

Abstract
Metabotropic glutamate receptors (mGluRs) are expressed throughout the mammalian central nervous system and integrate a host of signal transduction pathways that determine cellular function, plasticity and injury. Yet, one of the more unique regulatory functions of this family of GTP-binding proteins involves cytoprotection in the nervous system. Here, we demonstrate that activation of group I metabotropic glutamate receptors (mGluRIs) in primary hippocampal neurons not only provides intrinsic cellular protection for the maintenance of genomic DNA integrity, but also prevents inflammatory microglial activation and specific neuronal cell engulfment during free radical oxidative stress. Loss of cellular membrane asymmetry and exposure of membrane phosphatidylserine (PS) residues were necessary and sufficient to result in microglial activation and proliferation, since administration of an antibody to the PS receptor could block microglial activity. Through the continuous assessment of individual neurons in real time, activation of mGluRIs was documented to block neuronal PS exposure and prevented subsequent neuronal cell engulfment by microglia seeking “PS tagged” neurons. Furthermore, regulation of both cellular integrity and microglial activity by mGluRI activation was dependent upon the activation and phosphorylation of protein kinase B (Akt1), prevention of mitochondrial membrane depolarization with associated permeability transition pore complex formation, and the down regulation of caspase 9-like activity. Our work defines a significant role of mGluRIs for the modulation of cellular survival and inflammation in the nervous system during oxidative stress.
Keywords: Bromodeoxyuridine, caspase 9, cytochrome c, metabotropic glutamate receptor, microglia, mitochondria, nitric oxide, protein kinase B
INTRODUCTION
As a group, metabotropic glutamate receptors (mGluRs) are expressed throughout the brain and are present in regions such as the cerebral cortex (Berger, MA et al., 2001), cerebellum (Berthele, A et al., 1999), thalamus (Saugstad, JA et al., 1997), and spinal cord (Aronica, E et al., 2001). Recent investigations continue to focus on the “anti-apoptotic” mechanisms controlled by mGluRs (Chong, ZZ et al., 2003). In particular, group I mGluRs (mGluRIs) may have a prominent role as therapeutic agents against neurodegenerative disease. mGluRIs can regulate the metabolism of amyloid precursor protein (APP) and accelerate the processing of APP into non-amyloidogenic APP (Lee, RK et al., 1996). Modulation of synaptic activity by mGluRs during chronic neuronal disorders, such as in Huntington’s disease, has been suggested to alter cellular susceptibility to injury (Calabresi, P et al., 1999). During models of spinal cord trauma, mGluRIs are up regulated (Mills, CD et al., 2001). Neuronal protection through the activation of mGluRIs has been extended to several other models of neuronal injury such as glucose deprivation (Sagara, Y and Schubert, D, 1998), combined oxygen and glucose deprivation (Kalda, A et al., 2000), nitric oxide (NO) (Maiese, K et al., 1996, Maiese, K et al., 2000), and anoxia (Lin, SH and Maiese, K, 2001, Vincent, AM and Maiese, K, 2000).
Cytoprotection by mGluRIs may occur at two distinct levels of injury. At one level, intrinsic protection occurs through the prevention of genomic DNA degradation and the modulation of endonuclease activity (Vincent, AM et al., 1999) in both neuronal and vascular cell populations (Lin, SH et al., 2002, Rong, R et al., 2003, Sagara, Y et al., 1998, Vincent, AM et al., 1997). However, a second significant level of protection may involve the prevention of inflammatory injury through the control of microglial activation. Microglial activation can directly influence the progression of several neurodegenerative disorders. In Huntington’s disease and amyotrophic lateral sclerosis (ALS), significant microglial activation has been reported in regions of the nervous system that are specific for these disease entities (Obal, I et al., 2001, Singhrao, SK et al., 1999). In addition, microglial activation has been observed to occur in concert with the evolution of amyloid plaques (Chong, ZZ et al., 2005, Maiese, K and Chong, ZZ, 2004). Furthermore, work that has examined the effect of mGluRs on microglia, has demonstrated that activation of microglia mGluRs can confer protection to neurons (Taylor, DL et al., 2003).
Closely tied to the modulation of microglial activity is protein kinase B, also referred to as PKBα or Akt1. Enhanced Akt activity can promote cell survival against several toxic insults that include matrix detachment, DNA damage, anti-Fas antibody administration, oxidative stress, and transforming growth factor-β (TGF-β) application (Chong, ZZ et al., 2005). Although more relevant in regards to the activation of microglia, Akt activity also can prevent cellular membrane phosphatidylserine (PS) externalization in both neurons and vascular cells to prevent microglial activation during a variety of insults that involve anoxia, free radical exposure, and oxygen-glucose deprivation (Chong, ZZ et al., 2002, Maiese, K and Chong, ZZ, 2003).
Ultimately, regulation of microglial activity by Akt may require the maintenance of mitochondrial membrane potential (ΔΨm) and caspase 9 regulation. Akt can prevent mitochondrial membrane depolarization and cytochrome c release (Kang, JQ et al., 2003, Kennedy, SG et al., 1999). Akt also phosphorylates caspase 9 on its serine196 residue and inhibits its protease activity. Akt enhances cell survival through the modulation of mitochondrial membrane permeability and caspase 9 activity during oxidative stress (Chong, ZZ et al., 2005, Kang, JQ et al., 2003, Maiese, K et al., 2005) and protects against toxic insults such as TNF-α administration (Hermann, C et al., 2000). Furthermore, over expression of Akt has been shown to block microglial activation and proliferation that requires the inhibition of caspase 9 (Kang, JQ et al., 2003). Here, we demonstrate that activation of group I mGluRs can prevent apoptotic injury through not only the maintenance of genomic DNA integrity, but also through the novel modulation of microglial activity. Regulation of microglial activity by mGluRIs is dependent upon the enhanced activity of Akt1, the prevention of mitochondrial membrane depolarization with associated permeability transition pore complex (PTPC) formation, and the subsequent reduction in caspase 9-like activity.
MATERIALS AND METHODS
Primary Hippocampal Neuronal Cultures
Hippocampi were obtained from E-19 Sprague-Dawley rat pups and incubated in dissociation medium (90 mmol/L Na2SO4, 30 mmol/L K2SO4, 5.8 mmol/L MgCl2, 0.25 mmol/L CaCl2, 10 mmol/L kynurenic acid, and 1 mmol/L HEPES with the pH adjusted to 7.4) containing papain (10 U/ml) and cysteine (3 mmol/L) for two 20-minute periods. The hippocampi were then rinsed in dissociation medium and incubated in dissociation medium containing trypsin inhibitor (10-20 U/ml) for three 5-minute periods. The cells were washed in growth medium (Leibovitz’s L-15 medium, Invitrogen, Carlsbad, CA) containing 6% sterile rat serum (ICN, Aurora, OH), 150 mmol/L NaHCO3, 2.25 mg/ml of transferrin, 2.5 μg/ml of insulin, 10 nmol/L progesterone, 90 μmol/L putrescine, 15 nmol/L selenium, 35 mmol/L glucose, 1 mmol/L L-glutamine, penicillin and streptomycin (50 μg/ml), and vitamins. The dissociated cells were plated at a density of ∼1.5 ×103 cells/mm2 in 35 mm polylysine/laminin-coated plates (Falcon Labware, Lincoln Park, NJ). Neurons were maintained in growth medium at 37 °C in a humidified atmosphere of 5% CO2 and 95% room air for 2 weeks. Non-neuronal cells were negligible.
Microglia Cell Cultures and the Assessment of Microglial Activation
Per our prior protocols (Kang, JQ et al., 2003), microglia were obtained from the cerebral cortex of E-19 Sprague-Dawley rat pups, mechanically dissociated, and seeded in 75 cm2 plastic flasks at a density of 8.5×106 cells per flask. Microglia were purified from mixed cultures with reciprocal shaking at 180 rpm for 15 hours and then re-seeded at 105 cells/ml for cell adhesion of 3 hours duration to yield an almost pure preparation of microglia (>98%). Microglial cells were identified by α-naphthyl acetate esterase, OX-42, and isolectin B4 from griffonia simplicifolia (Sigma, St. Louis, MO). The cells did not stain for glial fibrillary acidic protein (GFAP).
For the assessment of microglial activation, microglia were conditioned for 3 hours with phosphatidylserine (PS) (10 μM) and phosphatidylcholine (PC) (10 μM) (Sigma, St. Louis, MO) or by media from either untreated neurons or neurons receiving (S)-3,5-dihydroxyphenylglycine (DHPG) (750 μM) 24 hours following NO exposure. Twelve hours later, proliferating cell nuclear antigen (PCNA) staining for microglial activation (Williams, K et al., 2002) was performed with anti-mouse monoclonal antibody PCNA (1:100) conjugated with biotinylated anti-mouse IgG (1:50) and visualized through fluorescein avidin (1:50) (Vector Laboratories, Burlingame, CA). Bromodeoxyuridine (BrdU) uptake for microglial proliferation (Martinez-Contreras, A et al., 2002) was performed with BrdU (1:100) conjugated with biotinylated anti-mouse IgG (1:50) and visualized through Texas Red streptavidin (Vector laboratories, Burlingame, CA). BrdU (10 μM) and fluorodeoxyuridine (1 μM) (Sigma, St. Louis, MO) were applied 1 hour prior to the time of fixation.
Experimental Treatments
NO administration was performed by replacing the culture media with media containing 6-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-hexanamine (NOC-9, 300 μM) (Calbiochem, San Diego, CA) or sodium nitroprusside (SNP, 300 μM) (Sigma, St. Louis, MO) as per the experimental paradigm. More than one NO generator was used as a control to demonstrate that cells were responding to NO rather than to other by-products of these agents (Maiese, K and Vincent, AM, 2000). Data for the two NO donors was combined, since no significant differences in cell injury were present among the agents. The mGluRI agonist (S)-3,5-dihydroxyphenylglycine (DHPG) (750 μM) (previously determined as the duration and concentration at which a plateau is achieved on survival curves for the mGluR agonists) (Lin, SH et al., 2001, Maiese, K et al., 1999, Vincent, AM et al., 1999) and the mGluR antagonist (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA) (750 μM) (Tocris, Ellisville, MO) were applied to neuronal cultures 1 hour prior to NO exposure and the treatment was continuous. For phosphatidylinositide-3-kinase (PI 3-K) inhibition, wortmannin (W) (Calbiochem, La Jolla, CA) was added directly to the cultures 1 hour prior to NO application and the treatment of PI 3-K inhibition was continuous.
Assessment of Cell Survival and DNA Fragmentation
Neuronal injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 hours following NO exposure as per our previous protocols, and genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay (Kang, JQ et al., 2003).
Assessment of Membrane Phosphatidylserine (PS) Residue Externalization
As per our prior protocols (Kang, JQ et al ., 2003), a 30 μg/ml stock solution of annexin V conjugated to phycoerythrin (PE) (R&D Systems, Minneapolis, MN) was prepared and plates were incubated with 500 μl of diluted annexin V for 10 min. Images were acquired by “blinded” assessment with a Leitz DMIRB microscope (Leica, McHenry, IL) and a Fuji/Nikon Super CCD (6.1 megapixels) using transmitted light and fluorescent single excitation light at 490 nm and detected emission at 585 nm.
For repetitive examination of single cells, the annexin V label was detached by washing three times in dissociation buffer (10 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM KCl, 2.8 mM MgCl2), which differed from binding buffer in that the calcium was replaced with magnesium. Plates could then be re-examined to confirm that the annexin V was completely removed, then returned to the incubator for a further specified period. By drawing a grid on the bottom of the culture dish, the same fields of neurons and microglia could be relocated for sequential imaging (Maiese, K et al., 2000).
Modulation of Membrane Phosphatidylserine (PS) Residue Exposure
The enzyme that removes phosphatidylserine (PS) from the extracellular leaflet of the plasma membrane, aminophospholipid translocase, may be inhibited under mild oxidizing conditions, leading to the externalization of PS (Deckwerth, TL and Johnson, E, Jr., 1993). Therefore, PS externalization was achieved through application of the sulfhydryl oxidizing agents N-ethylmaleimide (NEM) at a concentration of 0.1 mM for 2 minutes or diazine-dicarboxylic acid bis[N,N-dimethylamide] (diamide) at a concentration of 0.5 mM for 10 minutes. Both NEM and diamide were obtained from Sigma (St Louis, MO). These agents were applied alone or concurrent with exposure to NO as indicated in individual treatment paradigms.
Modulation and Assessment of Mitochondrial Permeability Transition pore Membrane Potential (ΔΨm)
The fluorescent probe JC-1 (Molecular Probes, Eugene, OR), a cationic membrane potential indicator, was used to assess ΔΨm. Neuronal cells in 35 mm2 plates were incubated with 2 μg/ml JC-1 in growth medium for 30 minutes. After washing, cells were then analyzed immediately under a Leitz DMIRB microscope (Leica, McHenry, IL) with a dual emission fluorescence filter with 515-545 nm for green fluorescence and emission at 585-615 nm for red fluorescence. For modulation of mitochondrial membrane permeability, the exposure of atractyloside (Atr) (Sigma, St. Louis, MO) and cyclosporin A (CsA) (Sigma, St. Louis, MO) were continuous.
Assessment of Cysteine Protease Activity
The activity of caspase 9 was assessed by determination of the cleavage of the colorimetric substrates (Ac-LEHD-pNA, caspase 9) (Calbiochem, San Diego, CA) as previously described (Chong, ZZ et al., 2004).
Modulation of Cysteine Protease Activity
Modulation of cysteine protease activity in neuronal cells was performed by using the irreversible and cell permeable caspase inhibitors (50 μmol/L 1 hour prior to NO) Z-LEHD-FMK for caspase 9 (LEHD) (BD Pharmingen, San Diego, CA).
Western Blot Analysis for Akt1 Phosphorylation and Cytochrome c Release
Cells were homogenized and, following protein determination, each sample (50 μg/lane) was then subjected to 7.5% (Akt1) or 12.5% (cytochrome c) SDS-polyacryl-amide gel electrophoresis. Membranes were incubated with primary mouse monoclonal antibodies against phosphorylated Akt1 (p-Akt1, Ser473, 1:1000) (Active-Motif, Carlsbad, CA) or cytochrome c (1:2000) (BD Pharmingen, San Diego, CA), and subsequently with the horseradish peroxidase, conjugated secondary antibodies. The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ).
Preparation of Mitochondria for the Analysis of Cytochrome c Release
As per our prior protocols (Kang, JQ et al., 2003), cells were harvested and supernatants were centrifuged at 10,000 g for 10 minutes and the cytosolic fraction was centrifuged at 50,000 g for 60 minutes at 4 °C.
Statistical Analysis
For each experiment involving assessment of neuronal survival, DNA degradation, membrane PS exposure, microglial activation, mitochondrial membrane potential, and caspase activity, the mean and standard error (M ± SEM) were determined from 4 to 6 replicate experiments. Statistical differences between groups were assessed by means of analysis of variance (ANOVA) with the post-hoc Student’s t-test.
RESULTS
Activation of mGluRI Increases Neuronal Survival and Prevents DNA Fragmentation
Following NO exposure (NOC-9 or SNP, 300 μM), neurons were observed to undergo cell injury and apoptosis manifested by decreased cell density, permeability to trypan blue dye (Fig. 1A, TB), chromatin condensation, and nuclear fragmentation (Fig. 1A, TUNEL). In contrast, neurons following DHPG (750 μM) administration had significantly reduced trypan blue staining (Fig. 1A, TB) and nuclear fragmentation (Fig. 1B, TUNEL). As shown in Fig. 1B, cells following DHPG administration increased neuronal survival during NO exposure from 37 ± 3% to 64 ± 3%. In a similar manner, DNA fragmentation was significantly reduced from 69 ± 3% to 28 ± 3% in cells following DHPG administration during NO exposure (Fig. 1C). In contrast, blockade of group I mGluR activity with AIDA did not improve cell survival or decrease TUNEL expression during NO exposure (Fig. 1B and 1C).
Fig. (1). Activation of mGluRI prevents neuronal injury against NO toxicity.
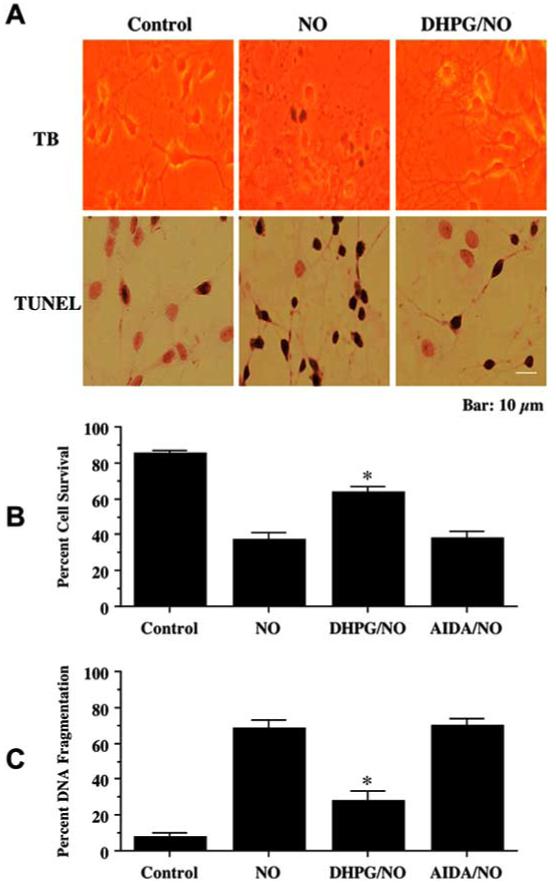
Neuronal survival and DNA fragmentation were assessed by trypan blue dye exclusion (TB) and TUNEL 24 hours following exposure to a NO donor (NOC-9 or SNP, 300 μM). The mGluRI agonist (DHPG, 750 μM) or antagonist (AIDA, 750 μM) was applied 1 hour prior to NO exposure. (A) Representative images illustrate that cultures with NO exposure were stained with TB and TUNEL. Application of DHPG significantly reduced TB staining and TUNEL labeling during NO exposure. (B) Application of DHPG (750 μM), but not AIDA, 1 hour prior to NO significantly increased neuronal survival 24 hours following NO exposure (*P<0.01 vs. NO treated alone). (C) DHPG, applied 1 hour prior to NO exposure significantly reduced DNA fragmentation 24 hours following NO exposure (*P<0.01 vs. NO treated alone). In B and C, to simplify the figures, the results for the two NO donors were combined. Each data point represents the mean ± SEM.
mGluRIs Activation Prevents Membrane Phosphatidylserine (PS) Externalization
Neurons were exposed to NO and cellular membrane PS exposure was determined by annexin V 24 hours later. In Fig. 2A, representative pictures demonstrate that NO resulted in membrane PS externalization in neurons. In cells exposed to NO, significant induction of PS exposure is seen with annexin V label. In contrast, administration of DHPG, 1 hour prior to NO, significantly reduced membrane PS externalization. To quantitatively determine the ability of DHPG to prevent NO-induced membrane PS externalization, DHPG or AIDA was administered 1 hour prior to NO and assessment was performed 24 hours later. As shown in Fig. 2B, an increase in annexin V label was observed in neurons 24 hours following NO exposure that reached a level of 72 ± 4% when compared to untreated control cultures of 17 ± 4%. Cells treated with DHPG had a significant reduction in annexin V label to approximately 35% 24 hours following NO exposure.
Fig. (2). mGluRI activation prevents membrane phosphatidylserine (PS) exposure that independently does not alter cell survival.
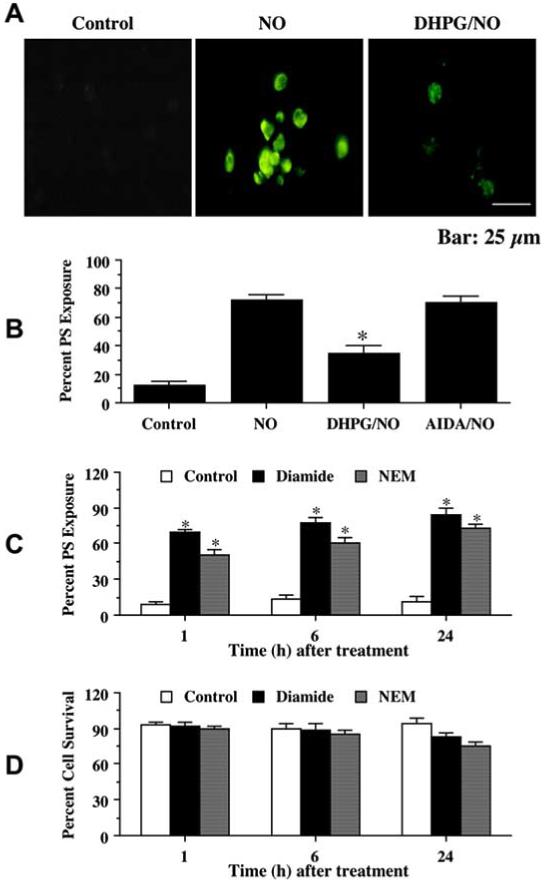
(A) Neurons were pretreated with DHPG (750 μM) 1 hour prior to NO application. PS exposure was determined by annexin V phycoerythrin labeling 24 hours following NO. Representative images illustrate membrane PS externalization in neurons using fluorescence light of the same microscopy field with 490 nm excitation and 585 nm emission wavelengths. DHPG significantly prevented membrane PS exposure following NO. (B) Pretreatment with DHPG (750 μM) 1 hour prior to NO significantly prevented membrane PS externalization (*P< 0.01 vs. NO treated alone). In all cases, control = untreated neurons. (C) Application of diamide (0.5 mM) or NEM (0.1 mM) alone significantly increased PS exposure in neurons over a 24 hour period (*P<0.01 vs. control). (D) Administration of diamide (0.5 mM) or NEM (0.1 mM) alone did not change neuronal survival over a 24 hour period (*P<0.01 vs. control). Each data point represents the mean ± SEM.
Externalization of Membrane PS Alone does not alter Cell Survival
We developed a model of independent PS externalization in our neuronal cultures to determine whether PS exposure alone was necessary to alter neuronal survival. Since mild oxidizing conditions may inhibit the aminophospholipid translocase that is responsible for the maintenance of membrane PS asymmetry (Le, DT et al., 1994), we selected the agents N-ethylmaleimide (NEM) and diazine-dicarboxylic acid bis[N,N-dimethylamide] (diamide) that can oxidize cell surface proteins without causing cell damage (Daleke, DL et al., 1994). Application of the agent diamide (0.5 mM) or NEM (0.1 mM) resulted in a significant exposure of PS to 69 ± 3% and 50 ± 5%, respectively, in neurons within 1 hour when compared with untreated control cultures (Fig. 2C). Exposure to diamide alone increased surface PS from 5 ± 3% (control) in untreated neurons to 83 ± 3% over a 24 hour period (Fig. 2C). Following application of NEM alone, the proportion of neurons with PS at the surface increased from 5 ± 3% (control) to 73 ± 4% (NEM) over a 24 hour period (Fig. 2C). Yet, application of either diamide or NEM that lead to membrane PS exposure did not result in the loss of cellular membrane integrity at 1, 6, or 24 hours following treatment with either agent (Fig. 2D).
Membrane PS Exposure is Necessary for Microglial Activation and Proliferation
Microglial activation was assessed through the expression of PCNA and the uptake of BrdU 3 hours following incubation with media from neuronal cultures exposed to NO (NOC-9 or SNP, 300 μM) for 24 hours. Administration of an antibody to PS receptor (PSR Ab) alone in a series of concentrations of 0.001-1.00 μg/ml did not alter microglial activation when compared to untreated control cultures (data not shown). Application of the PSR Ab during the application of NO-exposed media could prevent microglial activation as evidenced by the significant decrease in the expression of PCNA and the uptake of BrdU (Fig. 3A). The concentrations of PSR Ab of 0.10 and 1.00 μg/ml significantly decreased microglial activation and proliferation. Direct application of PS to microglial cultures resulted in a significant increase in the expression of PCNA and the uptake of BrdU (Fig. 3B). Application of the PSR Ab also prevented PS induced PCNA expression and BrdU uptake.
Fig. (3). Membrane PS exposure leads to microglial activation and proliferation.
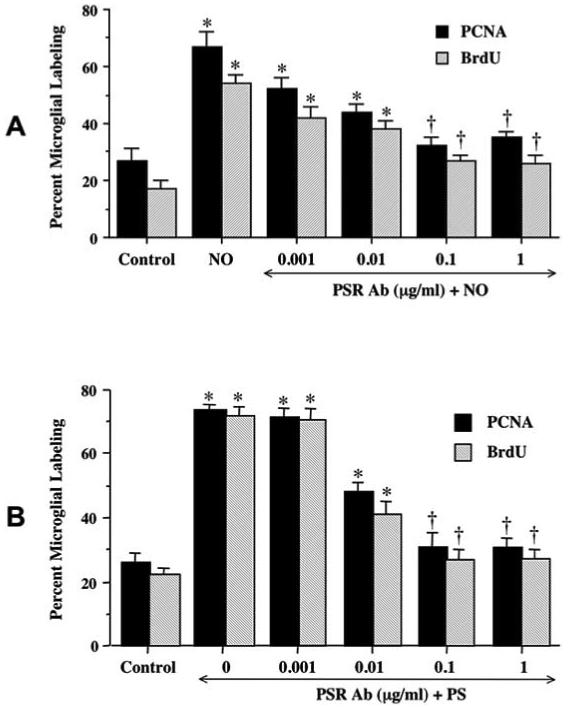
(A) Twenty-four hours following exposure to NO (NOC-9, or SNP, 300 μM), media from neuronal cultures was applied to pure microglia cultures for 3 hours treated with increasing concentrations of the PS receptor Ab (PSR, 0.001 - 1 μg/ml). Twelve hours later, microglial activation was assessed through PCNA expression and BrdU uptake. Antagonism against the microglial PSR receptor with the PSR Ab during the application of NO exposed media was observed to block microglial activation and proliferation. (B) PS was directly applied to pure microglia cultures for 3 hours co-treated with increasing concentrations of the PS receptor Ab (PSR, 0.001 - 1 μg/ml). Twelve hours later, microglial activation was assessed through PCNA expression and BrdU uptake. Antagonism against the microglial PSR receptor with the PSR Ab during the application of PS prevented microglial activation and proliferation as evidenced by decreased PCNA expression and BrdU uptake. In all cases, control = untreated microglial cultures.
mGluRIs Activation Blocks Microglial Activation
In Fig. 4A, representative microglial cultures illustrate a marked expression of PCNA and uptake of BrdU following treatment with media from NO treated neurons. In contrast, minimal PCNA and BrdU expression in microglia is present during a 1 hour pre-treatment with media from sister cultures with DHPG (750 μM). As shown in Fig. 4B, the quantitative results of PCNA and BrdU labeling revealed that NO significantly increased PCNA expression (66 ± 3%) and BrdU uptake (51 ± 4%), when compared to untreated control cultures (29 ± 3%, PCNA; 20 ± 2%, BrdU). Application of media from cultures receiving pre-treatment with DHPG during NO exposure resulted in significantly reduced PCNA (39 ± 4%) expression and BrdU (36 ± 2%) uptake. In contrast, application of media from cultures with AIDA during NO exposure did not change PCNA expression and BrdU uptake, when compared to cultures treated with NO only.
Fig. (4).
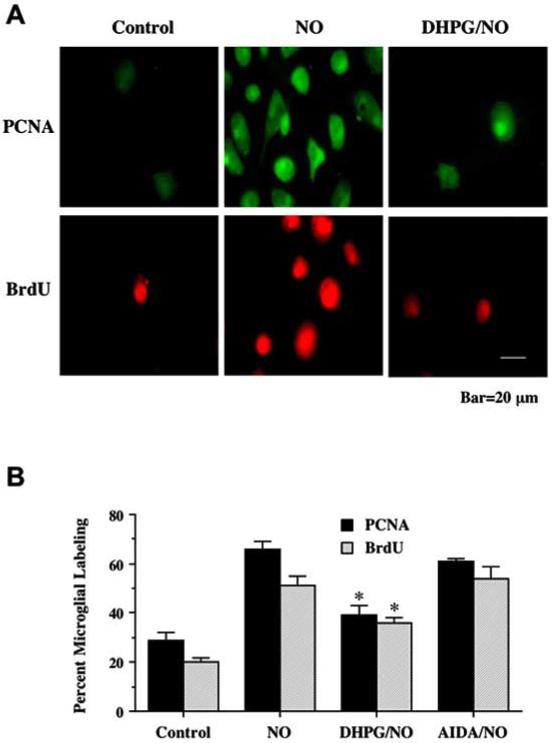
Activation of mGluRIs in neurons prevents microglia activation. Pure microglial cultures were treated for 3 hours with media from neuronal cultures conditioned with 24 hours of NO (NOC-9, 300 μM) exposure. (A) Representative images illustrate BrdU uptake and PCNA expression was significantly less in microglia treated with media from cells receiving DHPG (750 μM). (B) Application of DHPG significantly decreased microglial activation and proliferation following treatment with media exposed to NO (*P<0.01 vs. NO alone). In all cases, control = untreated neurons.
mGluRI Activation Selectively Protects Individual Neurons from Microglial Engulfment Through Maintenance of Cellular Membrane Asymmetry
To further investigate the relationship between PS exposure in neurons and microglial activation, microglia were co-cultured for 3 hours with neurons after neuronal cultures were exposed to a NO donor (NOC-9, 300 μM)) for 12 hours. Individual neurons and microglia were recorded at 10 minute intervals in real time. As shown in Fig. 5A, following exposure to NO, microglia began to migrate towards a neuron that had membrane PS exposure on its cellular surface. Once the target neuron was reached, microglia engulfment of the neuron ensued. In contrast, in sister neuronal cultures receiving DHPG (750 μM) pretreatment prior to NO exposure, a microglial cell migrated to check a neuron that had no exposed PS and subsequently traveled on leaving the neuron intact (Fig. 5B). The results suggest that PS exposure in neurons allows individualized identification by microglia for the eventual engulfment of neuron during cell injury and that mGluRI activation can prevent cell engulfment.
Fig. (5).
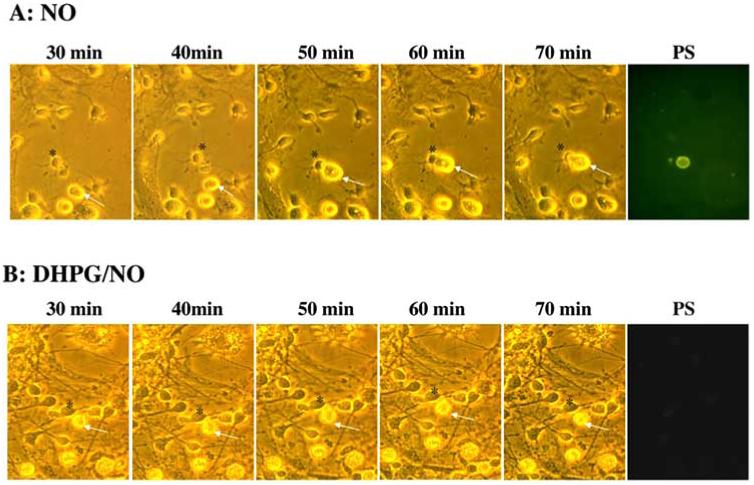
mGluRI activation selectively protects individual neurons from microglial engulfment through maintenance of cellular membrane asymmetry. (A) Microglia and neurons were obtained from the same animal pup litter and were subsequently co-cultured in a ratio of 1:5 (microglia: neurons). Twelve hours after NO (NOC-9, 300 μM) exposure, individual cells were examined over the time points indicated using transmitted light (panels 1-5 in A and B) and fluorescence light with 490 nm excitation and 585 nm emission wavelengths for PS (panel 6 in A and B) of the same microscopy field. A microglial cell is designated by the white arrow and a neuronal cell is identified by a black asterisk. Representative images from 30 minutes (min) to 70 minutes illustrate that NO leads to the specific “tagging” of a neuron with PS externalization and the subsequent engulfment of this cell by a microglial cell. In contrast, application of DHPG (750 μM) 1 hour prior to NO application prevents neuronal PS exposure and the engulfment by microglia.
mGluRI Activation Requires Akt1 Activation to Limit Membrane PS Exposure and Increase cell Survival
DHPG (750 μM) was applied to neuronal cultures 1 hour prior to NO exposure (NOC-9, 300 μM) and Western blot analysis for phospho-Akt1 (activated form of Akt1, p-Akt1) was performed 12 hours following NO exposure. In Fig. 6A, NO increased the expression of p-Akt1. The expression of p-Akt1 was increased to a greater degree by DHPG pretreatment alone or in conjunction with NO exposure than with application of NO alone. This increased expression of p-Akt1 by DHPG with NO was attenuated by the agent wortmannin (W, 500 nM). In Fig. 6B, application of DHPG 1 hour prior to NO exposure significantly decreased PS exposure to 29 ± 4%. Yet, co-application of W (500 nM)during NO exposure (Fig. 6B) with DHPG significantly reduced the ability of DHPG to prevent membrane PS externalization in neurons. Application of W also attenuated the ability of DHPG to increase neuronal survival during NO exposure (Fig. 6C).
Fig. (6). mGluRI prevents PS exposure and cell injury through Akt1.
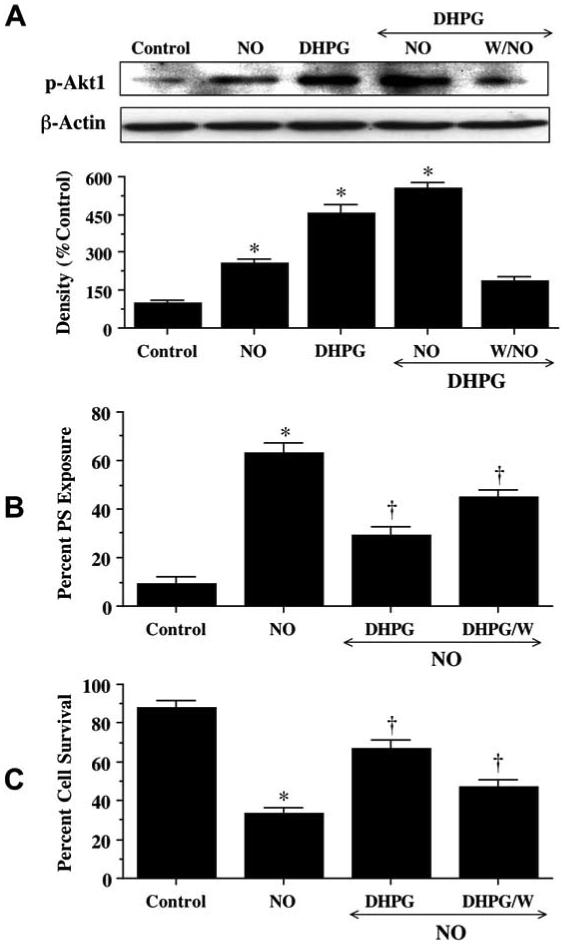
(A) Equal amounts of neuronal protein extracts (50 μg/lane) were immunoblotted 12 hours after exposure to NO, DHPG (750 μM), or DHPG (750 μM) with NO with anti-phospho-Akt1 (p-Akt1) antibody. Exposure to NO enhanced p-Akt1 expression, but exposure to DHPG (750 μM) alone or DHPG (750 μM)/NO increased p-Akt1 expression to a greater degree than NO alone. Application of the specific PI 3-K inhibitor wortmannin (W, 500 nM) 1 hour prior to NO exposure was sufficient to block the expression of p-Akt1 in the presence of DHPG 12 hours following NO. Quantitative analysis of the Western immunoblots for the data presented in (A) was performed using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/). (B, C) At a concentration that blocks activation of p-Akt1 during NO (NOC-9 or SNP, 300 μM), wortmannin (W, 0.5 μmol/L) applied 1 hour prior to NO significantly reduced the capacity of DHPG (750 μM) to prevent PS exposure of cell injury during NO (*P<0.01 vs. Control; †P<0.01 vs. NO). When applied alone, wortmannin was not toxic. In all cases, control = untreated neurons. In (B) and (C), to simplify the figures, the results of the two NO donors were combined. In all cases, control = untreated neurons.
mGluRI Activation Modulates Mitochondrial Membrane Depolarization and the Release of Cytochrome c
Exposure to NO resulted in a significant decrease in the red/green fluorescence intensity ratio using a cationic membrane potential indicator JC-1, within 3 hours when compared with untreated control cultures (Fig. 7A, B), suggesting that NO alters mitochondrial permeability transition pore membrane potential (ΔΨm) and results in mitochondrial membrane potential depolarization. Administration of DHPG 1 hour prior to NO exposure significantly increased the red/green fluorescence intensity of the neurons (Fig. 7A, B) and prevented mitochondrial cytochrome c release as demonstrated by Western blot analysis (Fig. 7C), suggesting that ΔΨm was restored to baseline.
Fig. (7). Activation of mGluRIs prevents mitochondrial membrane depolarization and cytochrome c release.
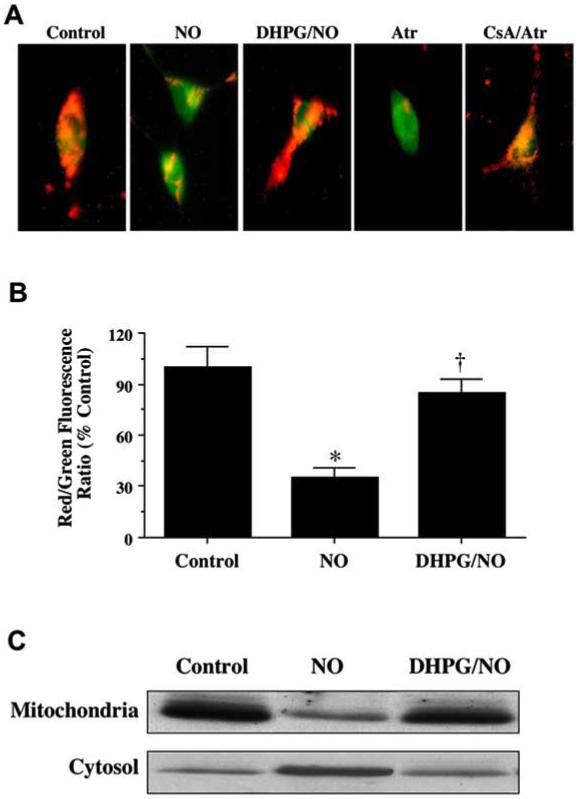
(A) Exposure to NO produced a significant decrease in the red/green fluorescence intensity ratio using a cationic membrane potential indicator JC-1 within 3 hours when compared with untreated control cultures (A, B), suggesting that NO results in mitochondrial membrane depolarization. Application of DHPG (750 μM) 1 hour prior to NO exposure significantly increased the red/green fluorescence intensity of neurons, indicating that ΔΨm was restored to baseline (A, B). Application of Atr (5 mmol/L) produced a significant decrease in the red/green fluorescence intensity ratio within 3 hours when compared with untreated control cultures. Application of CsA (5 μmol/L) with Atr administration significantly increased the red/green fluorescence intensity of neurons, indicating that ΔΨm was restored to baseline. (B) The relative ratio of red/green fluorescent intensity of mitochondrial staining in both untreated (control) neurons and neurons exposed to NO or DHPG (750 μM) plus NO 3 hours following the initial insult was measured in 4 independent experiments with analysis performed using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/) (Control vs. NO, *p<0.01; NO vs. DHPG/NO, †p<0.01). (C) A representative Western blot with equal amounts of mitochondrial protein and cytosol extracts (50 μg/lane) illustrates that cells treated with DHPG (750 μM) prior to NO significantly prevents cytochrome c release from mitochondria to the cytosol when compared to untreated control neurons.
In further control experiments, cyclosporin A (CsA), an agent that inhibits the assembly of the PTPC to prevent mitochondrial membrane potential depolarization (Halestrap, AP et al., 1997), was employed as an experimental control to modulate ΔΨm (Fig. 7A). CsA (5 μmol/L) prevented mitochondrial membrane potential depolarization during application of Atr (5 mmol/L) (Fig. 7A), an agent that binds to the mitochondrial adenosine nucleotide translocator to elicit pore formation (Brown, J et al., 1997).
mGluRI Inhibits Caspase 9-Like Activity that is Linked to the Loss of Mitochondrial Membrane Permeability
In Fig. 8 A. B, and C, Atr (1 or 5 mmol/L), CsA (5 μmol/L), or DHPG (750 μM) was applied to neuronal cultures alone or 1 hour prior to NO, and data for caspase 9 were obtained at 12 hours following NO exposure, since this time period represented the peak activities for this caspase (Chong, ZZ et al., 2004). Atr either alone or in conjunction with NO exposure significantly increased caspase 9-like activity (Fig. 8A). Interestingly, Atr at 1 mmol/L activated caspase 9 to a significantly lesser degree than with the combined application of NO, suggesting that NO exposure may employ alternate pathways that induce Atr to lead to caspase 9 activity. Administration of DHPG significantly decreased caspase 9-like activity during Atr administration (Fig. 8B) and during NO exposure (Fig. 8C), suggesting that DHPG functions at the level of the PTPC. CsA independently prevented caspase 9 activity during NO exposure, further supporting a role for the PTPC and mitochondrial membrane potential depolarization during caspase 9 activity.
Fig. (8). mGluRI prevents neuronal injury through caspase 9 by the maintenance of mitochondrial potential.
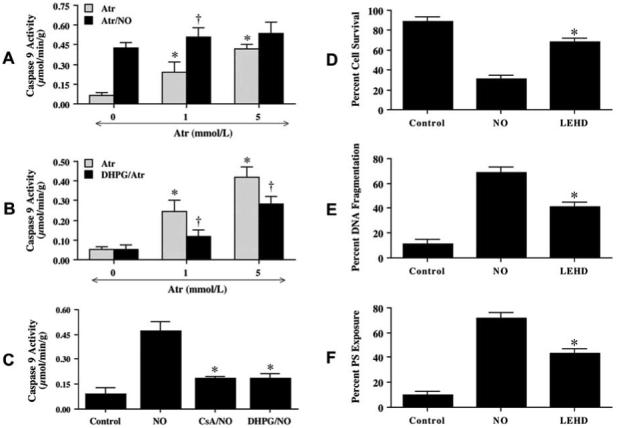
(A) DHPG (750 μM) was applied directly to neuronal cultures 1 hour prior to treatment with atractyloside (Atr, 1 mM or 5 mM) and caspase 9-like activity was determined at 12 hours following Atr treatment. DHPG prevented caspase 9-like activity following Atr (*P< 0.01 vs. Control; †p< 0.01 vs. Atr treated alone). (B) Atractyloside (Atr, 5 mM) was applied to neuronal cultures 1 hour prior to NO exposure (NOC-9 or SNP, 300 μM) and the activity of caspase 9 was determined 12 hours later (*P< 0.01 vs. NO Control; †P< 0.01 vs. Atr treated alone). (C) DHPG (750 μM) and cyclosporin A (CsA, 5 μM) were applied 1 h prior to NO (NOC-9 or SNP, 300 μM) exposure and significantly prevented the activation of caspase 9 (*P< 0.01 vs. NO treated alone). (D) Neurons were pretreated with a caspase 9 inhibitor (LEHD, 50 μM) 1 hour prior to NO (NOC-9 or SNP, 300 μM) and neuronal survival was determined 24 hours following NO exposure (*P<0.01 vs. NO). (E) Neurons were pretreated with an inhibitor of caspase 9 (LEHD, 50 μM) and DNA fragmentation was determined by TUNEL assay 24 hours following NO exposure (*P<0.01 vs. NO). (F) Neurons were pretreated with an inhibitor of caspase 9 (LEHD, 50 μM) and PS exposure was determined by annexin V labeling 24 hours following NO exposure (*P<0.01 vs. NO). In all cases, to simplify the figures, the results of the two NO donors were combined and control = untreated neurons.
We next examined whether the induction of caspase 9-like activity was required for NO-induced cell injury, DNA fragmentation and membrane PS exposure. As shown in Fig. 8D, neurons exposed to NO (NOC-9 or SNP, 300 μM) resulted in a significant decrease in cell survival. Pre-treatment of neurons with 50 μM of LEHD to inhibit caspase 9 - like activity significantly increased cell survival (Fig. 8D) and decreased DNA fragmentation from 69 ± 4% (NO alone) to 41 ± 4% (Fig. 8E). In addition, inhibition of caspase 9-like activity maintained cellular membrane asymmetry and significantly prevented PS externalization (Fig. 8F).
DISCUSSION
Neuroprotection through the activation of mGluRIs has been demonstrated during a variety of insults, such as N-methyl-D-aspartate (NMDA) toxicity (Blaabjerg, M et al., 2003), staurosporine (Rong, R et al., 2003), NO exposure (Vincent, AM et al., 2000, Vincent, AM et al., 1999), and anoxia (Lin, SH et al., 2001). Yet, elucidation of the cellular pathways that determine cytoprotection by mGluRIs may hold the greatest promise for the transition of this G-protein receptor into effective therapeutic programs. As a mGluRI agonist, DHPG provides protection against two independent components of neuronal apoptosis through the maintenance of intact genomic DNA and the prevention of cellular membrane PS exposure. Antagonism of the mGluRI receptor with AIDA does not confer protection against DNA degradation or membrane PS exposure.
Agonists of the mGluRs may use different cellular mechanisms to block genomic DNA degradation. mGluRIs may prevent the induction of a magnesium-dependent endonuclease following free radical injury (Vincent, AM et al., 1999). In addition to endonucleases, cysteine proteases become activated and may in turn activate a cysteine protease-dependent DNase (Enari, M et al., 1998). Agonists of mGluRIs appear to down-regulate two independent caspase pathways that involve caspase 3 (Lin, SH et al., 2001) as well as caspase 9, as illustrated in our new work. Other signal transduction pathways, such as intracellular calcium (Maiese, K et al., 1999) or protein kinases (Li, F et al., 2004, Li, F et al., 2004), can also be differentially regulated through the mGluR system and impact upon the maintenance of genomic DNA integrity. For example, mGluRIs prevent cell death during oxidative stress by altering protein kinase C activity, but other mGluRs, such as group III subtypes, depend upon the regulation of protein kinase A (Maiese, K et al., 1996, Pizzi, M et al., 1999).
Exposure of membrane PS residues can lead to the “tagging” of cells for removal by microglia (Maiese, K et al., 2004). It is interesting to note that although agonists of the mGluRIs, such as DHPG, prevent membrane PS exposure, agonists for group II mGluR subtypes do not prevent PS externalization during some toxic environments (Vincent, AM et al., 2000), suggesting that specific cell mechanisms must be initiated to control membrane PS exposure. Externalization of PS may involve the inhibition of aminophospholipid translocase that transfers PS from the outer to the inner leaflet of the plasma membrane (Bevers, EM et al., 1999). Apart from altering the expression of the aminophospholipid translocase, its activity is decreased following an increase in intracellular calcium concentration (Vincent, AM and Maiese, K, 1999) or a decrease in cellular energy metabolism (Auland, ME et al., 1994). These two regulatory mechanisms for aminophospholipid translocase may provide targets for mGluRIs to block PS externalization. In addition, the prevention of the externalization of membrane PS residues by mGluRs may result from the direct inhibition of cysteine protease activity, since caspase 3 and possibly caspase 9 can be tied to membrane PS externalization (Kang, JQ et al., 2003, Li, F et al., 2004, Li, F et al., 2004). In contrast, the specific activation of mGluR subtype 2 induces an intracellular calcium transient through the release of calcium from intracellular stores (Maiese, K et al., 1999). The release of intracellular calcium stores via the group II mGluR subtype may assist in the progression of PS externalization.
In addition to examining the ability of mGluRI activation to alter membrane PS exposure, we employed a model of independent PS externalization to investigate the biological role of membrane PS externalization in our system. Oxidizing agents have been reported to inhibit aminophospholipid translocase (Le, DT et al., 1994), thereby inducing the externalization of PS residues. Application of the sulfhydryl oxidizing agents NEM or diamide externalized PS in approximately 70% of neurons within 1 hour. This externalization of PS did not change neuronal cell survival, suggesting that membrane PS exposure does not immediately impact upon cell injury. Yet, PS externalization can signal for the phagocytosis of cells (Maiese, K et al., 2005). We show that microglial activation occurs during NO exposure in neurons and illustrate that application of an antibody to the PSR prevents microglial activation during NO or PS exposure, suggesting that membrane PS residue exposure is both necessary and sufficient to induce microglial activation. We also show that media taken from neurons treated with DHPG during NO exposure prevent microglial activation and the externalization of membrane PS residues. Furthermore, we show for the first time that microglial engulfment of neurons is specific for cells that express PS residues and that cell engulfment can be prevented during mGluRI agonism. Taken together, our work illustrates that activation of mGluRIs blocks microglial activation through membrane PS exposure as well as possibly prevent the shedding of membrane PS residues into the extracellular environment that is known to occur during apoptosis (Simak, J et al., 2002).
Downstream from the prevention of inflammatory neuronal demise and cellular engulfment is a series of signal transduction pathways that determine protection for mGluRI agonism. These include the protein Akt1, mitochondrial membrane permeability and caspase 9-like activity. Akt1 is necessary, at least in part, for activation of mGluRI to foster DNA integrity and cellular membrane asymmetry during NO exposure. In primary neuronal cultures, the mGluRI agonist DHPG independently increases the phosphorylation and activation of Akt1 to a greater degree than NO alone, suggesting that DHPG may employ Akt1 to provide neuroprotection. Subsequent work illustrates that DHPG is dependent upon the activation of Akt1 to prevent neuronal injury, since prevention of Akt1 phosphorylation with an inhibitor of PI-3K activity significantly reduces the ability of DHPG to prevent cell death and membrane PS exposure. These observations coincide with other studies that link phospoinositide 3 kinase activity and Akt with mGluRI hippocampal synaptic function (Hou, L and Klann, E, 2004) and cytoprotection (Rong, R et al., 2003).
Akt may foster cell survival through a variety of cellular pathways that involve the increased expression of Bcl-xL, modulation of Forkhead transcription factors, as well as the release of cytochrome c (Chong, ZZ et al., 2005). Our studies illustrate that NO acutely alters ΔΨm and results in mitochondrial membrane depolarization, but that administration of the mGluRI agonist DHPG maintains ΔΨm and prevents the release of cytochrome c. Closely associated with the disruption in ΔΨm and the release of cytochrome c is the induction of caspase activity (Chong, ZZ et al., 2004, Henry, MK et al., 2001, Kang, JQ et al., 2003). Although some “anti-apoptotic” proteins, such as erythropoietin (Chong, ZZ et al., 2003) and heat-shock protein 70 (Beere, HM et al., 2000), appear to modulate both Apaf-1 expression and cytochrome c release, DHPG may function directly at the level of mitochondrial pore formation and the prevention of caspase 9-like activity. Application of DHPG significantly decreased caspase 9-like activity during Atr administration and during NO exposure, suggesting that DHPG functions at the level of the PTPC. Furthermore, the regulation of caspase 9-like activity appears to determine cell survival, DNA fragmentation, and cellular membrane PS exposure. These observations provide further support that caspase 9 is associated with the independent apoptotic pathways of genomic DNA cleavage and cellular membrane PS exposure.
In summary, we show that the mGluRI agonist DHPG enhances neuronal cell survival during free radical injury through pathways that involve intrinsic DNA integrity and extrinsic microglial activation. Prevention of neuronal injury requires the activation of Akt1, the modulation of mitochondrial membrane permeability, prevention of cytochrome c release, and the reduction in caspase 9-like activity. Following mGluRI activation, these cellular pathways can converge to block microglial activation and the specific engulfment of injured neurons that express cellular membrane PS residues (Fig 9).
Fig. (9). mGluRI maintains neuronal integrity and fosters a novel capacity to block microglial activation.
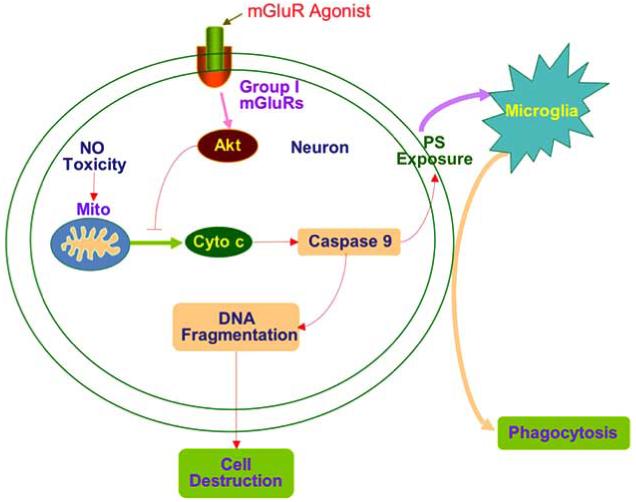
Free radical nitric oxide (NO) exposure results in mitochondrial (Mito) membrane depolarization followed by cytochrome c (Cyto c) release and activation of caspase 9. The activation of caspase 9 is not only responsible for the induction of DNA fragmentation, but also for cellular membrane PS exposure and subsequent microglial activation and phagocytosis. Activation of mGluRIs prevents neuronal apoptosis and subsequent cell engulfment through pathways that involve Akt activation and the maintenance of mitochondrial membrane potential.
ACKNOWLEDGEMENTS
This research was supported by the following grants (KM): American Heart Association (National), Janssen Neuroscience Award, Johnson and Johnson Focused Investigator Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
REFERENCES
- Aronica E, Catania MV, Geurts J, Yankaya B, Troost D. Immunohistochemical localization of group I and II metabotropic glutamate receptors in control and amyotrophic lateral sclerosis human spinal cord: upregulation in reactive astrocytes. Neuroscience. 2001;105(2):509–20. doi: 10.1016/s0306-4522(01)00181-6. [DOI] [PubMed] [Google Scholar]
- Auland ME, Roufogalis BD, Devaux PF, Zachowski A. Reconstitution of ATP-dependent aminophospholipid translocation in proteoliposomes. Proc Natl Acad Sci USA. 1994;91(23):10938–42. doi: 10.1073/pnas.91.23.10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2(8):469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Berger MA, Defagot MC, Villar MJ, Antonelli MC. D4 dopamine and metabotropic glutamate receptors in cerebral cortex and striatum in rat brain. Neurochem Res. 2001;26(4):345–52. doi: 10.1023/a:1010990812840. [DOI] [PubMed] [Google Scholar]
- Berthele A, Platzer S, Laurie DJ, Weis S, Sommer B, Zieglgansberger W, Conrad B, Tolle TR. Expression of metabotropic glutamate receptor subtype mRNA (mGluR1-8) in human cerebellum. Neuroreport. 1999;10(18):3861–7. doi: 10.1097/00001756-199912160-00026. [DOI] [PubMed] [Google Scholar]
- Bevers EM, Comfurius P, Dekkers DW, Zwaal RF. Lipid translocation across the plasma membrane of mammalian cells. Biochim Biophys Acta. 1999;1439(3):317–30. doi: 10.1016/s1388-1981(99)00110-9. [DOI] [PubMed] [Google Scholar]
- Blaabjerg M, Fang L, Zimmer J, Baskys A. Neuroprotection against NMDA excitotoxicity by group I metabotropic glutamate receptors is associated with reduction of NMDA stimulated currents. Exp Neurol. 2003;183(2):573–80. doi: 10.1016/s0014-4886(03)00204-8. [DOI] [PubMed] [Google Scholar]
- Brown J, Higo H, McKalip A, Herman B. Human papillomavirus (HPV) 16 E6 sensitizes cells to atractyloside-induced apoptosis: role of p53, ICE-like proteases and the mitochondrial permeability transition. J Cell Biochem. 1997;66(2):245–55. doi: 10.1002/(sici)1097-4644(19970801)66:2<245::aid-jcb11>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Bernardi G. Metabotropic glutamate receptors and cell-type-specific vulnerability in the striatum: implication for ischemia and Huntington’s disease. Exp Neurol. 1999;158(1):97–108. doi: 10.1006/exnr.1999.7092. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106(23):2973–9. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J Cereb Blood Flow Metab. 2003;23(3):320–30. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Metabotropic glutamate receptors promote neuronal and vascular plasticity through novel intracellular pathways. Histol Histopathol. 2003;18(1):173–89. doi: 10.14670/HH-18.173. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp Cell Res. 2004;296(2):196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005;20(1):299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Employing New Cellular Therapeutic Targets for Alzheimer’s Disease: A Change for the Better? Curr Neurovasc Res. 2005;2(1):55–72. doi: 10.2174/1567202052773508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daleke DL, Huestis WH, Newton AC. Protein kinase C as a measure of transbilayer phosphatidylserine asymmetry. Anal Biochem. 1994;217(1):33–40. doi: 10.1006/abio.1994.1080. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson E., Jr. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123(5):1207–22. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391(6662):43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997;272(6):3346–54. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- Henry MK, Lynch JT, Eapen AK, Quelle FW. DNA damage-induced cell-cycle arrest of hematopoietic cells is overridden by activation of the PI-3 kinase/Akt signaling pathway. Blood. 2001;98(3):834–41. doi: 10.1182/blood.v98.3.834. [DOI] [PubMed] [Google Scholar]
- Hermann C, Assmus B, Urbich C, Zeiher AM, Dimmeler S. Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20(2):402–9. doi: 10.1161/01.atv.20.2.402. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24(28):6352–61. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalda A, Kaasik A, Vassiljev V, Pokk P, Zharkovsky A. Neuroprotective action of group I metabotropic glutamate receptor agonists against oxygen-glucose deprivation-induced neuronal death. Brain Res. 2000;853(2):370–3. doi: 10.1016/s0006-8993(99)02166-6. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003;74(1):37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003;64(3):557–69. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19(8):5800–10. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Rapaport SI, Rao LV. Studies of the mechanism for enhanced cell surface factor VIIa/tissue factor activation of factor X on fibroblast monolayers after their exposure to N-ethylmaleimide. Thromb Haemost. 1994;72(6):848–55. [PubMed] [Google Scholar]
- Lee RK, Jimenez J, Cox AJ, Wurtman RJ. Metabotropic glutamate receptors regulate APP processing in hippocampal neurons and cortical astrocytes derived from fetal rats. Ann N Y Acad Sci. 1996;777:338–43. doi: 10.1111/j.1749-6632.1996.tb34443.x. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Erythropoietin on a Tightrope: Balancing Neuronal and Vascular Protection between Intrinsic and Extrinsic Pathways. Neurosignals. 2004;13(6):265–89. doi: 10.1159/000081963. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Navigating novel mechanisms of cellular plasticity with the NAD+ precursor and nutrient nicotinamide. Front Biosci. 2004;9:2500–2520. doi: 10.2741/1412. [DOI] [PubMed] [Google Scholar]
- Lin SH, Chong ZZ, Maiese K. The metabotropic glutamate receptor system: G-Protein mediated pathways that modulate neuronal and vascular cellular injury. Curr Med Chem -Central Nervous System Agents. 2002;2(1):17–28. [Google Scholar]
- Lin SH, Maiese K. The metabotropic glutamate receptor system protects against ischemic free radical programmed cell death in rat brain endothelial cells. J Cereb Blood Flow Metab. 2001;21(3):262–75. doi: 10.1097/00004647-200103000-00010. [DOI] [PubMed] [Google Scholar]
- Maiese K, Ahmad I, TenBroeke M, Gallant J. Metabotropic glutamate receptor subtypes independently modulate neuronal intracellular calcium. J Neurosci Res. 1999;55:472–485. doi: 10.1002/(SICI)1097-4547(19990215)55:4<472::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol Sci. 2003;24(5):228–32. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Insights into oxidative stress and potential novel therapeutic targets for Alzheimer disease. Restor Neurol Neurosci. 2004;22(2):87–104. [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. Erythropoietin in the brain: can the promise to protect be fulfilled? Trends Pharmacol Sci. 2004;25(11):577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005;293(1):90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Swiriduk M, TenBroeke M. Cellular mechanisms of protection by metabotropic glutamate receptors during anoxia and nitric oxide toxicity. J Neurochem. 1996;66(6):2419–28. doi: 10.1046/j.1471-4159.1996.66062419.x. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent A, Lin SH, Shaw T. Group I and Group III metabotropic glutamate receptor subtypes provide enhanced neuroprotection. J Neurosci Res. 2000;62(2):257–272. doi: 10.1002/1097-4547(20001015)62:2<257::AID-JNR10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Maiese K, Vincent AM. Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J Neurosci Res. 2000;59(4):568–80. doi: 10.1002/(SICI)1097-4547(20000215)59:4<568::AID-JNR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Martinez-Contreras A, Huerta M, Lopez-Perez S, Garcia-Estrada J, Luquin S, Beas Zarate C. Astrocytic and microglia cells reactivity induced by neonatal administration of glutamate in cerebral cortex of the adult rats. J Neurosci Res. 2002;67(2):200–10. doi: 10.1002/jnr.10093. [DOI] [PubMed] [Google Scholar]
- Mills CD, Fullwood SD, Hulsebosch CE. Changes in metabotropic glutamate receptor expression following spinal cord injury. Exp Neurol. 2001;170(2):244–57. doi: 10.1006/exnr.2001.7721. [DOI] [PubMed] [Google Scholar]
- Obal I, Jakab JS, Siklos L, Engelhardt JI. Recruitment of activated microglia cells in the spinal cord of mice by ALS IgG. Neuroreport. 2001;12(11):2449–52. doi: 10.1097/00001756-200108080-00032. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Boroni F, Bianchetti KM, Memo M, Spano P. Reversal of glutamate excitotoxicity by activation of PKC-associated metabotropic glutamate receptors in cerebellar granule cells relies on NR2C subunit expression. Eur J Neurosci. 1999;11(7):2489–96. doi: 10.1046/j.1460-9568.1999.00669.x. [DOI] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6(11):1153–61. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J Neurosci. 1998;18(17):6662–71. doi: 10.1523/JNEUROSCI.18-17-06662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugstad JA, Kinzie JM, Shinohara MM, Segerson TP, Westbrook GL. Cloning and expression of rat metabotropic glutamate receptor 8 reveals a distinct pharmacological profile. Mol Pharmacol. 1997;51(1):119–25. doi: 10.1124/mol.51.1.119. [DOI] [PubMed] [Google Scholar]
- Simak J, Holada K, Vostal JG. Release of annexin V-binding membrane microparticles from cultured human umbilical vein endothelial cells after treatment with camptothecin. BMC Cell Biol. 2002;3(1):11. doi: 10.1186/1471-2121-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol. 1999;159(2):362–76. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Pocock JM. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J Neurosci. 2003;23(6):2150–60. doi: 10.1523/JNEUROSCI.23-06-02150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. Direct temporal analysis of apoptosis induction in living adherent neurons. J Histochem Cytochem. 1999;47(5):661–72. doi: 10.1177/002215549904700508. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. The metabotropic glutamate system promotes neuronal survival through distinct pathways of programmed cell death. Exp Neurol. 2000;166(1):65–82. doi: 10.1006/exnr.2000.7487. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Mohammad Y, Ahmad I, Greenberg R, Maiese K. Metabotropic glutamate receptors prevent nitric oxide-induced programmed cell death. J Neurosci Res. 1997;50(4):549–64. doi: 10.1002/(SICI)1097-4547(19971115)50:4<549::AID-JNR6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Vincent AM, TenBroeke M, Maiese K. Metabotropic glutamate receptors prevent programmed cell death through the modulation of neuronal endonuclease activity and intracellular pH. Exp Neurol. 1999;155(1):79–94. doi: 10.1006/exnr.1998.6966. [DOI] [PubMed] [Google Scholar]
- Williams K, Schwartz A, Corey S, Orandle M, Kennedy W, Thompson B, Alvarez X, Brown C, Gartner S, Lackner A. Proliferating cellular nuclear antigen expression as a marker of perivascular macrophages in simian immunodeficiency virus encephalitis. Am J Pathol. 2002;161(2):575–85. doi: 10.1016/S0002-9440(10)64213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]