplete, in part, because it appears that some MAP3K function redundantly. Thus, it is possible that different MAP3K isoforms may function cooperatively or in a temporally sequential manner. For example, TAK1 plays an essential role in the early phase of JNK activation caused by tumor necrosis factor (TNF) and ASK1 plays an important role in the late phase of TNF-stimulated JNK activation (Sato et al., 2005; Shim et al., 2005; Tobiume et al., 2001). Nevertheless, unique functions for some MAP3K have been established, including a requirement of MEKK1 for JNK activation caused by activin in keratinocytes (Zhang et al., 2005) and a requirement of MEKK2 for JNK activation caused by FGF2 in fibroblasts (Kesavan et al., 2004).
The role of other MAP3K in JNK regulation is poorly understood. For example, the role of the mixed-lineage protein kinase (MLK) group of MAP3K has not been established. Three sub-groups of MLK can be defined based on their primary structure (Gallo and Johnson, 2002). First, the MLK1 - 4 sub-group is characterized by an NH2-terminal-terminal SH3 domain, a kinase domain, a leucine zipper domain, and a Cdc42/Rac1 binding (CRIB) motif. Second, the DLK and LZK sub-group consists of a kinase domain and a leucine zipper domain that is interrupted by a 31 amino acid insertion. Third, the ZAK sub-group is characterized by a kinase domain, a leucine zipper domain, and a sterile-α-motif (SAM). The physiological functions of these MLK isoforms have not been defined (Gallo and Johnson, 2002), although a role for DLK in radial migration of neurons during development has been proposed (Hirai et al., 2006) and it has been suggested that MLK3 may cause microtubule instability, regulate B-Raf/Raf-1 signaling complexes, and promote proliferation (Chadee and Kyriakis, 2004; Chadee et al., 2006; Swenson et al., 2003). MLK3 may also play a minor role in regulating the amplitude of TNF-stimulated JNK activation (Brancho et al., 2005).
The goal of this study was to identify the MAP3K isoform that mediates the effect of FFA on JNK activation. We report that MLK plays a central role in FFA-induced JNK activation. This function of MLK as a mediator of FFA signaling represents a mechanism that enables metabolic regulation of signal transduction.
Results
JNK is activated by saturated FFA
We examined FFA-stimulated JNK activation in mouse embryonic fibroblasts (MEF). Treatment with palmitate caused JNK activation in a time- and dose-dependent manner that was detected by an in vitro kinase assay using [γ-32P]ATP and cJun as substrates (Figure 1A,B). The delayed time course of JNK activation may reflect the proposal that saturated FFA may signal indirectly by increasing the cellular pools of diacylglyerol (Montell et al., 2001) and ceramide (Schmitz-Peiffer et al., 1999). Importantly, the concentration of FFA that is sufficient to activate JNK in these cells is within the physiological range for blood FFA concentrations in wild-type mice (Kim et al., 2004).
Figure 1. JNK is activated by saturated free fatty acids.
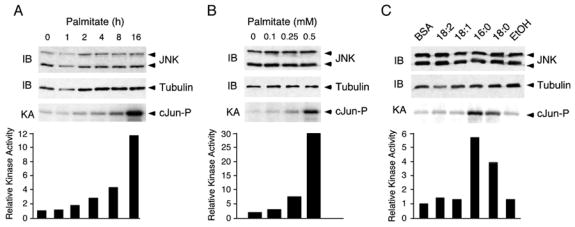
A) MEF were treated with 0.5mM palmitate for the indicated times. The expression of JNK was examined by immunoblot analysis. JNK activity was measured in a kinase assay (KA) using [γ-32P]ATP and cJun as substrates.
B) MEF were treated with different concentrations of palmitate for 16h. The expression of JNK was examined by immunoblot analysis. JNK activity was measured in a kinase assay (KA) using [γ-32P]ATP and cJun as substrates.
C) MEF were treated with 0.5mM linoleate (18:2), oleate (18:1), palmitate (16:0) and stearic acid (18:0) for 16h. A solvent control (ethanol, Etoh) for the treatment with steric acid is shown. The expression of JNK was examined by immunoblot analysis. JNK activity was measured in a kinase assay (KA) using [γ-32P]ATP and cJun as substrates.
We tested the ability of different FFA to activate JNK in MEF. Incubation with saturated FFA, including palmitate and stearate, resulted in JNK activation, while addition of mono- and poly-unsaturated FFA (oleate and linoleate) had no effect (Figure 1C). The inability of unsaturated FFA to cause JNK activation correlates with the finding that unsaturated FFA can increase the cellular pool of triacylglyerol rather than diacylglycerol (Montell et al., 2001). Together, these data demonstrate that exposure of MEF to saturated FFA, but not unsaturated FFA, causes JNK activation.
FFA-stimulated JNK activation is mediated by MKK4 and MKK7
Two different MAP2K (MKK4 and MKK7) are implicated in JNK activation (Davis, 2000). Indeed, compound mutants that lack expression of both MKK4 and MKK7 are resistant to stress-induced activation of JNK (Tournier et al., 2001). We therefore tested the effect of MKK4 and MKK7-deficiency on FFA-stimulated JNK activation. We found that, as expected, compound mutant Mkk4−/− Mkk7−/− MEF failed to exhibit JNK activation in response to treatment with palmitate (Figure 2C). Similarly, Mkk4−/− MEF and Mkk7−/− MEF failed to respond to palmitate with increased JNK activation (Figure 2A,B). These data indicate that both MKK4 and MKK7 are required for FFA-stimulated JNK activity in MEF. This combined requirement for MKK4 and MKK7 may reflect the co-operative phosphorylation of the JNK Thr-Pro-Tyr dual phosphorylation motif on Tyr by MKK4 and on Thr by MKK7 (Lawler et al., 1998; Tournier et al., 2001).
Figure 2. FFA-induced JNK activation is mediated by MKK4 and MKK7.
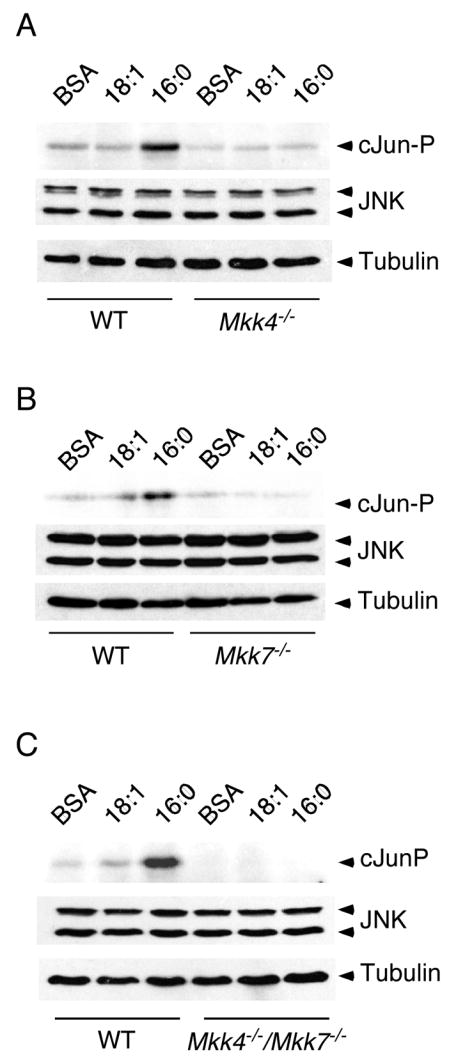
WT or MKK-deficient MEF were treated with 0.5 mM oleic acid (18:1) or 0.5 mM palmitic acid (16:0) for 16 h. The expression of JNK was examined by immunoblot analysis. JNK activity was measured in an in vitro kinase assay (KA) using [γ-32P]ATP and cJun as substrates. A) WT and Mkk4−/− MEF. B) WT and Mkk7−/− MEF. C) WT and Mkk4−/− Mkk7−/− MEF.
MLK3 is required for FFA-stimulated JNK activation
The MAP2K isoforms MKK4 and MKK7 are activated by several different classes of MAP3K (Davis, 2000). However, the MAP3K that mediates FFA-induced JNK activation has not been defined. Recent studies have implicated a role for Toll-like receptor 4 (TLR4) in the response to saturated FFA (Kim et al., 2007; Kim, 2006; Poggi et al., 2007; Shi et al., 2006; Song et al., 2006). It is established that the MAP3K isoform TAK1 is required for TLR4-stimulated activation of both NF-κB and JNK (Sato et al., 2005; Shim et al., 2005). TAK1 therefore represents a candidate MAP3K isoform that may mediate the effects of saturated FFA on JNK activity. However, we found that saturated FFA caused JNK activation in Tak1−/− MEF (Figure S1). These data indicate that the TLR4/TAK1 pathway is not essential for JNK activation caused by saturated FFA.
A second candidate class of MAP3K that may mediate the effects of saturated FFA on JNK activation is represented by the mixed-lineage protein kinase (MLK) group (Gallo and Johnson, 2002). Indeed, the ubiquitously expressed isoform MLK3 has been reported to be activated by ceramide (Sathyanarayana et al., 2002), a possible lipid mediator of FFA signaling (Schmitz-Peiffer et al., 1999). To test whether MLK3 is a component of a FFA-induced signaling pathway, we examined the effect of FFA on MLK3 regulation. Immunoblot analysis using an antibody to the MLK3 T-loop phosphorylation sites Thr-277 and Ser-281 demonstrated that treatment with saturated FFA caused increased T-loop phosphorylation of MLK3 (Figure 3A). This observation indicates that MLK3 is activated by FFA and that MLK3 is a component of a FFA-stimulated signaling pathway.
Figure 3. JNK activation by saturated FFA is MLK3 dependent.
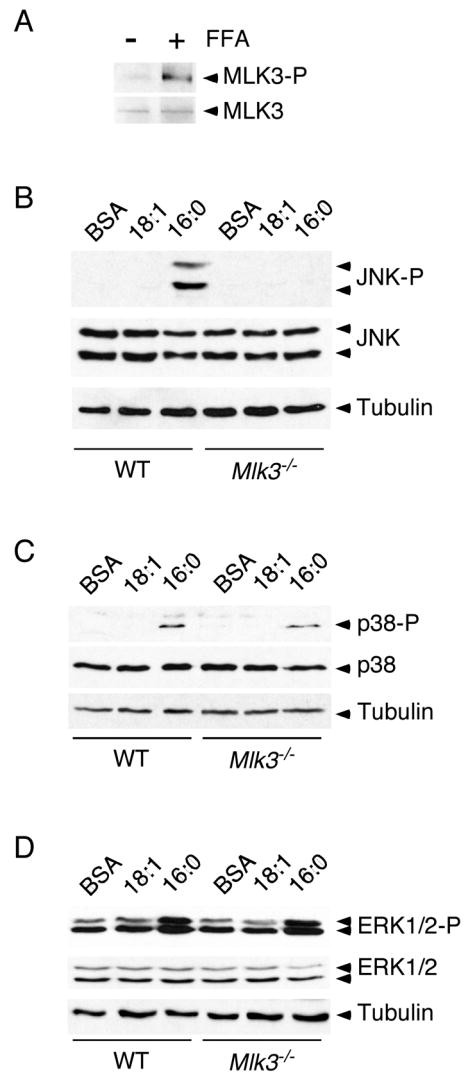
(A) Wild-type MEF were treated (16 h) with 0.5 mM palmitic acid. The expression of MLK3 and phosphorylation of the MLK3 T-loop (Thr-277 and Ser281) was examined by immunoblot analysis.
(B - D) Wild-type (WT) or Mlk3−/− MEF were treated (16 h) with 0.5 mM oleic acid (18:1) or 0.5 mM palmitic acid (16:0). The phosphorylation and expression of JNK (B), p38 MAPK (C), and ERK1/2 (D) was examined by immunoblot analysis.
To test whether MLK3 may be required for FFA-stimulated JNK activation, we prepared MEF from wild-type and Mlk3−/− mice. Treatment of wild-type MEF with saturated FFA (palmitate), but not unsaturated FFA (oleate), caused increased JNK activation (Figure 3B). In contrast, Mlk3−/− MEF were unresponsive to saturated FFA (Figure 3B). This analysis demonstrated that MLK3 is essential for FFA-stimulated JNK activation in MEF.
Previous studies have indicated that MLK3 may regulate multiple MAPK signaling pathways (Gallo and Johnson, 2002). It is therefore possible that MLK3 may mediate the effects of FFA on the p38 MAPK and ERK1/2 signaling pathways. Indeed, p38 MAPK and ERK1/2 were activated by treatment with saturated FFA, although the response of these MAPK pathways was modest compared with the robust effect of FFA to activate JNK (Figure 3B-D). Comparative studies indicated that MLK3-deficiency selectively blocked the effect of FFA on JNK activation. These studies demonstrated that MLK3 is required for FFA-stimulated JNK activation and that it plays either no role or a redundant role in FFA-stimulated activation of other MAPK pathways.
MLK3-deficient cells are protected against FFA-induced insulin resistance
Dysregulated lipid metabolism and increased levels of FFA are a major cause of insulin resistance (Delarue and Magnan, 2007). Indeed, FFA-stimulated JNK activation may play a central role in obesity-induced insulin resistance (Hirosumi et al., 2002). We therefore examined the effect of FFA on insulin signaling in MEF. Since MEF express only low levels of insulin receptors, we used a high concentration of insulin (100nM) that binds both IGF-1 and insulin receptors. Control studies demonstrated that treatment with FFA caused JNK activation (Figure 4A) and markedly decreased insulin-stimulated AKT activation (Figure 4B). In contrast, MLK3-deficient cells did not exhibit FFA-stimulated JNK activation (Figure 4A) and FFA did not inhibit insulin-stimulated AKT activation (Figure 4B). Together, these data indicate that MLK3 is essential for FFA-stimulated JNK activation and insulin resistance.
Figure 4. MLK3-deficient cells are protected against FFA-induced insulin resistance.
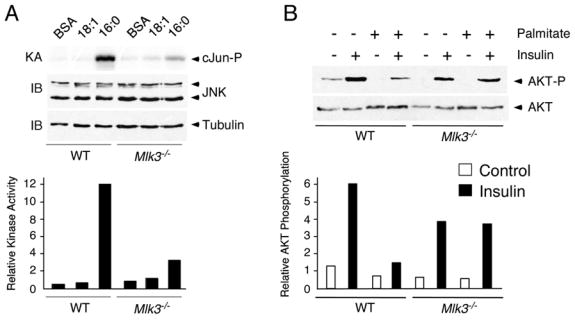
A) WT or Mlk3−/− MEF were treated with 0.5 mM oleic acid (18:1) or 0.5 mM palmitic acid (16:0) for 16 h. The expression of JNK was examined by immunoblot analysis. JNK activity was measured in a kinase assay (KA) using [γ-32P]ATP and cJun as substrates.
B) WT or Mlk3−/− MEF were pretreated (16 h) with BSA or 0.5mM palmitate. After incubation with 100nM insulin for 30 min, the cells were harvested and AKT expression and phosphorylation at Ser-473 were examined by immunoblot analysis.
Role of MLK3 in obesity-induced JNK activation
Studies using cultured cells indicated that MLK3 is essential for FFA-stimulated JNK activation (Figures 2 – 4). These observations suggest that MLK3 may be important for JNK regulation by FFA in vivo. To test this hypothesis, we examined the effect of MLK3-deficiency in an animal model that is associated with increased concentrations of blood FFA. Feeding mice a high fat diet causes increased blood FFA and JNK activation (Hirosumi et al., 2002). We found that the high fat diet also caused MLK3 activation that was detected by increased MLK3 T-loop phosphorylation in both epididymal white adipose tissue and interscapular brown adipose tissue (Figure 5A,B).
Figure 5. FFA causes MLK3 and JNK activation.
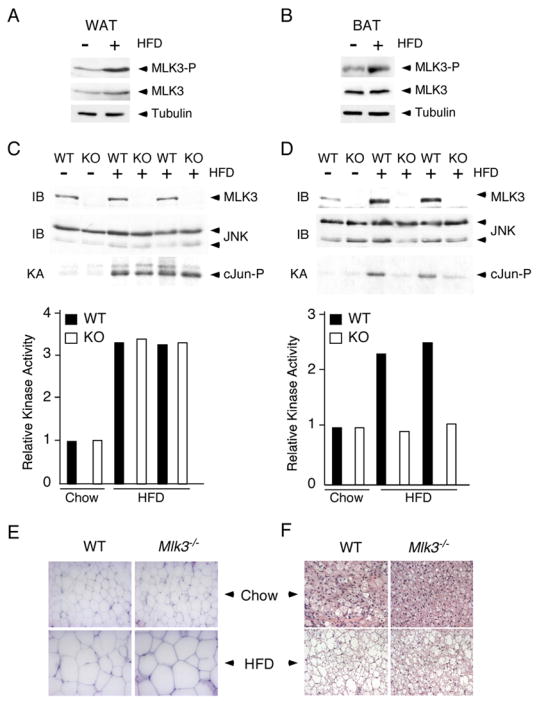
A, B) Wild-type mice were maintained (16 weeks) on a standard diet or on a high fat diet (HFD). MLK3 expression, MLK3 T-loop phosphorylation (Thr-277 and Ser-281), and Tubulin expression in white (epididymal) fat (WAT) and brown (interscapular) fat (BAT) was examined by immunoblot analysis.
C,D) White adipose tissue (C) and brown adipose tissue (D) of wild-type mice (WT) and Mlk3−/−mice (KO) maintained (16 weeks) on a standard diet (chow) and on a high fat diet (HFD) was examined by immunoblot analysis (IB) using antibodies to JNK and MLK3. JNK activity was measured in a kinase assay (KA) using [γ-32P]ATP and cJun as substrates.
E,F) Representative histological sections of white adipose tissue (E) and brown adipose tissue (F) stained with hematoxylin and eosin from wild-type and Mlk3−/− mice fed a standard or high fat diet for 16 wk.
To test whether MLK3 contributes to JNK activation in vivo, we examined adipose tissue from mice fed either a control diet (chow) or a high fat diet. JNK was activated in both the white fat (Figure 5C) and the brown fat (Figure 5D) of mice fed a high fat diet. Studies of Mlk3−/− mice demonstrated that MLK3 was required for obesity-induced JNK activation in brown fat (Figure 5D), but not in white fat (Figure 5C). The non-essential role of MLK3 in white fat may reflect the expression of other members of the MLK group in this tissue. These data indicate that MLK3 is essential for obesity-induced JNK activation in brown fat and that MLK3 may play only a redundant role in white fat.
Histological analysis of adipose tissue demonstrated that feeding a high fat diet caused hypertophy of both white and brown adipose tissue (Figure 5E,F). No differences between white fat from wild-type and Mlk3−/− mice were detected. However, MLK3-deficiency did cause markedly reduced lipid accumulation in the brown fat of mice fed a high fat diet.
MLK3 is required for inhibitory phosphorylation of IRS1
The adapter protein IRS1, an important mediator of signaling by the insulin receptor, is phosphorylated on tyrosine and serves to recruit multiple insulin-regulated signaling modules, including PI-3 kinase (White, 2006). JNK can suppress IRS1 function by phosphorylating the inhibitory site Ser-307 (Aguirre et al., 2000; Aguirre et al., 2002; Lee et al., 2003). This observation suggests that IRS1 phosphorylation may be an important target of JNK signaling in mice fed a high fat diet (Hirosumi et al., 2002). Indeed, immunoblot analysis demonstrated that feeding a high fat diet to wild-type mice caused increased phosphorylation of IRS1 on the JNK phosphorylation site Ser-307 in both white and brown adipose tissue (Figure 6A). Studies of Mlk3−/− mice demonstrated that MLK3-deficiency slightly reduced IRS1 phosphorylation on Ser-307 in white adipose tissue and markedly suppressed IRS1 Ser-307 phosphorylation in brown adipose tissue (Figure 6A). These defects in IRS1 Ser-307 phosphorylation are consistent with the effect of MLK3-deficiency to reduce obesity-induced JNK activation (Figure 5C,D).
Figure 6. MLK3 is required for inhibitory phosphorylation of IRS1 on Ser 307.
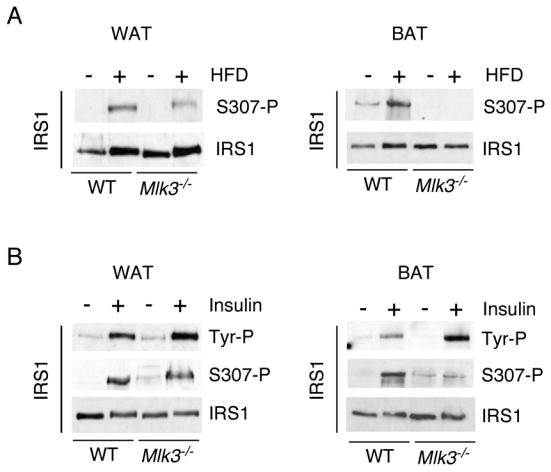
A) IRS1 expression and phosphorylation on Ser-307 in white epididymal adipose tissue (WAT) and brown interscapular adipose tissue (BAT) of wild-type and Mlk3−/− mice maintained (16 weeks) on a standard diet and on a high fat diet (HFD) was examined by immunoblot analysis.
B) Wild-type and Mlk3−/− mice were fasted overnight and then treated (30 mins) with insulin (1.5 units/Kg). Extracts prepared from the fat pads (WAT and BAT) were examined by immunoblot analysis using antibodies to IRS1, tyrosine phosphorylated IRS1 (Tyr-P), and IRS1 phosphorylated on Ser-307.
To test whether these changes in inhibitory IRS1 phosphorylation are functionally relevant, we examined the effect of insulin to cause tyrosine phosphorylation of IRS1. Studies of white adipose tissue demonstrated a similar increase in phosphorylation of IRS1 on Tyr and Ser-307 in wild-type and Mlk3−/− mice (Figure 6B). In contrast, studies of brown adipose tissue demonstrated that MLK3-deficiency caused decreased Ser-307 phosphorylation and markedly increased Tyr phosphorylation of IRS1 in brown adipose tissue (Figure 6B). These data provide biochemical evidence of increased insulin sensitivity of brown fat in MLK3-deficient mice. Together, these data indicate that MLK3 is required for JNK-induced insulin resistance in brown adipose tissue.
The observation that insulin signaling is altered in Mlk3−/− mice prompted us to test whether MLK3-deficiency, like JNK-deficiency, might protect against diet-induced obesity and insulin resistance. We found that feeding a high fat diet caused similar changes in body mass, glucose tolerance, insulin tolerance, and fasting blood insulin and glucose concentrations in wild-type and Mlk3−/− mice (Figure S2 and data not shown). The lack of systemic protection against diet-induced obesity and insulin resistance caused by MLK3-deficiency is most likely the result of the finding that MLK3 is required for obesity-induced JNK activation in brown adipose tissue (Figures 5 & 6) and liver (Figure S3) of Mlk3−/− mice, but MLK3 is not essential in other tissues, including white adipose tissue (Figure 5 & 6) and muscle (data not shown). It is possible that the function of MLK3 is redundant in white fat and other tissues because of the expression of other members of the MLK group.
PKC is required for MLK3-dependent JNK activation caused by FFA
The mechanism of activation of the MLK3 signaling pathway by FFA is unclear. Previous studies have established a role for protein kinase C (PKC) in FFA-induced insulin resistance (Dey et al., 2006). Obesity or treatment with FFA causes activation of novel PKC isoforms, including PKCθ, PKCδ, and PKC ε (Griffin et al., 1999; Kim et al., 2004; Lam et al., 2002; Samuel et al., 2007; Yu et al., 2002), probably by elevating the intracellular concentration of diacylglycerol (Montell et al., 2001) and ceramide (Schmitz-Peiffer et al., 1999).
We examined the effect of FFA to activate PKC in MEF by immunoblot analysis using a PKC phosphospecific antibody. This analysis demonstrated that saturated FFA, but not unsaturated FFA, caused PKC activation in both wild-type and Mlk3−/− MEF (Figure 7A). These data indicate that FFA can activate PKC independently of MLK3. To test whether PKC might act as an upstream component of a FFA-stimulated pathway that activates JNK, we compared the effect of constitutively activated PKC and kinase-negative PKC on JNK activity. These data demonstrated that PKC can activate JNK (Figure 7B). PKC may therefore function as a mediator of FFA signaling to MLK3.
Figure 7. PKC is required for MLK3 and JNK activation by FFA.
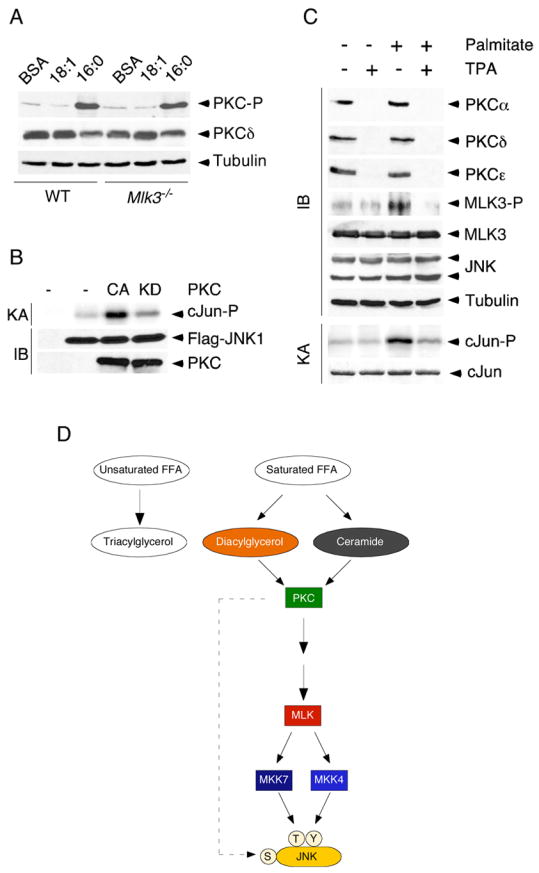
A) Wild-type or Mlk3−/− MEF were treated (16 h) with BSA or with 0.5 mM oleic acid (18:1) or 0.5 mM palmitic acid (16:0). The phosphorylation and expression of PKCδ was examined by immunoblot analysis.
B) Flag-tagged JNK1 and constitutively active or kinase-inactive PKCε were co-expressed in HEK 293 cells. The expression of JNK1 and PKC was examined by immunoblot analysis. JNK activity was measured in an IP-kinase assay (KA) using [γ-32P]ATP and cJun as substrates.
C) PKCζ−/− MEF were pre-treated without and with 1 μM TPA (24 h) and then treated without and with 0.5 mM palmitate (16 h). The expression of PKCα, PKCδ, PKCε, MLK3, phospho-MLK3, JNK, phospho-JNK, and Tubulin was examined by immunoblot analysis. JNK activity was measured in a kinase assay (KA) using [γ-32P]ATP and cJun as substrates.
D) Schematic illustration of a JNK signaling pathway that is activated by saturated FFA and is mediated by PKC, MLK, and MKK4/7.
To test the requirement of PKC for FFA-stimulated MLK3 and JNK activation, we examined the effect of PKC down-regulation. In initial studies, we used siRNA to down-regulate the PKC isoforms expressed in MEF (PKCα, PKCδ, PKCε, and PKCζ). Down-regulation of individual PKC isoforms caused no change in FFA-induced MLK3 or JNK activation (data not shown). This observation suggested that PKC isoforms may serve redundant functions in this pathway. Efficient simultaneous down-regulation of multiple PKC isoforms using siRNA was not obtained. We therefore took an alternative approach to obtain MEF lacking multiple PKC isoforms. Treatment with the phorbol ester TPA caused down-regulation of diacylglycerol-responsive PKC isoforms (α, δ, andε), but did not affect FFA-induced MLK3 or JNK activation (data not shown). This observation suggested an important role for PKCζ, although Pkcζ−/− MEF exhibited no defect FFA-induced MLK3 and JNK activation (Figure 7C). We therefore examined the effect of pan-PKC deficiency by treating Pkcζ−/− MEF with TPA (Figure 7C). This approach demonstrated that PKC was essential for FFA-induced activation of both MLK3 and JNK (Figure 7C). These data indicate that multiple PKC isoforms (α, δ, ε and ζ) in MEF serve redundant functions as upstream components of a FFA-stimulated signaling pathway that regulates MLK3-dependent activation of JNK (Figure 7D).
Discussion
Obesity can cause an increase in the level of blood FFA and it is thought that this increase in FFA mediates the effects of obesity on insulin resistance and type 2 diabetes (Arner, 2002; Boden, 2003; Boden, 2006; Kahn et al., 2006; Kovacs and Stumvoll, 2005). The FFA in the blood can be incorporated into cellular lipids. Indeed, unsaturated FFA accumulate as triacylglycerol whereas saturated FFA increase the diacylglycerol pool (Montell et al., 2001). In addition, saturated FFA may also lead to accumulation of ceramide (Schmitz-Peiffer et al., 1999). This increase in diacylglycerol and ceramide caused by saturated FFA may mediate the effects of FFA on insulin resistance. Possible targets of diacylglycerol signaling include members of the PKC family and ceramide may activate the diacylglycerol-insensitive isoform PKCζ. Indeed, several PKC isoforms have been implicated in insulin resistance in models of elevated blood FFA caused by lipid infusion or obesity (Dey et al., 2006; Griffin et al., 1999; Kim et al., 2004; Lam et al., 2002; Samuel et al., 2007). PKCθ and PKCδ appear to be relevant to FFA-induced insulin resistance in muscle while PKCε may be the important isoform in liver (Morino et al., 2006).
PKC is required for FFA-stimulated JNK activation
Our studies of MEF demonstrate that saturated FFA cause JNK activation. In contrast, unsaturated FFA cause no change in JNK activity. The selective effect of saturated FFA to activate JNK suggests that PKC may contribute to JNK activation. MEF express several PKC isoforms (PKCα, PKCδ, PKCε, and PKCζ) that appear to function redundantly in this pathway. Nevertheless, knockdown of these PKC isoforms prevents the activation of JNK by saturated FFA (Figure 7C). These data establish that PKC is a required component of a FFA-induced signaling pathway that activates JNK (Figure 7D).
MLK is required for FFA-stimulated JNK activation
We demonstrate that a MLK isoform (MLK3) is essential for FFA-stimulated JNK activation in MEF (Figures 3 & 4). The most direct evidence for this conclusion was obtained from the finding that FFA did not activate JNK in MEF prepared from Mlk3−/− mice. This observation indicated that MLK3 is a component of a FFA-stimulated pathway that leads to JNK activation. To test the relationship between PKC and MLK3, we performed epistatic analysis of the FFA signaling pathway. Importantly, FFA caused PKC activation in MLK3-deficient MEF (Figure 7A), but FFA did not cause MLK3 activation in PKC-deficient MEF (Figure 7C). These data indicate that the FFA signaling pathway is organized with MLK3 acting down-stream of PKC (Figure 7D).
The molecular mechanism of MLK3 regulation by PKC remains to be elucidated. Biochemical studies indicate that a major mechanism of MLK3 regulation is auto-inhibition mediated by the binding of the NH2-terminal SH3 domain to a proline-rich domain located between the leucine zipper and the CRIB motif. Cdc42 binding to the CRIB motif may promote de-inhibition and leucine zipper-mediated dimerization of MLK3 (Gallo and Johnson, 2002). This mechanism may contribute to the effect of PKC to activate MLK3. Alternatively, PKC may regulate MLK3 directly by phosphorylation. Indeed, MLK3 is phosphorylated on many sites in vivo (Vacratsis et al., 2002) that regulate MLK3 activity and sub-cellular localization (Barthwal et al., 2003; Schachter et al., 2006). An important goal for future studies will be to define the molecular mechanism of MLK3 regulation by PKC (Figure 7D).
MKK4 and MKK7 are required for FFA-stimulated JNK activation
It is established that MLK3 can phosphorylate and activate the MAP2K isoforms MKK4 and MKK7 (Gallo and Johnson, 2002). Compound deficiency of MKK4 plus MKK7 prevents stress-induced JNK activation (Tournier et al., 2001). Interestingly, FFA did not activate JNK in MEF prepared from Mkk4−/− mice or Mkk7−/− mice (Figure 2). This observation indicated that both MKK4 and MKK7 are required for FFA-stimulated JNK activation in MEF. The combined requirement for MKK4 and MKK7 may reflect the co-operative phosphorylation of the JNK Thr-Pro-Tyr dual phosphorylation motif on Tyr by MKK4 and on Thr by MKK7 (Lawler et al., 1998; Tournier et al., 2001).
PKC is an upstream component of the FFA signaling pathway that is required for the activation of MLK3 (Figure 7D). However, PKC may also contribute to JNK activation caused by MKK4 and MKK7. This is because PKC can phosphorylate JNK at a site (Ser-129) that may facilitate the activation of JNK by MKK4 and MKK7 (Lopez-Bergami et al., 2005). Thus, FFA-stimulated PKC may contribute to JNK activation at two different steps (Figure 7D).
Role of mixed-lineage protein kinases
Previous studies have not established biological functions for the MLK sub-group of mixed-lineage kinases. However, some studies of the ubiquitously expressed isoform MLK3 have been reported. Thus, over-expression of MLK3 promotes microtubule instability (Swenson et al., 2003). It has also been reported that RNAi-mediated knockdown of MLK3 destabilizes B-Raf/Raf-1 complexes and suppresses proliferation (Chadee and Kyriakis, 2004; Chadee et al., 2006). In contrast, targeted disruption of the Mlk3 gene in mice did not reveal defects in microtubule stability, Raf signaling to the ERK1/2 group of MAP kinases, or cellular proliferation (Brancho et al., 2005). Similarly, detailed studies of Mlk3−/− MEF did not indicate major defects in MAP kinase signaling pathways when these cells were exposed to many hormones and cytokines or physical and chemical stresses, although a small reduction in the amplitude of TNF-stimulated JNK activation was noted (Brancho et al., 2005). None of these observations provide compelling insight into the biological function of MLK. Nevertheless, many studies have implicated possible roles of MLK isoforms (Gallo and Johnson, 2002; Handley et al., 2007), including studies using the small molecule inhibitor CEP-1347 that can inhibit all members of the mixed-lineage protein kinase family (Maroney et al., 2001). Studies using CEP-1347 have primarily focused on the neuroprotective effect of this drug to inhibit JNK signaling (Saporito et al., 2002), although this drug may also decrease pancreatitis (Wagner et al., 2000) and pulmonary fibrosis (Hashimoto et al., 2001). These studies indicate that members of the MLK family may have a broad role in cellular physiology, although the kinome specificity of CEP-1347 has not been formally established.
The results of this study demonstrate an essential role of MLK3 in the JNK signal transduction pathway that is activated by FFA. This signaling pathway is implicated in FFA-induced insulin resistance that is caused, in part, by JNK-mediated inhibitory phosphorylation of IRS1 on Ser-307 (Weston and Davis, 2007). This FFA signaling pathway is also implicated in steatosis and the progression to steatohepatitis that is mediated, in part, by JNK-dependent apoptosis (Malhi et al., 2006; Schattenberg et al., 2006). These observations suggest that drugs that target MLK enzymes (like CEP-1347) that may have therapeutic benefits for neurodegenerative disease (Saporito et al., 2002) may also be useful for the treatment of FFA-induced insulin resistance and steatohepatitis. It is likely that inhibition of the specific group of MAP3K that mediates FFA signaling may provide greater specificity for therapeutic intervention than inhibition of JNK directly.
Recent studies have demonstrated that FFA can activate JNK in many cell types, including adipocytes, hepatocytes and β-cells (Gao et al., 2004; Malhi et al., 2006; Nguyen et al., 2005; Solinas et al., 2006). Here we show that the ubiquitously expressed MLK isoform MLK3 is essential for JNK activation caused by FFA and obesity in MEF, brown fat, and liver (Figures 3 –5 & Supplementary Figure S3). However, MLK3 is not essential in some other tissues, including white fat (Figure 5) and muscle (data not shown). It is likely that other MLK isoforms that are not expressed ubiquitously (e.g. MLK1, MLK2, and MLK4) may function redundantly with MLK3 in specific tissues. Indeed, markedly different expression of MLK isoforms in individual tissues has been reported (Gallo and Johnson, 2002; Shmueli et al., 2003 and Figure S4). These members of the MLK family may therefore contribute to the metabolic phenotype. An important goal for future studies will therefore be to characterize single and compound mutant mice with defects in the expression of all members of the MLK family.
Experimental procedures
Mice
Mlk3−/− mice (Brancho et al., 2005) were back-crossed 10 generations to the C57BL/6J strain (Jackson Laboratories) and were housed in a facility accredited by the American Association for Laboratory Animal Care. The animal studies were approved by the Institutional Animal Care and Use Committee of the University of Massachusetts. Male mice were fed a high fat diet ad libitum (Diet F3282, BioServ) or a standard diet for 16 wk and their body mass was recorded weekly. Blood samples were collected from the tail vain after an overnight fast after 6, 12, and 16 weeks on the diet. Blood glucose concentrations were measured with a DEX-Glucometer (Bayer), and plasma insulin was measured by ELISA kit for rat insulin (Crystal Chem). Tissues were removed and rapidly frozen in liquid nitrogen for biochemical analysis. Histology was performed using tissue fixed in 10% formalin, dehydrated, and embedded in paraffin. Sections were stained with hematoxylin and eosin.
Cell culture
We have previously described wild-type, Mkk4−/−, Mkk7−/−, and Mkk4−/− Mkk7−/− MEF (Tournier et al., 2001) and wild-type and Mlk3−/− MEF (Brancho et al., 2005). Wild-type and Pkcζ−/− MEF (Leitges et al., 2001) were provided by Dr. Jorge Moscat (University of Cincinnati). Wild-type and Tak1−/− MEF (Shim et al., 2005) were provided by Dr. Sankar Ghosh (Yale University Medical School). MEF and HEK293 (American Type Culture Collection) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen). HEK293 cells were transfected with Lipofectamine (Invitrogen). Sodium salts of fatty acids (Sigma) were dissolved in PBS and mixed with FFA-free BSA (Roche). Stearic acid (Sigma) was dissolved in ethanol and added to serum-free DMEM supplemented with 2% FFA-free BSA. After 1h incubation in serum-free DMEM, cells were treated with 0.5mM fatty acid/0.5%BSA for 1–16h at 37°C.
Plasmids
Plasmid expression vectors for wild-type and kinase-negative PKCε were provided by Dr. Jorge Moscat (University of Cincinnati). The plasmid expression vector for Flag-tagged JNK1 was described previously (Derijard et al., 1994).
Biochemical assays
Protein extracts were prepared using Triton lysis buffer [20 mM Tris (pH 7.4), 1% Triton X-100, 10% glycerol, 137 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/mL of aprotinin and leupeptin]. Extracts (50 μg of protein) were examined by imunoblot analysis with antibodies obtained from Cell Signaling (MLK3, phosphoThr277,Ser281-MLK3, ERK, phospho-ERK, p38, phospho-p38, AKT, phosphoSer473-AKT, PKCα, PKCδ, phospho-T-loop-PKC, phospho-JNK, and TAK1), Tranduction Labs (PKCε), PharMingen (JNK), Sigma (α-Tubulin), and Upstate Biotechnology (IRS1, phosphoSer307-IRS1 and phospho-MKK7). JNK activity was measured in an in vitro kinase assay using [γ-32P]ATP and cJun as substrates (Whitmarsh and Davis, 2001).
Supplemental Data
Supplemental Data include Figures S1–S4 and can be found with this article on-line at http//www.molecule.org/cgi/content/full/*****.
Acknowledgments
We thank Dr. Jorge Moscat for providing Pkcζ−/− MEF, Dr. Sankar Ghosh for providing Tak1−/−MEF, Judy Reilly, Jian-Hua Liu, and Beth Doran for expert technical assistance, and Kathy Gemme for administrative assistance. These studies were supported by a grant from the National Institutes of Health. R.J.D. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- Arner P. Insulin resistance in type 2 diabetes: role of fatty acids. Diabetes Metab Res Rev. 2002;18(Suppl 2):S5–9. doi: 10.1002/dmrr.254. [DOI] [PubMed] [Google Scholar]
- Barthwal MK, Sathyanarayana P, Kundu CN, Rana B, Pradeep A, Sharma C, Woodgett JR, Rana A. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J Biol Chem. 2003;278:3897–3902. doi: 10.1074/jbc.M211598200. [DOI] [PubMed] [Google Scholar]
- Boden G. Effects of free fatty acids (FFA) on glucose metabolism: significance for insulin resistance and type 2 diabetes. Exp Clin Endocrinol Diabetes. 2003;111:121–124. doi: 10.1055/s-2003-39781. [DOI] [PubMed] [Google Scholar]
- Boden G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr Diab Rep. 2006;6:177–181. doi: 10.1007/s11892-006-0031-x. [DOI] [PubMed] [Google Scholar]
- Brancho D, Ventura JJ, Jaeschke A, Doran B, Flavell RA, Davis RJ. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol Cell Biol. 2005;25:3670–3681. doi: 10.1128/MCB.25.9.3670-3681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadee DN, Kyriakis JM. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat Cell Biol. 2004;6:770–776. doi: 10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- Chadee DN, Xu D, Hung G, Andalibi A, Lim DJ, Luo Z, Gutmann DH, Kyriakis JM. Mixed-lineage kinase 3 regulates B-Raf through maintenance of the B-Raf/Raf-1 complex and inhibition by the NF2 tumor suppressor protein. Proc Natl Acad Sci U S A. 2006;103:4463–4468. doi: 10.1073/pnas.0510651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care. 2007;10:142–148. doi: 10.1097/MCO.0b013e328042ba90. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Dey D, Basu D, Roy SS, Bandyopadhyay A, Bhattacharya S. Involvement of novel PKC isoforms in FFA induced defects in insulin signaling. Mol Cell Endocrinol. 2006;246:60–64. doi: 10.1016/j.mce.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, Ye J. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18:2024–2034. doi: 10.1210/me.2003-0383. [DOI] [PubMed] [Google Scholar]
- Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- Handley ME, Rasaiyaah J, Chain BM, Katz DR. Mixed lineage kinases (MLKs): a role in dendritic cells, inflammation and immunity? Int J Exp Pathol. 2007;88:111–126. doi: 10.1111/j.1365-2613.2007.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S, Gon Y, Takeshita I, Matsumoto K, Maruoka S, Horie T. Transforming growth Factor-beta1 induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c-Jun-NH2-terminal kinase-dependent pathway. Am J Respir Crit Care Med. 2001;163:152–157. doi: 10.1164/ajrccm.163.1.2005069. [DOI] [PubMed] [Google Scholar]
- Hirai S, Cui de F, Miyata T, Ogawa M, Kiyonari H, Suda Y, Aizawa S, Banba Y, Ohno S. The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J Neurosci. 2006;26:11992–12002. doi: 10.1523/JNEUROSCI.2272-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kesavan K, Lobel-Rice K, Sun W, Lapadat R, Webb S, Johnson GL, Garrington TP. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J Cell Physiol. 2004;199:140–148. doi: 10.1002/jcp.10457. [DOI] [PubMed] [Google Scholar]
- Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, Hawn TR, Raines EW, Schwartz MW. Toll Like Receptor-4 Mediates Vascular Inflammation and Insulin Resistance in Diet-Induced Obesity. Circ Res. 2007 doi: 10.1161/CIRCRESAHA.106.142851. [DOI] [PubMed] [Google Scholar]
- Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, Liu ZX, Soos TJ, Cline GW, O’Brien WR, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004;114:823–827. doi: 10.1172/JCI22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK. Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab. 2006;4:417–419. doi: 10.1016/j.cmet.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Kovacs P, Stumvoll M. Fatty acids and insulin resistance in muscle and liver. Best Pract Res Clin Endocrinol Metab. 2005;19:625–635. doi: 10.1016/j.beem.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Lam TK, Yoshii H, Haber CA, Bogdanovic E, Lam L, Fantus IG, Giacca A. Free fatty acid-induced hepatic insulin resistance: a potential role for protein kinase C-delta. Am J Physiol Endocrinol Metab. 2002;283:E682–691. doi: 10.1152/ajpendo.00038.2002. [DOI] [PubMed] [Google Scholar]
- Lawler S, Fleming Y, Goedert M, Cohen P. Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr Biol. 1998;8:1387–1390. doi: 10.1016/s0960-9822(98)00019-0. [DOI] [PubMed] [Google Scholar]
- Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem. 2003;278:2896–2902. doi: 10.1074/jbc.M208359200. [DOI] [PubMed] [Google Scholar]
- Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J. Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol Cell. 2001;8:771–780. doi: 10.1016/s1097-2765(01)00361-6. [DOI] [PubMed] [Google Scholar]
- Lopez-Bergami P, Habelhah H, Bhoumik A, Zhang W, Wang LH, Ronai Z. RACK1 mediates activation of JNK by protein kinase C. Mol Cell. 2005;19:309–320. doi: 10.1016/j.molcel.2005.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu Z, Meyer SL, Savage MJ, Greene LA, et al. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276:25302–25308. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- Montell E, Turini M, Marotta M, Roberts M, Noe V, Ciudad CJ, Mace K, Gomez-Foix AM. DAG accumulation from saturated fatty acids desensitizes insulin stimulation of glucose uptake in muscle cells. Am J Physiol Endocrinol Metab. 2001;280:E229–237. doi: 10.1152/ajpendo.2001.280.2.E229. [DOI] [PubMed] [Google Scholar]
- Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Suppl 2):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MT, Satoh H, Favelyukis S, Babendure JL, Imamura T, Sbodio JI, Zalevsky J, Dahiyat BI, Chi NW, Olefsky JM. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J Biol Chem. 2005;280:35361–35371. doi: 10.1074/jbc.M504611200. [DOI] [PubMed] [Google Scholar]
- Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, Peiretti F, Verdier M, Juhan-Vague I, Tanti JF, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. 2007;50:1267–1276. doi: 10.1007/s00125-007-0654-8. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporito MS, Hudkins RL, Maroney AC. Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem. 2002;40:23–62. doi: 10.1016/s0079-6468(08)70081-x. [DOI] [PubMed] [Google Scholar]
- Sathyanarayana P, Barthwal MK, Kundu CN, Lane ME, Bergmann A, Tzivion G, Rana A. Activation of the Drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3. Mol Cell. 2002;10:1527–1533. doi: 10.1016/s1097-2765(02)00734-7. [DOI] [PubMed] [Google Scholar]
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- Schachter KA, Du Y, Lin A, Gallo KA. Dynamic positive feedback phosphorylation of mixed lineage kinase 3 by JNK reversibly regulates its distribution to Triton-soluble domains. J Biol Chem. 2006;281:19134–19144. doi: 10.1074/jbc.M603324200. [DOI] [PubMed] [Google Scholar]
- Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem. 1999;274:24202–24210. doi: 10.1074/jbc.274.34.24202. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmueli O, Horn-Saban S, Chalifa-Caspi V, Shmoish M, Ophir R, Benjamin-Rodrig H, Safran M, Domany E, Lancet D. GeneNote: whole genome expression profiles in normal human tissues. C R Biol. 2003;326:1067–1072. doi: 10.1016/j.crvi.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci U S A. 2006;103:16454–16459. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MJ, Kim KH, Yoon JM, Kim JB. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun. 2006;346:739–745. doi: 10.1016/j.bbrc.2006.05.170. [DOI] [PubMed] [Google Scholar]
- Swenson KI, Winkler KE, Means AR. A new identity for MLK3 as an NIMA-related, cell cycle-regulated kinase that is localized near centrosomes and influences microtubule organization. Mol Biol Cell. 2003;14:156–172. doi: 10.1091/mbc.E02-02-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001;15:1419–1426. doi: 10.1101/gad.888501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacratsis PO, Phinney BS, Gage DA, Gallo KA. Identification of in vivo phosphorylation sites of MLK3 by mass spectrometry and phosphopeptide mapping. Biochemistry. 2002;41:5613–5624. doi: 10.1021/bi016075c. [DOI] [PubMed] [Google Scholar]
- Wagner AC, Mazzucchelli L, Miller M, Camoratto AM, Goke B. CEP-1347 inhibits caerulein-induced rat pancreatic JNK activation and ameliorates caerulein pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2000;278:G165–172. doi: 10.1152/ajpgi.2000.278.1.G165. [DOI] [PubMed] [Google Scholar]
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- White MF. Regulating insulin signaling and beta-cell function through IRS proteins. Can J Physiol Pharmacol. 2006;84:725–737. doi: 10.1139/y06-008. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Davis RJ. Analyzing JNK and p38 mitogen-activated protein kinase activity. Methods Enzymol. 2001;332:319–336. doi: 10.1016/s0076-6879(01)32212-7. [DOI] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Deng M, Parthasarathy R, Wang L, Mongan M, Molkentin JD, Zheng Y, Xia Y. MEKK1 transduces activin signals in keratinocytes to induce actin stress fiber formation and migration. Mol Cell Biol. 2005;25:60–65. doi: 10.1128/MCB.25.1.60-65.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include Figures S1–S4 and can be found with this article on-line at http//www.molecule.org/cgi/content/full/*****.