

Abstract
Recognized as a robust cytoprotectant for multiple tissues of the hematopoietic, vascular, cardiac, and nervous systems, erythropoietin (EPO) also is considered to be an attractive therapeutic candidate to modulate inflammatory cell function and survival during neurodegenerative disorders. To this end, microglia of the central nervous system serve a complex function not only to dispense of foreign organisms and injured cells of the brain, but also to foster tissue repair and reorganization during neuronal and vascular cell insults. We therefore examined the ability of EPO to modulate microglial cell survival and the underlying signal transduction pathways that govern microglial integrity during oxygen-glucose deprivation (OGD) - induced oxidative stress. We demonstrate in the microglial cell line EOC 2 that EPO provides direct microglial protection against early and late apoptotic programs of membrane phosphatidylserine exposure and genomic DNA degradation. Furthermore, expression and activation of Akt1 is vital to the cytoprotective capacity of EPO, since pharmacological inhibition of the PI 3-K pathway or gene silencing of Akt1 expression eliminates the ability of EPO to protect microglial cells. Through Akt1 dependent mechanisms that can be abrogated through the gene silencing of Akt1, maintenance of microglial cell integrity during OGD by EPO is closely integrated with the phosphorylation and inhibition of glycogen synthase kinase-3β activity as well as the intracellular trafficking of β-catenin and nuclear factor-κB. Further work that continues to elucidate the ability of EPO to target the intricate pathways that determine inflammatory cell function and integrity may lay the ground work for new therapeutic avenues for neurodegenerative disease.
Keywords: Akt1, apoptosis, gene silencing, GSK-3β, inflammation, microglia, NF-κB, oxygen-glucose deprivation, protein kinase B, oxidative stress, phosphatidylinositide-3-kinase, phosphatidylserine, programmed cell death, RelA, siRNA
INTRODUCTION
As monocyte derived cells that reside within the central nervous system, microglia can lead a complex life that can be responsible for an intense inflammatory response to remove injured cells and debris balanced by the ability to foster reparative processes that lead to new cell proliferation and tissue reorganization. For example, microglia can aggravate cellular injury through the generation of free radicals and oxidative stress (Sankarapandi, et al., 1998), promote the production of pro-inflammatory cytokines that may heighten cell sensitivity to free radical exposure (Combs, et al., 2001), and lead to the phagocytic removal of both neurons and vascular cells (Chong, et al., 2005a, Chong, et al., 2004, Kang, et al., 2003b). Furthermore, microglial activation has been correlated with several neurodegenerative disorders, such as Alzheimer’s disease with the co-localization of microglia and amyloid plaque development (Sheng, et al., 1997). On the converse side, microglia can form a barrier for the removal of foreign microorganisms from the central nervous system and promote tissue repair during neuronal and vascular cell injury (Dringen, 2005, Maiese, et al., 2005a, Neumann, et al., 2006).
Given the positive impact that microglia may have upon the progression or resolution of degenerative insults to the brain, it becomes essential to consider cellular pathways that serve to support microglial survival and proliferation. To this end, robust cytoprotective agents that are known to modulate inflammatory cell function may offer attractive therapeutic targets to promote microglial survival during neurodegenerative disorders. Erythropoietin (EPO) appears to fill such a need in consideration of its role during periods of cellular inflammation. EPO can modulate cytokine expression and release (Avasarala and Konduru, 2005, Savino, et al., 2006) as well as regulate the ability of microglia to engulf cells during early apoptotic injury (Chong, et al., 2003b, Chong, et al., 2005c). EPO also has been linked to microglial dependent brain parenchyma restructuring following cerebral injury (Bernaudin, et al., 1999, Chong, et al., 2002b, Maiese, et al., 2004) by preserving microglia integrity (Vairano, et al., 2002).
EPO can function through a number of cellular signaling pathways (Maiese, et al., 2005b), none being of less prominence than those involving Akt1, one of three family members of the protein kinase B family that is highly expressed in the brain (Chong, et al., 2005b) and serves as a central regulator for the proliferative and protective attributes of EPO (Ghezzi and Brines, 2004, Maiese, et al., 2004). Interestingly, the activation of Akt has been linked to the promotion of microglial cell proliferation (Ito, et al., 2005, Kim, et al., 2004, Suh, et al., 2005). In order for Akt1 to enhance cell survival, Akt1 must oversee the activity of several down-stream substrates that are necessary for the support of cell survival. In particular, inhibition of glycogen synthase kinase-3β (GSK-3β) activity through its phosphorylation is required to block apoptosis (Crowder and Freeman, 2000, Papkoff and Aikawa, 1998). With the inhibition of GSK-3β activity, “anti-apoptotic” pathways can be set in motion through β-catenin. Inhibition of GSK-3β activity promotes β-catenin integrity with its translocation to the cell nucleus to enhance cell survival (Chen, et al., 2001, Li, et al., 2006, Papkoff and Aikawa, 1998, You, et al., 2004). In addition to these pathways, cytoprotection of EPO also has been shown to require the expression and cellular trafficking of nuclear factor-κB (NF-κB) to the cell nucleus for cellular functions that foster neural stem cell growth (Shingo, et al., 2001), lead to the differentiation of glial cells (Lee, et al., 2004), prevent apoptotic cell death (Bittorf, et al., 2001, Chong, et al., 2005c, Sae-Ung, et al., 2005), and possibly assist with tissue repair by microglial cells (Sanz, et al., 2002).
We demonstrate that the broad and robust cytoprotective capacity of EPO for a variety of cell systems is conserved for the maintenance of genomic DNA integrity and cellular membrane asymmetry during oxygen-glucose deprivation (OGD)-induced oxidative stress in the microglial cell line EOC 2. EPO requires the activation of Akt1 not only for microglial cellular protection, but also to modulate specific downstream substrates of Akt1, since gene silencing of Ak1 protein expression eliminates the cytoprotective capacity and control of Akt1 substrate intracellular signaling by EPO. The ability of EPO to promote microglial survival is tightly aligned with the phosphorylation and inhibition of GSK-3β activity as well as the expression and intracellular trafficking of β-catenin and NF-κB from the cytoplasm to the nucleus.
MATERIALS AND METHODS
Microglia Cell Cultures
The microglial cell line EOC 2 was obtained from American Type Culture Collection (ATTC, Manassas, VA.). Cells were maintained in Dulbecco’s modified Eagle medium (ATTC, Manassas, VA), supplemented with 10% fetal bovine serum (SIGMA, St Louis, MO), 50 μg/ml penicillin and streptomycin and 20% media from the LADMAC cell line (ATCC, Manassas, VA) which contains colony stimulating factor-1 (CSF-1) secreted by LADMAC cells. Cells were seeded onto 24-well plates or 35 mm culture dishes at a density of 1.5 × 106 cells per well or 4 × 106 cells per dish.
Experimental Treatments
Oxygen-glucose deprivation (OGD)
OGD in microglial cultures was performed by replacing media with glucose-free HBSS containing 116 mmol/L NaCl, 5.4 mmol/L KCl, 0.8 mmol/L MgSO4, 1 mmol/L NaH2PO4, 0.9 mmol/L CaCl2, and 10 mg/L phenol red (pH 7.4) and cultures were maintained in an anoxic environment (95% N2 and 5% CO2) at 37 °C for 6 hours or per the experimental paradigm. During both pre-paradigm applications, EPO (R&D Systems, Minneapolis, MN) application was continuous. To inhibit Akt activation (phosphorylation), the PI 3-K inhibitor wortmannin (W) (Calbiochem, La Jolla, CA) or the Akt selective inhibitor D-2,3-dieoxy-myo inositol 1-(R)-2-methoxy-3-(octadecyloxy) propyl hydrogen phosphate (SH-6) (Alexis, San Diego, CA) were applied to microglial cultures 1 hour prior to OGD. The glycogen synthase kinase (GSK)-3β inhibitors SB216763 [3-(2,4-Dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione] (SB21) or SB415286 [3-[(3-Chloro-4-hydroxyphenyl)amino]-4-(2-nitrophenyl)-1H-pyrrole-2,5-dione] (SB41) (Tocris, Ellisville, MO) were applied continuously to the microglial cultures 1 hour prior to OGD.
Assessment of Microglial Cell Survival
Microglial cell injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 hours following treatment with OGD per our previous protocols (Chong, et al., 2005c). The mean survival was determined by counting eight randomly selected non-overlapping fields with each containing approximately 10-20 cells (viable + non-viable). Each experiment was replicated 6 times independently with different cultures.
Assessment of DNA Fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay (Chong, et al., 2002a, Kang, et al., 2003b). Briefly, microglial cells were fixed in 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde and the 3′-hydroxy ends of cut DNA were labeled with biotinylated dUTP using the enzyme terminal deoxytransferase (Promega, Madison, WI) followed by streptavidin-peroxidase and visualized with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA).
Assessment of Membrane Phosphatidylserine (PS) Residue Externalization
Phosphatidylserine (PS) exposure was assessed through the established use of annexin V. Per our prior protocols (Chong, et al., 2004, Kang, et al., 2003b), a 30 μg/ml stock solution of annexin V conjugated to phycoerythrin (PE) (R&D Systems, Minneapolis, MN) was diluted to 3 μg/ml in warmed calcium containing binding buffer (10 mmol/L Hepes, pH 7.5, 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2). Plates were incubated with 500 μl of diluted annexin V for 10 minutes. Images were acquired with “blinded” assessment with a Leitz DMIRB microscope (Leica, McHenry, IL) and a Fuji/Nikon Super CCD (6.1 megapixels) using transmitted light and fluorescent single excitation light at 490 nm and detected emission at 585 nm.
Gene Silencing of Akt1 with Small Interfering RNA (siRNA)
Cells were plated into 35 mm dishes or 24-well plates. To silence Akt1 gene expression, commercial reagents using the SMARTpool Akt1 siRNA kit (Upstate, Lake Placid, NY) were used. Transfection of siRNA duplexes were performed with Oligofectamine reagent according to manufacturer guidelines (Invitrogen, Carlsbad, CA). Experimental assays were performed 72 hours post-transfection. For each siRNA assay, negative controls contain multiple siRNAs including the target siRNA and positive controls are absent of the target siRNA.
Expression of Phosphorylated Akt1, Phosphorylated β-Catenin, and Phosphorylated Glycogen Synthase Kinase
Cells were homogenized and following protein determination, each sample (50 μg/lane) was then subjected to 7.5% SDS-polyacrylamide gel electrophoresis. After transfer, the membranes were incubated with a rabbit polyclonal antibody against a mouse monoclonal antibody against phosphorylated Akt1 (p-Akt1, Ser473, 1:1000, Active Motif, Carlsbad, CA), a rabbit antibody against phosphorylated-β-catenin (1:1000) (p-β-catenin, Ser33/37 Thr41, Cell Signaling, Beverly, MA), or a rabbit antibody against phosphorylated GSK-3β (p-GSK-3β, Ser9). Following washing, the membranes were incubated with a horseradish peroxidase (HRP) conjugated secondary antibody (goat anti-mouse IgG, 1:2000 (p-Akt1) or goat anti-rabbit IgG, 1:15000 (p-β-catenin, p-GSK-3β). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, NJ).
β-Catenin Immunocytochemistry
For immunocytochemical staining of β-catenin, microglial cells were fixed with 4% paraformaldehyde and permeabilized using 0.2% Triton X-100. Cells were then incubated with rabbit anti-β-catenin 1 (1:100, Cell Signaling Technology, Beverly, MA) over night at 4°C and then with biotinylated anti-rabbit IgG (1:50, Vector laboratories) for 2 hours followed by Texas Red streptavidin (1:50, Vector laboratories) for 1 hour. Cells are washed in PBS and then stained with DAPI (Sigma, St. Louis, MO) for nuclear identification. Protein for β-catenin was imaged with fluorescence at the wavelengths of 565 nm (red) and 400 nm (DAPI nuclear staining).
Western Blot Analysis for the NF-κB p65 Family Member Expression
Cells were homogenized and following protein determination, each sample (50 μg/lane) was then subjected to 7.5% SDS-polyacrylamide gel electrophoresis. Following transfer, the membranes were incubated with primary rabbit antibody against NF-κB p65 (1:200) (Santa Cruz Biotechnologies, Santa Cruz, CA). After washing, the membranes were incubated with a horseradish peroxidase conjugated secondary antibody (goat anti-rabbit IgG, 1:15000) (Pierce, Rockford, IL). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ). The cytoplasmic and nuclear proteins were prepared by using NE-PER nuclear and cytoplasmic extraction reagents according to manufacture’s instruction (purchased from Pierce, Rockford, IL). The expression of NF-κB p65 in nucleus and cytoplasm was determined by Western blot performed as described as above.
Statistical Analysis
For each experiment, the mean and standard error were determined. Statistical differences between groups were assessed by means of analysis of variance (ANOVA) from 6 replicate experiments with the post-hoc Student’s t-test. Statistical significance was considered at p<0.05.
RESULTS
OGD is Toxic to Primary Microglia
In Fig. 1A, microglial survival was significantly reduced over a 12 hour course following OGD application to 60 ± 2% (4 hours), 39 ± 3% (6 hours), 36 ± 2% (8 hours), and 31 ± 3% (12 hours) when compared with untreated control cultures (93 ± 3%, p<0.01). Since an OGD exposure period of 6 hours resulted in survival rate of approximately 40% (60% microglial cell loss), this duration of OGD toxicity was employed for the remainder of the experimental paradigms.
Fig. (1). OGD leads to microglial cell injury that is prevented by erythropoietin (EPO).
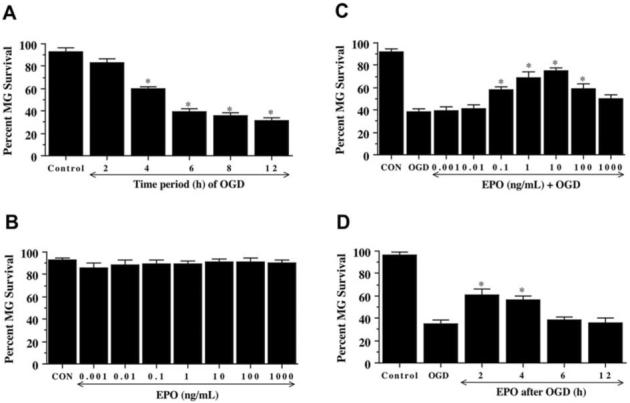
(A) Microglia were exposed to OGD for 2, 4, 6, 8, and 12 hours and cell survival was determined 24 hours after OGD. The cell survival was progressively decreased over a period of 2, 4, 6, 8, and 12 hours of OGD (* P < 0.01 vs. untreated control). CON = untreated control cultures. (B) Increasing concentrations of EPO from 0.001 to 1000 ng/mL were applied to microglia for a 24-hour period and cell survival was determined by using the trypan blue exclusion method. The cell survival of microglia in the presence of EPO at the concentration 0.001 to 1000 ng/mL was not different from that in the cultures in absence of EPO. (C) EPO, at concentrations of 0.001 to 1000 ng/mL, was applied to the cultures of microglia 1 hour prior to a 6 hour period of OGD and cell survival was assessed 24 hours later. Cytoprotection by EPO during OGD was evident in cultures with 0.1 to 100 ng/mL EPO when compared with cultures exposed to OGD alone (* P < 0.01 vs. OGD). CON = untreated control cultures. (D) EPO (10 ng/mL) was applied to microglia at 2, 4, 6, and 12 hours following a 6 hour period of OGD exposure. Cell survival was evaluated 24 hours following OGD. Application of EPO at 2 and 4 hours after OGD significantly increased microglial survival (* P < 0.01 vs. OGD). In all cases, each data point represents the mean and SEM. Control indicates untreated cultures.
EPO Pre- and Post-Treatment Increases Microglial Survival During OGD
As shown in Fig. 1B, no significant toxicity over a 24 hour period was present in microglia exposed to EPO alone in the concentrations of 0.001 ng/mL to 1000 ng/mL when compared with microglial survival in untreated control cultures (92 ± 3%). Administration of EPO 1 hour prior to OGD significantly increased survival with progressive increases in EPO concentration (Fig. 1C). Yet, EPO (10 ng/mL) administration reached the maximal microglial survival (75 ± 3%, p<0.01), but concentrations lower than 0.1 ng/mL or higher than 10 ng/mL did not improve microglial survival during OGD (Fig. 1C). In addition, concentrations greater than 10 ng/mL did not reduce microglial survival below the level for cultures exposed to OGD alone.
In post-treatment experiments (Fig. 1D), microglial cells received EPO (10 ng/mL) at 2, 4, 6, and 12 hours following OGD with cell survival determined at 24 hours after OGD administration. EPO applied at 2 and 4 hours following OGD exposure significantly increased microglial survival from 35 ± 3% (OGD alone) to 61 ± 5% (p<0.01) and 56 ± 4% (p<0.01) respectively. In contrast, post-treatment with EPO at 6 and 12 hours following OGD exposure did not significantly increase microglial survival.
EPO Prevents Apoptotic Genomic DNA Fragmentation and Membrane Phosphatidylserine Exposure in Microglia During OGD
Twenty-four hours following OGD exposure, apoptosis with chromatin condensation and nuclear fragmentation in microglia cells was evident by TUNEL labeling and cellular membrane PS exposure was present annexin V labeling (Fig. 2A). In contrast, microglia pretreated with EPO (10 ng/mL) 1 hour prior to OGD were without nuclear fragmentation or significant membrane PS exposure (Fig. 2A). In Fig. 2B, OGD lead to a significant increase in percent DNA fragmentation (65 ± 3%) when compared to untreated control cultures (7 ± 1%), but EPO significantly reduced DNA fragmentation to 33 ± 3% (p<0.01). Similarly, an increase in membrane PS exposure was observed in microglia 24 hours following OGD that reached a level of 66 ± 5% when compared to untreated control cultures of 11 ± 2% (Fig. 2B). Microglia treated with EPO displayed a significant reduction in membrane PS externalization to 34 ± 3% at 24 hours after OGD.
Fig. (2). Erythropoietin (EPO) prevents apoptotic genomic DNA fragmentation and membrane phosphatidylserine (PS) exposure during OGD in microglia.
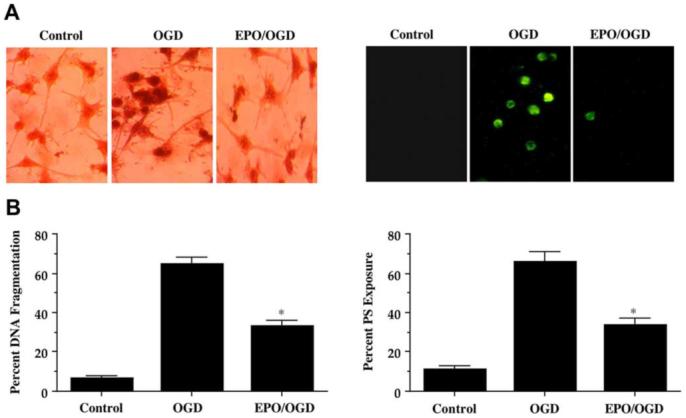
EPO (10 ng/mL) was applied to microglia 1 hour prior to a 6 hour period of OGD exposure. Apoptotic genomic DNA fragmentation and membrane PS exposure were assessed using TUNEL assay and annexin V phycoerythrin labeling respectively 24 hours after OGD. (A) Representative images for TUNEL illustrate DNA fragmentation in microglia following OGD, but prevention of DNA fragmentation during EPO (10 ng/mL) application. (B) Quantification of data demonstrates that EPO (10 ng/mL) applied to microglia 1 hour prior to a 6 hour period of OGD significantly decreased DNA fragmentation 24 hours after OGD (* P < 0.01 vs. OGD alone). (C) Representative images illustrate cellular membrane PS exposure in microglia by fluorescence (F) light field with 490-nm excitation and 585-nm emission wavelengths 24 hours following OGD exposure. EPO significantly reduced PS staining during OGD exposure. (C) Quantification of data demonstrates that EPO (10 ng/mL) applied to microglia 1 hour prior to a 6 hour period of OGD significantly decreased membrane PS exposure 24 hours later (* P < 0.01 vs. OGD). In all cases, control indicates untreated cultures.
Primary Microglia Rely Upon the PI 3-K Pathway and Akt1 Phosphorylation for EPO Protection Against OGD
Western blot assay was performed for phosphorylated Akt1 (p-Akt1) (activated form of Akt1) (Fig. 3) 12 hours following OGD. For Akt1 phosphorylation, Ser473 was examined since phosphorylation of Ser473 is considered to be a critical component for the complete activation of Akt1 (Bellacosa, et al., 1998). Both OGD alone and EPO (10 ng/mL) alone independently increased the expression of p-Akt1, but EPO, either alone or in the presence of OGD, elevated p-Akt1 expression significantly to a greater degree than application of OGD alone. This increased expression of p-Akt1 by either OGD or EPO was blocked by application of wortmannin (W, 500 nM), which forms a covalent link with the lysine residue of PI 3-K (Wymann, et al., 1996), and the specific Akt1 inhibitor SH-6 (20 μmol/L) (Kozikowski, et al., 2003).
Fig. (3). Erythropoietin (EPO) increases phosphorylation of Akt1 in microglia.
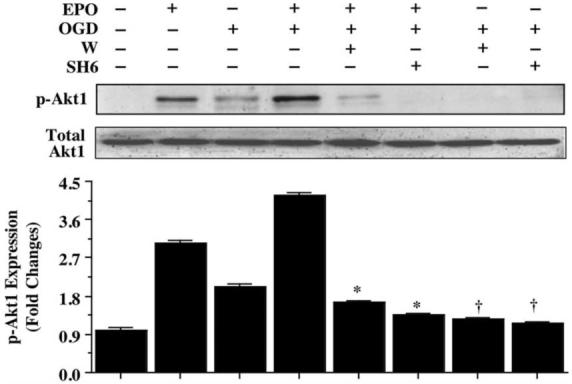
Equal amounts of microglial protein extracts (50 μg/lane) were immunoblotted with anti-phospho-Akt1 (p-Akt1, active form) antibody 12 hours following a 6 hour period of OGD exposure. EPO (10 ng/mL) treatment significantly increased the expression of p-Akt1 alone or to a greater degree than OGD alone. In contrast, application of the phosphatidylinositol-3-kinase (PI 3-K) inhibitor wortmannin (W, 1000 nM) or the specific Akt1 inhibitor SH6 (20 μM) diminished the expression of p-Akt1 in the presence of EPO (10 ng/mL) 12 hours after OGD (* P < 0.01 vs. EPO/OGD; †P <0.01 vs. OGD). Band density was performed using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image).
In Fig. 4A, application of EPO (10 ng/mL) 1 hour prior to OGD prevented microglial membrane injury and trypan blue uptake, but this protection by EPO was lost during application of wortmannin (W, 500 nM). Quantification of these observations reveal that EPO (10 ng/mL) significantly increased microglial survival from 31 ± 3% to 80 ± 3% (p<0.01). Yet, co-application of wortmannin (W, 500 nM) or SH-6 (20 μmol/L) at concentrations that block activation of Akt1 through phosphorylation (Fig. 3) during OGD with EPO (10 ng/mL) significantly reduced the ability of EPO to protect microglial cells against OGD, suggesting that microglial require the activation of Akt1 for EPO to exert its cytoprotective effects (Fig. 4B).
Fig. (4). Erythropoietin (EPO) protects microglia against OGD through the PI 3K pathway and Akt1.
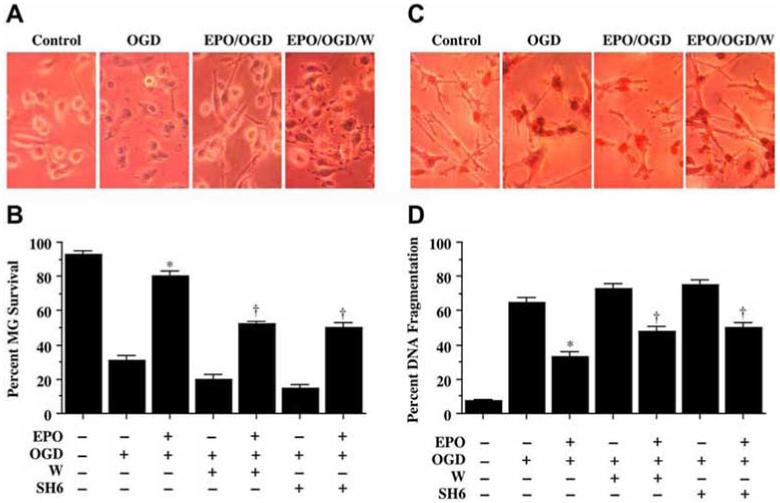
EPO (10 ng/mL) was applied to microglia 1 hour prior to a 6 hour period of OGD. Cell survival and DNA fragmentation were determined 24 hours following OGD using trypan blue dye exclusion method and TUNEL assay respectively. (A) Representative images are illustrated for trypan blue staining in cells following OGD. EPO application significantly reduced trypan blue uptake during OGD. In contrast, co-application of the phosphatidylinositol-3-kinase (PI 3-K) inhibitor wortmannin (W, 1000 nM) with EPO prevented cytoprotection during OGD. (B) Quantification of data demonstrates that wortmannin (W, 1000 nM) applied 1 hour prior to OGD decreased cell survival during OGD alone and significantly blocked protection by EPO (10 ng/mL) (* P < 0.01 vs. OGD; †P <0.01 vs. EPO/OGD). (C) Representative images illustrate DNA fragmentation in microglia following OGD. EPO (10 ng/mL) application significantly reduced TUNEL staining during OGD. In contrast, co-application of the phosphatidylinositol-3-kinase (PI 3-K) inhibitor wortmannin (W, 1000 nM) prevented EPO from blocking DNA fragmentation during OGD. (D) Quantification of data demonstrates that wortmannin (W, 1000 nM) applied 1 hour prior to OGD increased DNA fragmentation during OGD alone and significantly blocked the ability of EPO (10 ng/mL) to prevent DNA fragmentation (* P < 0.01 vs. OGD; †P <0.01 vs. EPO/OGD). In B and D, each data point represents the mean and SEM.
In a similar manner for analysis with DNA fragmentation, administration of wortmannin (W, 500 nM) led to significant DNA fragmentation during application of EPO (10 ng/mL) 1 hour prior to OGD and negated the cytoprotective effects of EPO (Fig. 4C). Quantification of these results further demonstrated that either wortmannin (W, 500 nM) or SH-6 (20 μmol/L) administration during OGD with EPO (10 ng/mL) significantly reduced the capacity of EPO to prevent DNA fragmentation in microglial cells during OGD, illustrating that microglial require the Akt1 activation for EPO preservation of cell survival and genomic DNA integrity (Fig. 4D). When administered in the absence of OGD, wortmannin (W, 500 nM) or SH-6 (20 μmol/L) were not toxic to microglia, but did slightly enhance injury during OGD, suggesting that endogenous Akt1 activation provides a small level of protection during toxic insults (data not shown).
Microglial Cytoprotection by EPO is Eliminated During the Gene Silencing of Akt1
Pharmacological inhibitors, such as wortmannin or SH-6, can have their limitations in modulating the specific activity of a protein. We therefore have incorporated into our studies siRNA gene silencing of Akt1 to specifically knockdown Akt1 activity. Microglia were transfected with Akt1 siRNA and the expression of total Akt1 and p-Akt1 was observed through Western blot analysis (Fig. 5A). Gene silencing of Akt1 during administration of EPO (10 ng/mL) with OGD exposure prevents phosphorylation of Akt1 (Figs. 5A and 5B). As shown in Figs. 5C and 5D, OGD significantly decreased microglial cell survival from 95 ± 4% to 36 ± 3% (p<0.01) and increased DNA fragmentation from 6 ± 2% to 66 ± 3% (p<0.01), but application of EPO (10 ng/mL) during OGD exposure prevented microglial injury and DNA fragmentation. Yet, protection by EPO was lost with gene silencing of Akt1 with decreased cell survival to 48 ± 3% (p<0.01) (Fig. 5C) and increased DNA fragmentation to 55 ± 4% (p<0.01) during OGD exposure (Fig. 5D), illustrating that activation of Akt1 is an essential component for EPO to prevent microglial cell injury and genomic DNA degradation. Transfection with siRNA for Akt1 alone did not significantly alter cell survival or DNA fragmentation when compared to controls.
Fig. (5). Gene silencing of Akt1 eliminates protection of erythropoietin (EPO) during OGD in microglia.
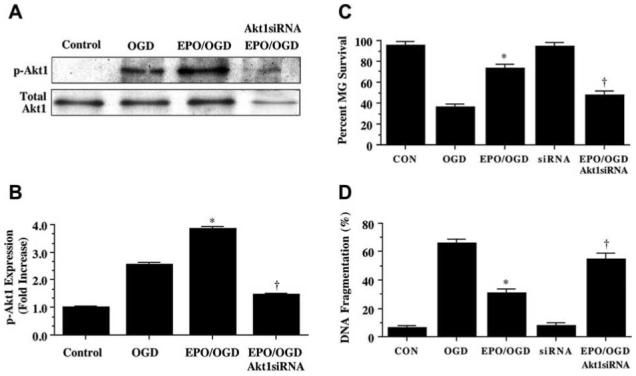
(A and B) Equal amounts of microglial protein extracts (50 μg/lane) were immunoblotted with anti-phospho-Akt1 (p-Akt1, active form) antibody 12 hours following a 6 hour period of OGD exposure. Application of EPO (10 ng/mL) in microglia 1 hour prior to OGD significantly increased the expression of p-Akt1. Transfection of Akt1 siRNA in microglia for 72 hours prior to OGD blocked the expression of p-Akt1 (* P < 0.01 vs. OGD; †P <0.01 vs. EPO/OGD). Band density was performed using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image). (C and D) Transfection of Akt1 siRNA in microglia for 72 hours prior to OGD was performed and cell survival and DNA fragmentation were determined 24 hours following OGD using trypan blue dye exclusion method and TUNEL assay respectively. Transfection of Akt1 siRNA in microglia prior to a 6 hour period of OGD prevented cytoprotection by EPO resulting in a significant decrease in cell survival and a significant increase in DNA fragmentation in microglia 24 hours following OGD. Akt1 siRNA alone did not alter cell survival and TUNEL staining (* P < 0.01 vs. OGD; †P <0.01 vs. EPO/OGD). In all cases, each data point represents the mean and SEM. CON = untreated control cultures.
EPO Phosphorylates Microglial Glycogen Synthase Kinase-3β (GSK-3β) through an Akt1 mediated pathway
We investigated the ability of EPO (10 ng/mL) to modulate phosphorylation of the Akt1 substrate GSK-3β during OGD at the conserved regulatory residue of Ser9 (Eldar-Finkelman, et al., 1996). Using western blot analysis, application of EPO (10 ng/mL) either alone or OGD significantly increased the phosphorylation of GSK-3β over a 12 hour course (Fig. 6). In addition, EPO (10 ng/mL) resulted in the phosphorylation of GSK-3β to a significantly greater degree than phosphorylation of GSK-3β by OGD. However, the ability of EPO to phosphorylate GSK-3β was lost during transfection with Akt1 siRNA, suggesting the EPO blockade of GSK-3β is closely aligned to the presence and activity of Akt1 (Fig. 6).
Fig. (6). Erythropoietin (EPO) phosphorylates glycogen synthase kinase-3β (GSK-3β) through Akt1.
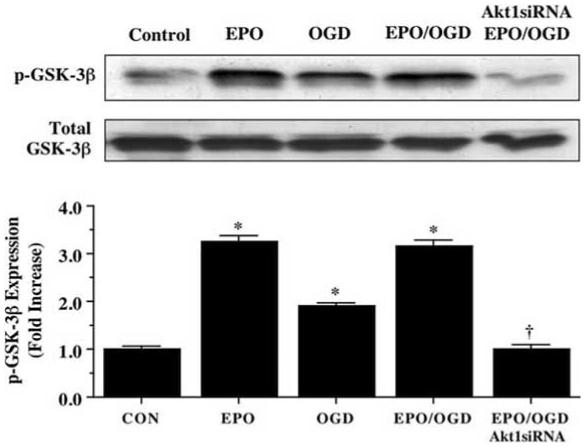
Equal amounts of microglial protein extracts (50 μg/lane) were immunoblotted with phospho-GSK-3β antibody 12 hours after a 6 hour period of OGD. EPO (10 ng/mL) applied 1 hour prior to a 6 hour period of OGD significantly increased the expression of p-GSK-3β (inactive form). Transfection of Akt1 siRNA in microglia for 72 hours prior to the OGD insult decreased the efficacy of EPO resulting in a significantly diminished expression of p-GSK-3β (* P < 0.01 vs. Control; †P <0.01 vs. EPO/OGD). Band density was performed using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image). Each data point represents the mean and SEM. CON = untreated control cultures.
Inhibition of Glycogen Synthase Kinase-3β (GSK-3β) Activity Prevents Microglial cell Injury During OGD Similar to EPO
In Fig. 7A, OGD leads to cellular injury and microglial trypan blue uptake 24 hours following OGD exposure. In contrast, application of the GSK-3β inhibitors (SB216763, (SB21), 5 μM or SB415286, (SB41), 25 μM) 1 hour prior to OGD in concentrations consistent with the current literature (Yoshimura, et al., 2005) enhance cell survival and block trypan blue uptake. Further analysis in Fig. 7B illustrates that inhibition of GSK-3β activity alone or in combination with the application of EPO (10 ng/mL) administered 1 hour prior to OGD significantly increase microglial survival when compared to OGD alone and to the same degree as during application of EPO alone or during co-administration with EPO, suggesting that EPO relies upon the inhibition of GSK-3β activity to exert microglial cytoprotection. Similarly, application of the GSK-3β inhibitors (SB216763, (SB21), 5 μM or SB415286, (SB41), 25 μM) 1 hour prior to OGD prevented microglial apoptotic DNA fragmentation (Fig. 7C) assessed by TUNEL without evidence of a synergistic increase in protection against DNA fragmentation application of EPO alone or during co-administration of EPO (10 ng/mL) (Fig. 7D), further supporting the premise that EPO utilizes inhibition of GSK-3β activity to prevent microglial cell injury and apoptotic demise.
Fig. (7). Inhibition of glycogen synthase kinase-3β (GSK-3β) protects microglia against OGD.
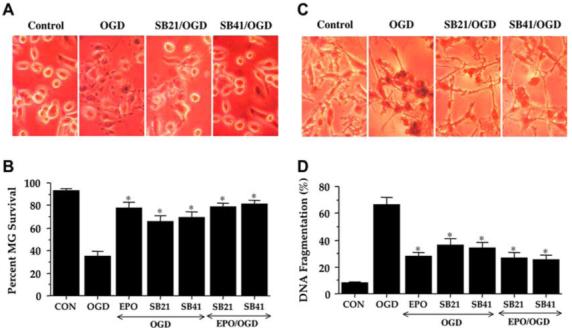
The GSK-3β inhibitors SB216763 (SB21, 5 μM) or SB415286 (SB41, 25 μM) were applied to microglia 1 hour prior to a 6 hour period of OGD. Cell survival and DNA fragmentation were determined 24 hours following OGD using trypan blue dye exclusion method and TUNEL assay respectively. (A) Representative images illustrate trypan blue staining in microglia following OGD, but application of SB216763 (SB21, 5 μM) or SB415286 (SB41, 25 μM) significantly decreased cell staining following OGD. (B) Quantification of data demonstrates that SB216763 (SB21, 5 μM) or SB415286 (SB41, 25 μM) applied 1 hour prior to OGD significantly increased microglial cell survival during OGD. Furthermore, the ability of EPO (10 ng/mL) to enhance cell survival during co-application of SB216763 or SB415286 was not increased (* P < 0.01 vs. OGD). (C) Representative images illustrate DNA fragmentation in microglia following OGD. SB216763 (SB21, 5 μM) or SB415286 (SB41, 25 μM) application significantly prevented TUNEL staining during OGD. (D) Quantification of data demonstrates that SB216763 (SB21, 5 μM) or SB415286 (SB41, 25 μM) applied 1 hour prior to OGD significantly reduced DNA fragmentation during OGD, but did not further increase the ability of EPO (10 ng/mL) to block DNA fragmentation during co-application of SB216763 or SB415286 (* P < 0.01 vs. OGD). In B and D, each data point represents the mean and SEM. CON = untreated control cultures.
EPO Employs Akt1 for the Prevention of β-Catenin Phosphorylation
Given that EPO is intimately linked to Akt1 for microglial cell protection and that β-catenin can be indirectly modulated by the Akt1 pathway through GSK-3β, we next examined the ability of EPO to modulate the phosphorylation of β-catenin following OGD exposure. Phosphorylation of β-catenin can occur sequentially with Ser45, Thr41, Ser37, and Ser33, but recent literature suggests that phosphorylation at Ser33, Ser37, and Thr41 occurs without the requirement of Ser45 (Wang, et al., 2003). We therefore examined phosphorylation of β-catenin at Ser33/37 and Thr41. EPO (10 ng/mL either alone or during OGD prevented the expression of phosphorylated β-catenin (p-β-catenin) 12 hours following OGD. However, expression of p-β-catenin was significantly increased during OGD exposure (Figs. 8A and 8B). Interestingly, gene silencing of Akt1 during OGD also led to a marked increase in the expression of p-β-catenin either during application of EPO (10 ng/mL) alone or during the combined administration of EPO (10 ng/mL) and OGD (Figs. 8C and 8D), demonstrating that EPO employs Akt1 to block the phosphorylation of β-catenin.
Fig. (8). Erythropoietin (EPO) prevents the phosphorylation of β-catenin through an Akt1 dependent mechanism in microglia during OGD.
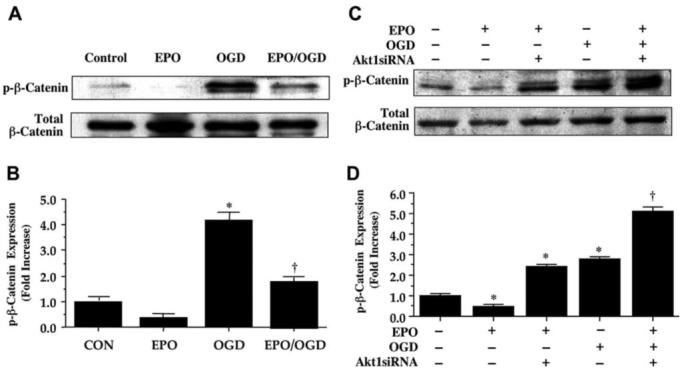
(A and B) Equal amounts of protein extracts (50μg/lane) from microglia were immunoblotted with phospho (p)-β-catenin (Ser33/37Thr41) antibody 12 hours following a 6 hour period of OGD exposure. Although OGD exposure alone increased the expression of p-β-catenin in microglia, EPO (10 ng/mL) applied 1 hour prior to OGD was able to significantly decrease the phosphorylation β-catenin (* P < 0.01 vs. CON; †P <0.01 vs. OGD). CON = untreated control cultures. (C and D) Transfection of Akt1 siRNA in microglia for 72 hours was performed prior to a 6 hour period of OGD. Subsequent protein extracts were obtained 12 hours following OGD and were immunoblotted with p-β-catenin antibody. Transfection of Akt1 siRNA was sufficient to block the ability of EPO (10 ng/mL) to prevent the expression of p-β-catenin 12 hours following OGD (* P < 0.01 vs. control untreated cultures; †P <0.01 vs. OGD). Band density was performed using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image).
EPO Maintains β-Catenin in the Nucleus of Microglia During OGD
In order for β-catenin to prevent apoptotic injury in cells, the protein must transfer to the cell nucleus and activate the transcription of its target genes that support cell survival (Chen, et al., 2001, Li, et al., 2006, Papkoff and Aikawa, 1998, You, et al., 2004). We therefore used immunofluorescent staining for β-catenin and DAPI nuclear staining to follow the subcellular translocation of β-catenin in microglia during OGD alone and during combined administration of EPO (10 ng/mL) and OGD (Figs. 9A and 9B). With OGD exposure alone, significant immunofluorescent staining for β-catenin in the cytoplasm of microglia with minimal nuclear staining is present. This is evident by the ability to detect significant DAPI nuclear staining (white in color) in cells during merged OGD images since prominent β-catenin staining is not present in the nucleus (Figs. 9A and 9B). In contrast, during application of EPO (10 ng/mL) with OGD, β-catenin is translocated to the nucleus with minimal cytoplasmic staining as shown by the pink nuclear regions on merged images of β-catenin and DAPI and subsequent quantification (Figs. 9A and 9B). In Fig. 9B, quantification of nuclear β-catenin staining reveals a high proportion of β-catenin in the nucleus during EPO and OGD application, but with minimal β-catenin in the nucleus during OGD exposure alone.
Fig. (9). Erythropoietin (EPO) maintains β-catenin in the nucleus of microglia during OGD.
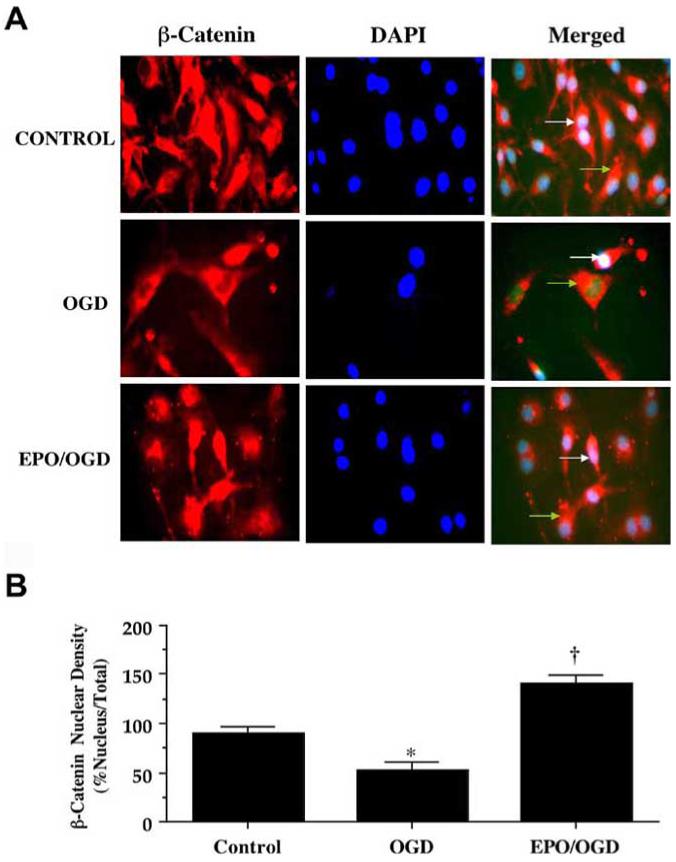
(A) EPO (10 ng/mL) with OGD was followed at 12 hours with immunofluorescent staining with a primary rabbit anti-β-catenin antibody and visualized with Texas-red streptavidin. Microglial cell nuclei were stained with DAPI. In merged images, cells with EPO and OGD with white arrows show neuronal nuclei with strong β-catenin staining (pink) and green arrows show neuronal cytoplasm with decreased β-catenin staining in contrast to cells with OGD alone with green arrow demonstrating minimal β-catenin nuclear staining (white), illustrating the ability of EPO to promote nuclear transfer of β-catenin. (B) EPO increased nuclear translocation of β-catenin during OGD. Intensity of β-catenin nuclear staining was performed using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image). Control = untreated microglia.
EPO Promotes the Transfer of NF-κB p65 from the Cell Cytoplasm to the Cell Nucleus in Microglial Cells
Although each of the family members of NF-κB (p65 (RelA), RelB, and c-Rel) contains a C-terminal transactivation domain that can lead to gene transcription, significant gene activation is triggered by the p65 member that consists of two potent transactivation domains within its C terminus (Schmitz and Baeuerle, 1991). As a result, we chose to examine the effect of EPO in microglial cells upon the expression of NF-κB p65 and whether EPO could modulate the nuclear translocation of NF-κB p65 in microglia during OGD. Upon activation, NF-κB p65 translocates to the nucleus and increases the expression of many of its target genes, which function to prevent apoptosis (Chen, et al., 2000, Chong, et al., 2005d, Chong, et al., 2005f, Li, et al., 1999). We determined the expression of NF-κB p65 in the cytoplasmic and nuclear fractions separately by Western blot analysis. EPO (10 ng/mL) was applied 1 hour prior to OGD and cytoplasmic and nuclear protein was extracted at 6 hours following OGD. In cytoplasmic fractions, NF-κB p65 expression was significantly reduced during EPO (10 ng/mL) administration alone or with combined EPO (10 ng/ml) and OGD administration. In contrast, significant expression of NF-κB p65 was present in the cytoplasmic fraction during OGD administration alone (Fig. 10A). Nuclear fractions revealed significant expression of NF-κB p65 during EPO (10 ng/ml) administration alone or with combined EPO (10 ng/ml) and OGD application, but minimal expression of NF-κB p65 during OGD alone, illustrating that EPO fosters the translocation of NF-κB p65 in microglia from the cytoplasm to the nucleus (Fig. 10B).
Fig. (10). Erythropoietin (EPO) facilitates the nuclear translocation of NF-κB in microglia during OGD.
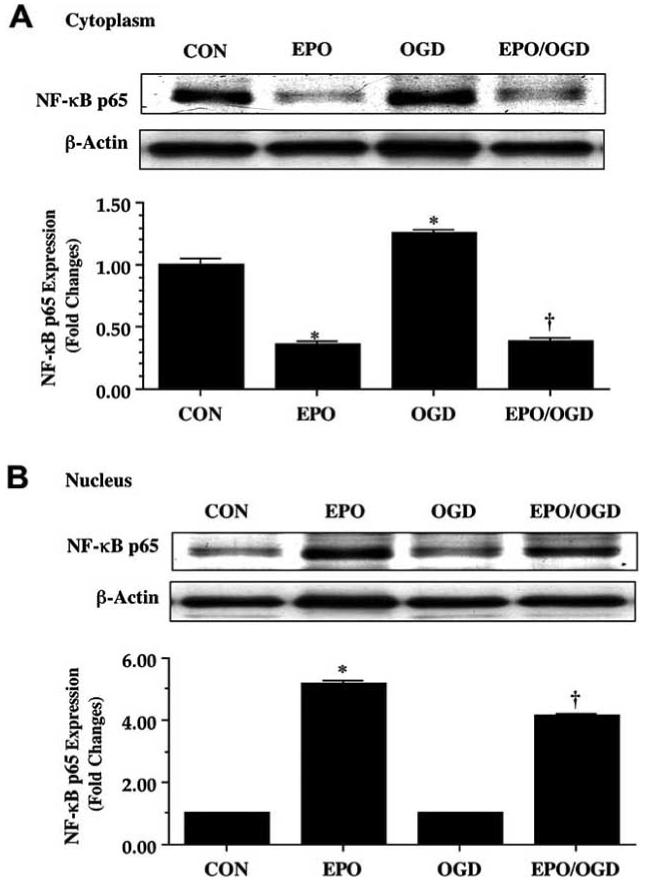
Equal amounts of protein extracts (50 μg/lane) of the cytoplasm (A) and the nucleus (B) were immunoblotted with anti NF-κB p65 antibody 6 hours following a 6 hour period of OGD alone, an application of EPO (10 ng/mL), or administration of EPO (10 ng/mL) 1 hour prior to OGD exposure. Exposure to OGD led to the retention of NF-κB p65 in the cytoplasm of microglia (A), but EPO (10ng/mL) alone or during OGD facilitated the maintenance NF-κB p65 in the nucleus of microglia (B) (*P<0.01 vs. CON; †P< 0.01 vs. OGD). Intensity of NF-κB p65 nuclear staining was performed using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image). In all cases, CON = untreated control cultures.
DISCUSSION
As a cytoprotectant against toxic insults, EPO supports the survival of a number of cell systems that include the brain and cardiovascular system. In neuronal or retinal cells, EPO can prevent injury from glutamate excitotoxicity (Yamasaki, et al., 2005), free radical exposure (Chong, et al., 2003a, Chong, et al., 2003c, Yamasaki, et al., 2005), ischemia (Chong, et al., 2003b, Chong, et al., 2002a, Liu, et al., 2006, Meloni, et al., 2006, Wei, et al., 2006, Yu, et al., 2005), amyloid toxicity (Chong, et al., 2005c), and dopaminergic cell injury (Demers, et al., 2005, Signore, et al., 2006). EPO also has an important role in the cardiac and vascular systems through the maintenance of endothelial cell function (Chong, et al., 2003a, Chong, et al., 2002a, van der Meer, et al., 2004), modulation of cerebral vasospasm (Santhanam, et al., 2005), the induction of angiogenesis (George, et al., 2005, Maiese, et al., 2005b, Sakamaki, 2004), the reduction in myocardial injury (Bullard, et al., 2005, Parsa, et al., 2003), and the promotion of cardiac remodeling (Miki, et al., 2005).
Yet, of equal importance to the functional preservation of neuronal and vascular cells is the role of EPO during cellular inflammation. EPO can reduce cytokine gene expression in endothelial cells exposed to tumor necrosis factor (Avasarala and Konduru, 2005), decrease cytokine production during experimental autoimmune encephalomyelitis (Savino, et al., 2006), and block primary microglial activation and proliferation during oxidative stress (Chong, et al., 2003b, Chong, et al., 2005c). In addition, EPO may foster the preservation of microglial cells for neuronal and vascular restructuring following cerebral injury (Bernaudin, et al., 1999, Chong, et al., 2002b, Maiese, et al., 2004) by preventing apoptotic injury in microglia (Vairano, et al., 2002). In regards to the capacity of EPO to maintain microglial cellular integrity, we demonstrate that EPO alone is not toxic to microglia in a concentration range of 0.001 - 1000 ng/L. Application of EPO from 0.1 - 100.0 ng/L with either 1 hour pre-treatment or within 4 hours post-treatment significantly increases microglial cell survival during OGD. Maximum survival was observed at 10.0 ng/L, but concentrations of EPO less than 0.1 ng/L or greater than 100 ng/L did not enhance neuronal survival during OGD and may reflect a potential toxic side of EPO during cellular injury (Maiese, et al., 2005b, Walrafen, et al., 2004). It also should be noted that EPO retains its capacity to prevent early apoptotic injury with membrane PS externalization as well as later stages of apoptotic injury involving DNA fragmentation in microglia similar to other cell systems of neurovascular origin (Chong, et al., 2003b, Chong, et al., 2002a, Chong, et al., 2005c, Parsa, et al., 2003, Sharples, et al., 2004).
Although EPO can rely on a variety of intracellular signals to foster cell proliferation and survival (Kilic, et al., 2005, Menon, et al., 2006a, Menon, et al., 2006b), cytoprotection by EPO in microglial cells is determined by a series of signal transduction pathways that are initiated by Akt1. Pathways that involve Akt1 enhance cell survival during oxidative stress (Kang, et al., 2003a, Kang, et al., 2003b, Um and Lodish, 2006), free radical exposure (Chong, et al., 2004, Wang and MacNaughton, 2005), neoplastic growth (Hardee, et al., 2006), and amyloid exposure (Chong, et al., 2005c, Du, et al., 2004, Martin, et al., 2001, Nakagami, et al., 2002). In addition, the Akt pathway has been associated with the maintenance of microglial cell proliferation (Ito, et al., 2005, Kim, et al., 2004, Suh, et al., 2005). We show that EPO can independently increase phosphorylation of Akt1 to a significantly greater degree than OGD alone. Yet, application of the PI 3-K inhibitor wortmannin or the specific Akt1 inhibitor SH-6 significantly blocked the ability of EPO to phosphorylate Akt1 and resulted in increased microglial cell injury and genomic DNA fragmentation during OGD (Fig. 4), suggesting that EPO depends upon Akt1 activation to protect microglial cells during OGD. In addition, both wortmannin and SH-6 alone slightly worsened microglial survival and DNA fragmentation (Fig. 4), illustrating that endogenous Akt1 activation in microglial cells during OGD may afford some protection during cellular insults. Loss of microglial cell protection with EPO during the gene silencing of Akt1 further supports our observations that Akt1 is necessary for EPO to protect against apoptosis during OGD (Fig. 5).
Downstream of Akt1 activation is the modulation and phosphorylation of GSK-3β (Chong, et al., 2005b, Chong, et al., 2005e). Inhibition of GSK-3β activity through its phosphorylation can prevent apoptotic cell death (Crowder and Freeman, 2000, Papkoff and Aikawa, 1998) and has been associated with disorders of the nervous system (Chong, et al., 2005d, Phiel, et al., 2003, Qin, et al., 2006). We show that GSK-3β becomes phosphorylated 12 hours after OGD exposure, but that the presence of EPO alone or during OGD significantly increases the phosphorylation of GSK-3β to a greater degree than OGD alone (Fig. 6). Furthermore, inhibition of GSK-3β activity by application of either SB216763 (SB21) or SB415286 (SB41) alone or in combination with EPO during OGD significantly and to the same degree prevents microglial cell injury and apoptotic DNA fragmentation (Fig. 7), suggesting that EPO uses inhibition of GSK-3β activity since co-application with GSK-3β inhibitors does not enhance cytoprotection by EPO in a synergistic fashion.
The integrity and ability of β-catenin to support an “anti-apoptotic” program is directly linked to whether the activation of GSK-3β and its ability to phosphorylate β-catenin that result in the ubiquitination and subsequent degradation of β-catenin is prevented (Chen, et al., 2001, Li, et al., 2006, Papkoff and Aikawa, 1998, You, et al., 2004). Given the dependence of EPO on the inhibition of GSK-3β activity, we assessed the ability of EPO to control β-catenin phosphorylation and its intracellular trafficking during OGD. We show that phosphorylation of β-catenin is increased during OGD, but EPO alone or during OGD prevents phosphorylation of β-catenin 12 hours following OGD (Fig. 8). The capacity of EPO to control the phosphorylation β-catenin and prevent its subsequent destruction is linked to Akt1, since gene silencing of Akt1 expression during EPO application results in the significant phosphorylation of β-catenin during OGD. Furthermore, we illustrate that OGD alone maintains β-catenin in the cytoplasm of microglia, but EPO prevents this retention in the cytoplasm and allows the translocation of β-catenin from the cytoplasm to the nucleus (Fig. 9).
In addition to centralized pathways such as Akt1, cytoprotection by EPO has been tied to the expression and nuclear translocation of NF-κB that occurs in erythroid progenitor cells (Sae-Ung, et al., 2005) and is responsible for the removal of reactive oxygen species (Nakata, et al., 2004), the blockade of apoptosis (Bittorf, et al., 2001, Sae-Ung, et al., 2005), and the prevention of amyloid toxicity (Chong, et al., 2005c). Although not all studies support a cytoprotective role for NF-κB including those that study EPO (Matsushita, et al., 2000, Xu, et al., 2005), some injury models suggest that NF-κB may be necessary for reparative processes in the brain that depend upon microglial cells (Sanz, et al., 2002). We have further clarified a potential role for NF-κB in microglial cells during the application of EPO by illustrating that EPO maintains the expression and integrity of NF-κB p65 in the nucleus of microglia at 6 hours following OGD. Without administration of EPO, OGD results in the loss of NF-κB p65 expression in the nucleus of microglia (Fig. 10). In addition, EPO leads to the translocation of NF-κB p65 from the cytoplasm to the nucleus consistent with prior work that demonstrates the requirement for subcellular translocation for NF-κB p65 to activate its “anti-apoptotic program” (Chong, et al., 2005c, Sae-Ung, et al., 2005).
Under conditions where microglial cell proliferation and survival may form an important inflammatory cell counter-part for tissue repair during neuronal and vascular cell injury, we show that EPO can play a critical role to insure cellular integrity and prevent apoptotic injury in microglial cells. Similar to cellular systems with neurons or vascular cells, EPO increased microglial cell survival and prevented both early apoptotic programs with membrane PS exposure and late apoptotic injury involving genomic DNA degradation during oxidative stress associated with OGD. Promotion of microglial cell survival during OGD by EPO is coupled to the presence and activation of Akt1 and closely integrated with the inhibition of GSK-3β activity and the intracellular trafficking of β-catenin and NF-κB p65. Future investigations that continue to tease apart the conditions and controls for fostering microglial survival to promote neuronal and vascular function through cellular mediators as EPO may further the development of strategies against a variety of disorders in the nervous and vascular systems.
ACKNOWLEDGEMENTS
This research was supported by the following grants (KM): American Diabetes Association, American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
REFERENCES
- Avasarala JR, Konduru SS. Recombinant erythropoietin down-regulates IL-6 and CXCR4 genes in TNF-alpha-treated primary cultures of human microvascular endothelial cells: implications for multiple sclerosis. J Mol Neurosci. 2005;25:183–9. doi: 10.1385/JMN:25:2:183. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–25. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, Petit E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:643–51. doi: 10.1097/00004647-199906000-00007. [DOI] [PubMed] [Google Scholar]
- Bittorf T, Buchse T, Sasse T, Jaster R, Brock J. Activation of the transcription factor NF-kappaB by the erythropoietin receptor: structural requirements and biological significance. Cell Signal. 2001;13:673–81. doi: 10.1016/s0898-6568(01)00189-9. [DOI] [PubMed] [Google Scholar]
- Bullard AJ, Govewalla P, Yellon DM. Erythropoietin protects the myocardium against reperfusion injury in vitro and in vivo. Basic Res Cardiol. 2005;100:397–403. doi: 10.1007/s00395-005-0537-4. [DOI] [PubMed] [Google Scholar]
- Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000;20:2687–95. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J Cell Biol. 2001;152:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang J, Li F, Maiese K. mGluRI Targets Microglial Activation and Selectively Prevents Neuronal Cell Engulfment Through Akt and Caspase Dependent Pathways. Curr Neurovasc Res. 2005a;2:197–211. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp Cell Res. 2004;296:196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J Cereb Blood Flow Metab. 2003a;23:320–30. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol. 2003b;138:1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002a;106:2973–9. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Hematopoietic factor erythropoietin fosters neuroprotection through novel signal transduction cascades. J Cereb Blood Flow Metab. 2002b;22:503–14. doi: 10.1097/00004647-200205000-00001. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005b;20:299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr Neurovasc Res. 2005c;2:387–99. doi: 10.2174/156720205774962683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005d;75:207–46. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Stress in the brain: novel cellular mechanisms of injury linked to Alzheimer’s disease. Brain Res Brain Res Rev. 2005e;49:1–21. doi: 10.1016/j.brainresrev.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li FQ, Maiese K. Employing new cellular therapeutic targets for Alzheimer’s disease: A change for the better? Curr Neurovasc Res. 2005f;2:55–72. doi: 10.2174/1567202052773508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Kang JQ, Maiese K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res. 2003c;71:659–69. doi: 10.1002/jnr.10528. [DOI] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–88. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Glycogen synthase kinase-3 beta activity is critical for neuronal death caused by inhibiting phosphatidylinositol 3-kinase or Akt but not for death caused by nerve growth factor withdrawal. J Biol Chem. 2000;275:34266–71. doi: 10.1074/jbc.M006160200. [DOI] [PubMed] [Google Scholar]
- Demers EJ, McPherson RJ, Juul SE. Erythropoietin protects dopaminergic neurons and improves neurobehavioral outcomes in juvenile rats after neonatal hypoxia-ischemia. Pediatr Res. 2005;58:297–301. doi: 10.1203/01.PDR.0000169971.64558.5A. [DOI] [PubMed] [Google Scholar]
- Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal. 2005;7:1223–33. doi: 10.1089/ars.2005.7.1223. [DOI] [PubMed] [Google Scholar]
- Du B, Ohmichi M, Takahashi K, Kawagoe J, Ohshima C, Igarashi H, Mori-Abe A, Saitoh M, Ohta T, Ohishi A, Doshida M, Tezuka N, Takahashi T, Kurachi H. Both estrogen and raloxifene protect against beta-amyloid-induced neurotoxicity in estrogen receptor alpha-transfected PC12 cells by activation of telomerase activity via Akt cascade. J Endocrinol. 2004;183:605–15. doi: 10.1677/joe.1.05775. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Argast GM, Foord O, Fischer EH, Krebs EG. Expression and characterization of glycogen synthase kinase-3 mutants and their effect on glycogen synthase activity in intact cells. Proc Natl Acad Sci USA. 1996;93:10228–33. doi: 10.1073/pnas.93.19.10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George J, Goldstein E, Abashidze A, Wexler D, Hamed S, Shmilovich H, Deutsch V, Miller H, Keren G, Roth A. Erythropoietin promotes endothelial progenitor cell proliferative and adhesive properties in a PI 3-kinase-dependent manner. Cardiovasc Res. 2005;68:299–306. doi: 10.1016/j.cardiores.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Ghezzi P, Brines M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ. 2004;11(Suppl 1):S37–44. doi: 10.1038/sj.cdd.4401450. [DOI] [PubMed] [Google Scholar]
- Hardee ME, Rabbani ZN, Arcasoy MO, Kirkpatrick JP, Vujaskovic Z, Dewhirst MW, Blackwell KL. Erythropoietin inhibits apoptosis in breast cancer cells via an Akt-dependent pathway without modulating in vivo chemosensitivity. Mol Cancer Ther. 2006;5:356–61. doi: 10.1158/1535-7163.MCT-05-0196. [DOI] [PubMed] [Google Scholar]
- Ito S, Sawada M, Haneda M, Fujii S, Oh-Hashi K, Kiuchi K, Takahashi M, Isobe K. Amyloid-beta peptides induce cell proliferation and macrophage colony-stimulating factor expression via the PI3-kinase/Akt pathway in cultured Ra2 microglial cells. FEBS Lett. 2005;579:1995–2000. doi: 10.1016/j.febslet.2005.02.048. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003a;74:37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003b;64:557–69. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005;19:2026–8. doi: 10.1096/fj.05-3941fje. [DOI] [PubMed] [Google Scholar]
- Kim WK, Hwang SY, Oh ES, Piao HZ, Kim KW, Han IO. TGF-beta1 represses activation and resultant death of microglia via inhibition of phosphatidylinositol 3-kinase activity. J Immunol. 2004;172:7015–23. doi: 10.4049/jimmunol.172.11.7015. [DOI] [PubMed] [Google Scholar]
- Kozikowski AP, Sun H, Brognard J, Dennis PA. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J Am Chem Soc. 2003;125:1144–5. doi: 10.1021/ja0285159. [DOI] [PubMed] [Google Scholar]
- Lee SM, Nguyen TH, Park MH, Kim KS, Cho KJ, Moon DC, Kim HY, Yoon do Y, Hong JT. EPO receptor-mediated ERK kinase and NF-kappaB activation in erythropoietin-promoted differentiation of astrocytes. Biochem Biophys Res Commun. 2004;320:1087–95. doi: 10.1016/j.bbrc.2004.06.060. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Winding through the Wnt pathway during cellular development and demise. Histol Histopathol. 2006;21:103–124. doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–45. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Suzuki A, Guo Z, Mizuno Y, Urabe T. Intrinsic and extrinsic erythropoietin enhances neuroprotection against ischemia and reperfusion injury in vitro. J Neurochem. 2006;96:1101–10. doi: 10.1111/j.1471-4159.2005.03597.x. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Li F. Driving cellular plasticity and survival through the signal transduction pathways of metabotropic glutamate receptors. Curr Neurovasc Res. 2005a;2:425–46. doi: 10.2174/156720205774962692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. Erythropoietin in the brain: can the promise to protect be fulfilled? Trends Pharmacol Sci. 2004;25:577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005b;293:90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Abeta(25-35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem. 2001;78:1000–8. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- Matsushita H, Morishita R, Nata T, Aoki M, Nakagami H, Taniyama Y, Yamamoto K, Higaki J, Yasufumi K, Ogihara T. Hypoxia-induced endothelial apoptosis through nuclear factor-kappaB (NF-kappaB)-mediated bcl-2 suppression: in vivo evidence of the importance of NF-kappaB in endothelial cell regulation. Circ Res. 2000;86:974–81. doi: 10.1161/01.res.86.9.974. [DOI] [PubMed] [Google Scholar]
- Meloni BP, Tilbrook PA, Boulos S, Arthur PG, Knuckey NW. Erythropoietin preconditioning in neuronal cultures: signaling, protection from in vitro ischemia, and proteomic analysis. J Neurosci Res. 2006;83:584–93. doi: 10.1002/jnr.20755. [DOI] [PubMed] [Google Scholar]
- Menon MP, Fang J, Wojchowski DM. Core erythropoietin receptor signals for late erythroblast development. Blood. 2006a;107:2662–72. doi: 10.1182/blood-2005-02-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon MP, Karur V, Bogacheva O, Bogachev O, Cuetara B, Wojchowski DM. Signals for stress erythropoiesis are integrated via an erythropoietin receptor-phosphotyrosine-343-Stat5 axis. J Clin Invest. 2006b;116:683–94. doi: 10.1172/JCI25227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Miura T, Yano T, Takahashi A, Sakamoto J, Tanno M, Kobayashi H, Ikeda Y, Nishihara M, Naitoh K, Ohori K, Shimamoto K. Alteration in erythropoietin-induced cardioprotective signaling by post-infarct ventricular remodeling. J Pharmacol Exp Ther. 2006;317(1):68–75. doi: 10.1124/jpet.105.095745. [DOI] [PubMed] [Google Scholar]
- Nakagami Y, Nishimura S, Murasugi T, Kubo T, Kaneko I, Meguro M, Marumoto S, Kogen H, Koyama K, Oda T. A novel compound RS-0466 reverses beta-amyloid-induced cytotoxicity through the Akt signaling pathway in vitro. Eur J Pharmacol. 2002;457:11–7. doi: 10.1016/s0014-2999(02)02657-2. [DOI] [PubMed] [Google Scholar]
- Nakata S, Matsumura I, Tanaka H, Ezoe S, Satoh Y, Ishikawa J, Era T, Kanakura Y. NF-kappaB family proteins participate in multiple steps of hematopoiesis through elimination of reactive oxygen species. J Biol Chem. 2004;279:55578–86. doi: 10.1074/jbc.M408238200. [DOI] [PubMed] [Google Scholar]
- Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. 2006;20:714–6. doi: 10.1096/fj.05-4882fje. [DOI] [PubMed] [Google Scholar]
- Papkoff J, Aikawa M. WNT-1 and HGF regulate GSK3 beta activity and beta-catenin signaling in mammary epithelial cells. Biochem Biophys Res Commun. 1998;247:851–8. doi: 10.1006/bbrc.1998.8888. [DOI] [PubMed] [Google Scholar]
- Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB, Thompson RB, Petrofski JA, Annex BH, Stamler JS, Koch WJ. A novel protective effect of erythropoietin in the infarcted heart. J Clin Invest. 2003;112:999–1007. doi: 10.1172/JCI18200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–9. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Qin W, Peng Y, Ksiezak-Reding H, Ho L, Stetka B, Lovati E, Pasinetti GM. Inhibition of cyclooxygenase as potential novel therapeutic strategy in N141I presenilin-2 familial Alzheimer’s disease. Mol Psychiatry. 2006;11:172–81. doi: 10.1038/sj.mp.4001773. [DOI] [PubMed] [Google Scholar]
- Sae-Ung N, Matsushima T, Choi I, Abe Y, Winichagoon P, Fucharoen S, Nawata H, Muta K. Role of NF-kappa B in regulation of apoptosis of erythroid progenitor cells. Eur J Haematol. 2005;74:315–23. doi: 10.1111/j.1600-0609.2004.00400.x. [DOI] [PubMed] [Google Scholar]
- Sakamaki K. Regulation of endothelial cell death and its role in angiogenesis and vascular regression. Curr Neurovasc Res. 2004;1:305–315. doi: 10.2174/1567202043362072. [DOI] [PubMed] [Google Scholar]
- Sankarapandi S, Zweier JL, Mukherjee G, Quinn MT, Huso DL. Measurement and characterization of superoxide generation in microglial cells: evidence for an NADPH oxidase-dependent pathway. Arch Biochem Biophys. 1998;353:312–21. doi: 10.1006/abbi.1998.0658. [DOI] [PubMed] [Google Scholar]
- Santhanam AV, Smith LA, Akiyama M, Rosales AG, Bailey KR, Katusic ZS. Role of endothelial NO synthase phosphorylation in cerebrovascular protective effect of recombinant erythropoietin during subarachnoid hemorrhage-induced cerebral vasospasm. Stroke. 2005;36:2731–7. doi: 10.1161/01.STR.0000190021.85035.5b. [DOI] [PubMed] [Google Scholar]
- Sanz O, Acarin L, Gonzalez B, Castellano B. NF-kappaB and IkappaBalpha expression following traumatic brain injury to the immature rat brain. J Neurosci Res. 2002;67:772–80. doi: 10.1002/jnr.10140. [DOI] [PubMed] [Google Scholar]
- Savino C, Pedotti R, Baggi F, Ubiali F, Gallo B, Nava S, Bigini P, Barbera S, Fumagalli E, Mennini T, Vezzani A, Rizzi M, Coleman T, Cerami A, Brines M, Ghezzi P, Bianchi R. Delayed administration of erythropoietin and its non-erythropoietic derivatives ameliorates chronic murine autoimmune encephalomyelitis. J Neuroimmunol. 2006;172:27–37. doi: 10.1016/j.jneuroim.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 1991;10:3805–17. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples EJ, Patel N, Brown P, Stewart K, Mota-Philipe H, Sheaff M, Kieswich J, Allen D, Harwood S, Raftery M, Thiemermann C, Yaqoob MM. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J Am Soc Nephrol. 2004;15:2115–24. doi: 10.1097/01.ASN.0000135059.67385.5D. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol (Berl) 1997;94:1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- Shingo T, Sorokan ST, Shimazaki T, Weiss S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci. 2001;21:9733–43. doi: 10.1523/JNEUROSCI.21-24-09733.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signore AP, Weng Z, Hastings T, Van Laar AD, Liang Q, Lee YJ, Chen J. Erythropoietin protects against 6-hydroxydopamine-induced dopaminergic cell death. J Neurochem. 2006;96:428–43. doi: 10.1111/j.1471-4159.2005.03587.x. [DOI] [PubMed] [Google Scholar]
- Suh HS, Kim MO, Lee SC. Inhibition of granulocyte-macrophage colony-stimulating factor signaling and microglial proliferation by anti-CD45RO: role of Hck tyrosine kinase and phosphatidylinositol 3-kinase/Akt. J Immunol. 2005;174:2712–9. doi: 10.4049/jimmunol.174.5.2712. [DOI] [PubMed] [Google Scholar]
- Um M, Lodish HF. Antiapoptotic Effects of Erythropoietin in Differentiated Neuroblastoma SH-SY5Y Cells Require Activation of Both the STAT5 and AKT Signaling Pathways. J Biol Chem. 2006;281:5648–56. doi: 10.1074/jbc.M510943200. [DOI] [PubMed] [Google Scholar]
- Vairano M, Dello Russo C, Pozzoli G, Battaglia A, Scambia G, Tringali G, Aloe-Spiriti MA, Preziosi P, Navarra P. Erythropoietin exerts anti-apoptotic effects on rat microglial cells in vitro. Eur J Neurosci. 2002;16:584–92. doi: 10.1046/j.1460-9568.2002.02125.x. [DOI] [PubMed] [Google Scholar]
- van der Meer P, Lipsic E, Henning RH, de Boer RA, Suurmeijer AJ, van Veldhuisen DJ, van Gilst WH. Erythropoietin improves left ventricular function and coronary flow in an experimental model of ischemia-reperfusion injury. Eur J Heart Fail. 2004;6:853–9. doi: 10.1016/j.ejheart.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Walrafen P, Verdier F, Kadri Z, Chretien S, Lacombe C, Mayeux P. Both proteasomes and lysosomes degrade the activated erythropoietin receptor. Blood. 2004;105(2):600–8. doi: 10.1182/blood-2004-03-1216. [DOI] [PubMed] [Google Scholar]
- Wang H, MacNaughton WK. Overexpressed beta-catenin blocks nitric oxide-induced apoptosis in colonic cancer cells. Cancer Res. 2005;65:8604–7. doi: 10.1158/0008-5472.CAN-05-1169. [DOI] [PubMed] [Google Scholar]
- Wang Z, Vogelstein B, Kinzler KW. Phosphorylation of beta-catenin at S33, S37, or T41 can occur in the absence of phosphorylation at T45 in colon cancer cells. Cancer Res. 2003;63:5234–5. [PubMed] [Google Scholar]
- Wei L, Han BH, Li Y, Keogh CL, Holtzman DM, Yu SP. Cell death mechanism and protective effect of erythropoietin after focal ischemia in the whisker-barrel cortex of neonatal rats. J Pharmacol Exp Ther. 2006;317:109–16. doi: 10.1124/jpet.105.094391. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996;16:1722–33. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Dong GH, Liu H, Wang YQ, Wu HW, Jing H. Recombinant human erythropoietin pretreatment attenuates myocardial infarct size: a possible mechanism involves heat shock Protein 70 and attenuation of nuclear factor-kappaB. Ann Clin Lab Sci. 2005;35:161–8. [PubMed] [Google Scholar]
- Yamasaki M, Mishima HK, Yamashita H, Kashiwagi K, Murata K, Minamoto A, Inaba T. Neuroprotective effects of erythropoietin on glutamate and nitric oxide toxicity in primary cultured retinal ganglion cells. Brain Res. 2005;1050:15–26. doi: 10.1016/j.brainres.2005.05.037. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–49. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- You L, He B, Uematsu K, Xu Z, Mazieres J, Lee A, McCormick F, Jablons DM. Inhibition of Wnt-1 signaling induces apoptosis in beta-catenin-deficient mesothelioma cells. Cancer Res. 2004;64:3474–8. doi: 10.1158/0008-5472.CAN-04-0115. [DOI] [PubMed] [Google Scholar]
- Yu YP, Xu QQ, Zhang Q, Zhang WP, Zhang LH, Wei EQ. Intranasal recombinant human erythropoietin protects rats against focal cerebral ischemia. Neurosci Lett. 2005;387:5–10. doi: 10.1016/j.neulet.2005.07.008. [DOI] [PubMed] [Google Scholar]