

Abstract
Understanding the role of nicotinamide (NIC) in different cell systems represents a significant challenge in several respects. Recently, NIC has been reported to have diverse roles during cell biology. In the absence of NIC, sirtuin protein activity is enhanced and pyrazinamidase/nicotinamidase 1 (PNC1) expression, an enzyme that deaminates NIC to convert NIC into nicotinic acid, is increased to lead to lifespan extension during calorie restriction, at least in yeast. Yet, NIC may be critical for cell survival as well as the modulation of inflammatory injury during both experimental models as well as in clinical studies. We therefore investigated some of the underlying signal transduction pathways that could be critical for the determination of the neuroprotective properties of NIC. We examined neuronal injury by trypan blue exclusion, DNA fragmentation, phosphatidylserine (PS) exposure, Akt1 phosphorylation, Bad phosphorylation, mitochondrial membrane potential, caspase activity, cleavage of poly(ADP-ribose) polymerase (PARP), and mitogen-activated protein kinases (MAPKs) phosphorylation. Application of NIC (12.5 mM) significantly increased neuronal survival from 38 ± 3% of anoxia treated alone to 68 ± 3%, decreased DNA fragmentation and membrane PS exposure from 67 ± 4% and 61 ± 5% of anoxia treated alone to 30 ± 4% and 26 ± 4% respectively. We further demonstrate that NIC functions through Akt1 activation, Bad phosphorylation, and the downstream modulation of mitochrondrial membrane potential, cytochrome c release, caspase 1, 3, and 8 - like activities, and PARP integrity to prevent genomic DNA degradation and PS externalization during anoxia. Yet, NIC does not alter the activity of either the MAPKs p38 or JNK, suggesting that protection by NIC during anoxia is independent of the p38 and JNK pathways. Additional investigations targeted to elucidate the cellular pathways responsible for the ability of NIC to modulate both lifespan extension and cytoprotection may offer critical insight for the development of new therapies for nervous system disorders.
Keywords: Apoptosis, caspase, Bad, Cytochrome c, Phosphatidylserine, MAP kinase, poly(ADP-ribose) polymerase (PARP), p38, JNK, Sir2
INTRODUCTION
Nicotinamide (NIC) is the precursor for the coenzyme ß-nicotinamide adenine dinucleotide (NAD+) (Li, F et al., 2004, Maiese, K and Chong, ZZ, 2003) and is utilized by the body for cellular metabolism through the generation of adenosine triphosphate in the mitochondrial electron transport chain (Ishaque, A and Al-Rubeai, M, 2002). Recently, NIC has been reported to have diverse roles during cell biology that involve potential lifespan reduction as well as enhanced cell survival during acute cellular injury (Li, F et al., 2004, Porcu, M and Chiarugi, A, 2005). In regards to cellular lifespan, increased longevity in yeast has been shown to be dependent upon sirtuin 2 (Sir2) protein expression and is associated with NIC and pyrazinamidase/nicotinamidase 1 (PNC1), an enzyme that deaminates NIC to convert NIC into nicotinic acid. Without NIC, Sir2 is activated and PNC1 expression is increased to lead to yeast lifespan extension during calorie restriction.
Yet, NIC may be critical for cell survival as well as the modulation of inflammatory injury (Chong, ZZ et al., 2005, Chong, ZZ et al., 2002, Chong, ZZ et al., 2004, Lin, SH et al., 2001, Lin, SH et al., 2000). For example, in pancreatic islet cells, NIC prevents cellular injury during free radical exposure (Kallmann, B et al., 1992) and blocks hydrogen peroxide induced necrosis in human β-cells (O’Brien, BA et al., 2000). Administration of NIC in non-obese diabetic mice also prevents apoptosis in β-cells during cyclophosphamide injections and delays the development of diabetes (O’Brien, BA et al., 2000). In neuronal and vascular cells, NIC has been demonstrated to protect cells against apoptosis induced by deoxycholate (Crowley, CL et al., 2000), free radical nitric oxide (NO) exposure (Chong, ZZ et al., 2002, Lin, SH et al., 2001, Lin, SH et al., 2000), and oxygen-glucose deprivation (Chong, ZZ et al., 2004). Clinical studies also support a role for NIC in the treatment of a variety of disorders, such as the resolution of lactic acidemia in the MELAS syndrome (Majamaa, K et al., 1997) with NADH:ubiquinone oxidoreductase (Smeitink, J et al., 2004).
Given the possible detrimental effects of NIC observed in the lifespan of yeast and adult metazoans (Porcu, M et al., 2005) that contrasts with the robust capacity of NIC to enhance cell survival in higher eukaryotic cells, we elected to elucidate some of the cellular pathways that would promote cytoprotection by NIC. In this regard, the serine/threonine protein kinase B (also known as Akt), a key target of phosphatidylinositide-3-kinase (PI 3-K) and the modulation of anti-apoptotic pathways (Chong, ZZ et al., 2005, Chong, ZZ et al., 2005), may be central to the cytoprotection by NIC, since activation of Akt occurs in the brain following either focal or global cerebral ischemia (Friguls, B et al., 2001, Yano, S et al., 2001), hypoxic preconditioning may be mediated by the activation of Akt (Wick, A et al., 2002), and loss of Akt activity leads to cell injury during oxidative stress (Chong, ZZ et al., 2004, Kang, JQ et al., 2003, Kang, JQ et al., 2003).
Akt may block neuronal apoptosis through the phosphorylation of Bad, modulation of mitochondrial permeability, and prevention of mitogen-activated protein kinases (MAPKs) and caspases. During periods of hypoxia, activation of platelet-derived growth factor β in the dorsocaudal brain stem also has been shown to block apoptosis through Akt activation and the subsequent phosphorylation of Bad (Simakajornboon, N et al., 2001). In the vascular system, the protection of cells during growth factor deprivation or oxidative stress can be fostered by activation of Akt and the direct inactivation of Bad (Chen, JH et al., 2004, Chong, ZZ et al., 2003). Furthermore, Akt may control mitochondrial permeability to block apoptosis (Yamaguchi, A et al., 2001), prevent the release of cytochrome c (Chong, ZZ et al., 2003, Kang, JQ et al., 2003), and inhibit the activation of MAPKs and caspases (Zhuang, S et al., 2000). Potentially closely linked to caspase activity and the protective ability of NIC is the maintenance of poly(ADP-ribose) polymerase (PARP) integrity and the preservation of cellular energy reserves, since NIC has been shown to prevent PARP degradation and allow for DNA repair through the direct inhibition of caspase 3 - like activity (Chong, ZZ et al., 2002, Lin, SH et al., 2000). We demonstrate that NIC protects against anoxic neuronal DNA degradation and membrane PS exposure through a series of pathways that require activation of Akt1, phosphorylation of Bad that is Akt dependent, prevention of mitochondrial permeability and subsequent cytochrome c release, and maintenance of PARP integrity. Yet, protection against anoxia injury is independent of MAPK p38 and JNK activity.
MATERIALS AND METHODS
Primary Hippocampal Neuronal Cultures
The hippocampi were obtained from E-19 Sprague-Dawley rat pups and incubated in dissociation medium (90 mM Na2SO4, 30 mM K2SO4, 5.8 mM MgCl2, 0.25 mM CaCl2, 10 mM kynurenic acid, and 1 mM HEPES with the pH adjusted to 7.4) containing papain (10 U/ml) and cysteine (3 mM) for two 20-minute periods. The hippocampi were then rinsed in dissociation medium and incubated in dissociation medium containing trypsin inhibitor (10-20 U/ml) for three 5-minute periods. The cells were washed in growth medium (Leibovitz’s L-15 medium, Invitrogen, Carlsbad, CA) containing 6% sterile rat serum (ICN, Aurora, OH), 150 mM NaHCO3, 2.25 mg/ml of transferrin, 2.5 μg/ml of insulin, 10 nM progesterone, 90 μM putrescine, 15 nM selenium, 35 mM glucose, 1 mM L-glutamine, penicillin and streptomycin (50 μg/ml), and vitamins. The dissociated cells were plated at a density of ∼1.5 ×103 cells/mm2 in 35 mm polylysine/laminin-coated plates (Falcon Labware, Lincoln Park, NJ). Neurons were maintained in growth medium at 37 °C in a humidified atmosphere of 5% CO2 and 95% room air for 2 weeks. Non-neuronal cells were negligible.
Experimental Treatments
To induce anoxia, neuronal cultures were deprived of oxygen by placing them into an anoxic chamber system (Sheldon Manufacturing, Cornelius, OR). The cultures were maintained in an anaerobic environment (95% N2, 5% CO2) at 37 °C per the experimental paradigms. In both pre- and postparadigm applications, NIC exposure was continuous.
To inhibit Akt activation (phosphorylation), PI3K inhibitor wortmannin or LY 294002 (Calbiochem, La Jolla, CA) and Akt selective inhibitor D-3-deoxy-2-O-methyl-myo inositol 1-(R)-2-methoxy-3-(octadecyloxy) propyl hydrogen phosphate (SH-5) or D-2,3-dieoxy-myo inositol 1-(R)-2-methoxy-3-(octadecyloxy) propyl hydrogen phosphate (SH-6) (Alexis, San Diego, CA) were applied to neuronal cultures 1 h (hour) prior to anoxia. To inhibit caspase activity, the irreversible and cell permeable caspase inhibitors Z-IETD-FMK, Z-YVAD-FMK, and Z-DEVD-FMK (all from Pharmingen Inc, Livermore, CA) were used. Inhibitors were added directly to the culture media 1 h prior to anoxic exposure.
Assessment of Neuronal Survival
Hippocampal neuronal injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 h following anoxia exposure per our previous protocols (Lin, SH et al., 2000). Neurons were identified by morphology and the mean survival was determined by counting eight randomly selected non-overlapping fields with each containing approximately 10-20 neurons (viable + non-viable) in each 35 mm Petri dish.
Assessment of DNA Fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay (Lin, SH et al., 2000, Maiese, K and Vincent, AM, 2000). Briefly, neurons were fixed in 4% para-formaldehyde/0.2% picric acid/0.05% glutaraldehyde and the 3′-hydroxy ends of cut DNA were labeled with biotinylated dUTP using the enzyme terminal deoxytransferase (Promega, Madison, WI) followed by streptavidin-peroxidase and visualized with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA).
Assessment of Membrane Phosphatidylserine (PS) Residue Externalization
Per our prior protocols (Vincent, AM and Maiese, K, 1999), a 30 μg/ml stock solution of annexin V conjugated to phycoerythrin (PE) (R&D Systems, Minneapolis, MN) was diluted to 3 μg/ml in warmed calcium containing binding buffer (10 mM Hepes, pH 7.5, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mMCaCl2). Plates were incubated with 500 μl of diluted annexin V for 10 min. Images were acquired with “blinded” assessment with a Leitz DMIRB microscope (Leica, McHenry, IL) and a Fuji/Nikon Super CCD (6.1 megapixels) using transmitted light and fluorescent single excitation light at 490 nm and detected emission at 585 nm.
Assessment of Mitochondrial Membrane Potential
The fluorescent probe JC-1 (Molecular Probes, Eugene, OR), a cationic membrane potential indicator, was used to assess the mitochondrial membrane potential. Neurons in 35 mm dishes were incubated with 2 μg/ml JC-1 in growth medium at 37 °C for 30 min. The cultures were washed three times using fresh growth medium. Neurons were then analyzed immediately under a Leitz DMIRB microscope (Leica, McHenry, IL, USA) with a dual emission fluorescence filter with 515-545 nm for green fluorescence and emission at 585-615 nm for red fluorescence.
Assessment of Cysteine Protease Activity
At specific times following anoxia, cysteine protease activities were determined as previously described (Chong, ZZ et al., 2003, Chong, ZZ et al., 2003). Cell suspensions were prepared and an aliquot of supernatant containing 30 μg protein was incubated with a 250 μM colorimetric substrate for caspase 8 (Ac-IETD-pNA), caspase 1 (Ac-YVAD-pNA), or for caspase 3 (Ac-DEVD-pNA) (Calbiochem, San Diego, CA). Absorbance was measured at 405 nm and substrate cleavage reported in micromoles per minute per gram protein (μmol/min/g) against standard p-nitroaniline solutions.
Western Blot Analysis
Akt1, Bad phosphorylation, Cytochrome c Release, p38, p-p38, JNK, and p-JNK
Cells were homogenized and following protein determination, each sample (50 μg/lane) was then subjected to SDS-polyacrylamide gel electrophoresis. The membranes were incubated with a mouse monoclonal antibody against the active form of Akt1 (phospho-Akt1, Ser 473, 1:1000) (Active-Motif, Carlsbad, CA), cytochrome c (1:2000) (Pharmingen, San Diego, CA), a goat polyclonal antibody against phosphorylated Bad (p-Bad, Ser 136, 1:100) (Santa Cruz Biotechnologies, Santa Cruz, CA), primary rabbit polyclonal antibody against p38 or phospho-(p)-p38(1: 2000) (Santa Cruz Biotechnologies, Santa Cruz, CA), rabbit ployclonal antibody against active forms of JNK or phospho(p)-JNK (1: 2000) (Santa Cruz Biotechnologies, Santa Cruz, CA). After washing, the membranes were incubated with a horseradish peroxidase conjugated secondary antibody (goat anti-mouse IgG, 1:2000) (Pierce, Rockford, IL), rabbit anti-goat IgG (1:5000) (Santa Cruz Biotechnologies, Santa Cruz, CA) or goat anti-rabbit IgG (1:15000, Pierce, Rockford, IL). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech).
Preparation of Mitochondria for the Analysis of Cytochrome c Release
Per our prior protocols (Chong, ZZ et al., 2002), after washing once with ice-cold PBS, cells were harvested and resuspended in buffer A (20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 0.1 phenylmethylsulfonylfluoride) containing 250 mM sucrose. The cells were homogenized and then centrifuged twice at 750 g for 10 min at 4 °C. The harvested supernatants were centrifuged at 10,000 g for 10 min and the cytosolic fraction was centrifuged at 50,000 g for 60 min at 4 °C.
PARP Cleavage
Following protein determination, equal protein concentration of cell extracts were added to a 2x sample buffer (62.5 mM Tris-HCl, pH 6.8; 6 M urea, 10% glycerol, 2% SDS, 0.00125% bromophenol blue, 5% β-mercaptoethanol) and incubated at 65 °C for 15 min. Each sample (50 μg/lane) was then subjected to 7.5% SDS-polyacrylamide gel electrophoresis followed by a transfer onto a nitrocellulose membrane with plate electrodes (BioRad, Hercules, CA). The membranes were blocked overnight at 4 °C in 5% nonfat dry milk in Tris-buffered saline (50 mM Tris-HCl pH 7.4, 150 mM NaCl) with 0.1% Tween 20 (TBST) and incubated with rabbit anti-PARP polyclonal antibody (1:1000) for 2.5 h at room temperature. The membranes were incubated with goat anti-rabbit IgG (1:15000, Pierce, Rockford, IL) coupled to horseradish peroxidase. The antibody-reactive bands were revealed by enhanced chemiluminescence (Amersham Pharmacia Biotech).
Statistical Analysis
For each experiment involving assessment of neuronal cell survival, DNA degradation, membrane PS exposure, mitochondrial membrane potential, and caspase activity, the mean and standard error were determined from 4 to 6 replicate experiments. Statistical differences between groups were assessed by means of analysis of variance (ANOVA) with the post-hoc Student’s t-test. Results are expressed as the mean ± the standard error. Statistical significance was considered at P< 0.05.
RESULTS
NIC Protects Neurons Against Anoxia-Induced Neuronal Injury
In order to examine the effect of anoxia on neuronal viability, we determined neuronal survival following different time periods of anoxia. Hippocampal neurons were exposed in an oxygen-free environment for a 2 h, 4 h, and 8 h period and neuronal survival was examined 24 h later following anoxia (Fig. 1A). A significant decrease in neuronal survival was observed in cultures following anoxia for the duration of 2 h (61 ± 4%), 4 h (39 ± 2%), and 8 h (21 ± 3%) when compared to untreated control cultures (86 ± 3%) over a 24 h period following anoxia.
Fig. (1). Nicotinamide (NIC) increases neuronal survival during anoxia.
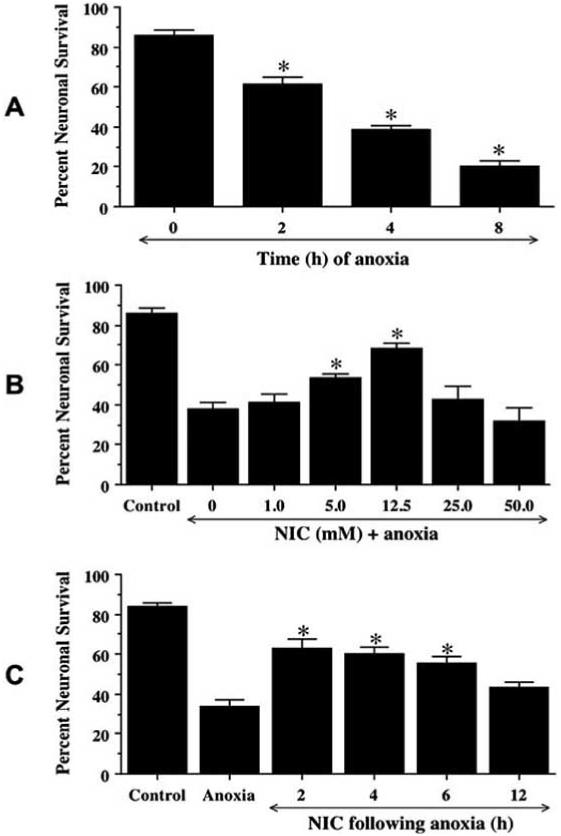
(A) To examine neuronal injury during anoxia, hippocampal neurons were exposed to anoxia for a period of 2, 4, and 8 h and neuronal survival was determined at 24 h following anoxia using a 0.4% trypan blue assay. Neuronal survival was progressively decreased over a 8 h period (*p<0.01 vs. untreated control). (B) NIC was applied directly to culture media 1 h prior to a 4 h period of anoxia at different concentrations from 1.0 to 50.0 mM. A significant increase in neuronal survival was seen in cultures pretreated with 5.0 mM and 12.5 mM of NIC when compared to cultures without NIC application during anoxic exposure (*p<0.01 vs anoxia only). (C) NIC (12.5 mM) was applied to neuronal cultures at 2, 4, 6, and 12 h following a 4 h period of anoxia. Post-treatment with NIC 2, 4, and 6 h following anoxia significantly increased neuronal survival (* p<0.01 vs. anoxia treated only). In A, B, and C, data represent the mean and SEM from 4 to 6 experimental preparations.
To examine the effect of NIC on neuronal viability during anoxia, NIC (1-50 mM) was applied directly to hippocampal cultures 1 h prior to a 4 h period of anoxia and cell survival was examined 24 h following anoxia. As shown in Fig. 1B, anoxic exposure directly decreased neuronal survival from 85 ± 3% in untreated control to 38 ± 3% in cultures exposed to anoxia. Treatment with NIC at concentrations of 5 mM and 12.5 mM significantly increased neuronal survival to 53 ± 2% and 68 ± 3% respectively. Treatment with NIC at concentrations of 1 mM, 25 mM, and 50 mM did not significantly protect neurons from anoxic insult.
To further examine whether NIC can rescue neurons from anoxic injury, cells were treated with 12.5 mM NIC at 2 h, 4 h, 6 h, and 12 h post anoxia, and cell survival was determined 24 h later. As shown in Fig. 1C, post-treatment of NIC at 2, 4, 6 h significantly increased neuronal survival from 34 ± 4% (anoxia only) to 63 ± 4%, 60 ± 4%, and 56 ± 3% respectively. Yet, post-treatment with NIC at 12 h following anoxia did not increase neuronal survival.
NIC Prevents Apoptotic Genomic DNA Fragmentation and Membrane Phosphatidylserine (PS) Externalization During Anoxia
Neurons were exposed to anoxia and either cellular genomic DNA fragmentation was assessed with TUNEL or cell membrane PS exposure was determined by annexin V labeling 24 h later. In Fig. 2A, representative pictures demonstrated that anoxia resulted in both DNA fragmentation and membrane PS externalization in neurons. In cells exposed to anoxia, cell injury is evident by chromatin condensation and significant induction of annexin V labeling. In contrast, pretreatment with NIC (12.5 mM) 1 h prior to anoxia significantly reduced nuclear condensation and membrane PS externalization.
Fig. (2). Nicotinamide (NIC) prevents DNA fragmentation and membrane PS externalization during anoxia.
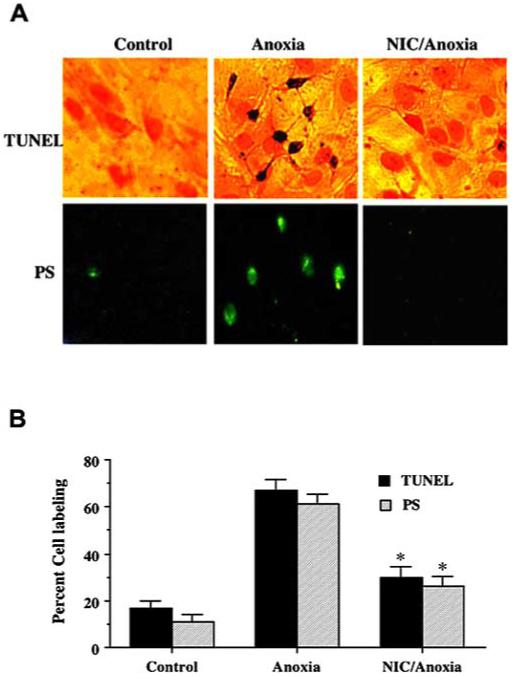
(A), Representative pictures for DNA fragmentation and PS exposure that were determined using the TUNEL assay and annexin V labeling respectively were demonstrated in neurons 24 h following a 4 h period of anoxia. (B) Pretreatment with NIC (12.5 mM) decreased DNA fragmentation and membrane PS externalization significantly during anoxia (*p<0.01 vs. anoxia). In B, data represent the mean and SEM from 4 to 6 experimental preparations. In all cases, control = untreated neurons.
To quantitatively determine the ability of NIC to prevent anoxia induced DNA fragmentation and membrane PS externalization, NIC (12.5 mM) was administered 1 h prior to anoxia and assessment was performed 24 h later. As shown in Fig. 2B, anoxia alone resulted in a significant increase in percent DNA fragmentation (67 ± 4%) in neurons when compared to untreated control cultures (16 ± 3%). DNA fragmentation was reduced to 30 ± 4% in cells treated with NIC following anoxia. An increase in annexin V label was observed in neurons 24 h following anoxia that reached a level of 61 ± 5%) when compared to untreated control cultures of 11 ± 3% (Fig. 2B). Cells treated with NIC displayed a significant reduction in annexin V label to 26 ± 4% 24 h following anoxia.
NIC Requires Enhanced Activity of Akt1 to Foster Neuronal Protection
Western blot analysis was performed for phospho-Akt1 (activated form of Akt1, p-Akt1) 12 h following anoxia. In Fig. 3A, NIC and anoxia independently increased the expression of p-Akt1. This increased expression of p-Akt1 was blocked by agents wortmannin (W, 500 nM) and LY294002 (LY, 10 μM), inhibitors of Akt phosphorylation. In combination with anoxia exposure, NIC continued to enhance the expression of p-Akt1. The increased expression of p-Akt1 by NIC was blocked by W and LY.
Fig. (3). Nicotinamide (NIC) protects neurons through Akt1 mediated pathways.
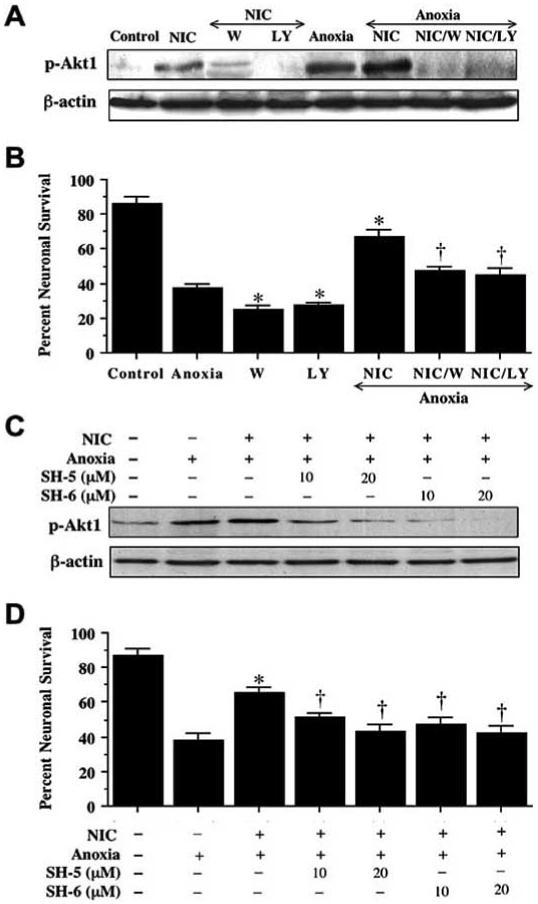
In (A), equal amounts of neuronal protein extracts (50 μg/lane) were immunoblotted with anti - phospho-Akt1 (p-Akt1, active Akt1) antibody. Exposure to NIC (12.5 mM) or anoxia significantly increased p-Akt1 expression and NIC application during anoxia further increased p-Akt1 expression. Application of the Akt1 phosphorylation inhibitor wortmannin (W, 500 nM) or LY294002 (LY, 10 μM) was sufficient to block the expression of p-Akt1 in the presence of NIC during anoxia. (B) At a concentration that blocks activation of p-Akt1 during anoxia, W (500 nM) or LY (10 μM) applied 1 h prior to anoxia significantly reduced the protective capacity of NIC (12.5 mM) during anoxia (*p<0.01 vs. anoxia; †p<0.01 vs. NIC/anoxia). (C) Application of the Akt1 phosphorylation selective Akt inhibitor SH-5 or SH-6 (10-20 μM) significantly attenuated p-Akt1 expression during NIC treatment following anoxia. (D) Application of SH-5 or SH-6 to block Akt1 activation 1 h prior to anoxia significantly reduced the protective capacity of NIC (12.5 mM) during anoxia (*p<0.01 vs. anoxia; †p<0.01 vs. NIC/anoxia). In B and D, data represent the mean and SEM from 4 to 6 experimental preparations. Control = untreated neurons.
In Fig. 3B, application of NIC (12.5 mM) 1 h prior to anoxia significantly increased neuronal survival to 67 ± 4%. Yet, co-application of wortmannin (W, 500 nM) or LY294002 (LY, 10 μM) at a concentration that blocks activation of p-Akt1 during anoxia (Fig. 3A) with NIC (12.5 mM) significantly reduced the ability of NIC to protect neurons against anoxia, suggesting that NIC required some level of Akt1 activation to offer neuroprotection. Application of wortmannin (500 nM) or LY294002 (10 μM) without anoxia was not toxic to neurons (data not shown), suggesting that endogenous Akt1 activation provides some level of protection during toxic insults.
To further define the contribution of Akt1 to neuroprotection of NIC during anoxia, Akt1 activation selective inhibitors SH-5 or SH-6 was applied to neuronal cultures 1 h prior to a 4 h period of anoxia. Representative Western blot result, as shown in Fig. 3C, indicated that application of SH-5 (10∼20 μM) or SH-6 (10∼20 μM) attenuated or eliminated the p-Akt1 expression during NIC treatment with anoxia. Co-application of SH-5 or SH-6 at a concentration that blocks activation of p-Akt1 during anoxia (Fig. 3C) with NIC (12.5 mM) also significantly reduced the ability of NIC to protect neurons against anoxia, further convincing that NIC required some level of Akt1 activation to offer neuroprotection (Fig. 3D).
NIC Induces Phosphorylation of Bad and Prevents Mitochondrial Membrane Depolarization and the Release of Cytochrome c During Anoxia
As phosphorylation of Bad, a downstream of Akt1, has been associated with initiation of apoptosis through its influence on the function of Bcl-2/Bcl-xL and mitochondria (Kennedy, SG et al., 1999), we investigated Bad phosphorylation following anoxia and NIC treatment. Western blot analysis was performed for p-Bad (phosphorylated Bad) 1-24 h following anoxia. In Fig. 4A, anoxic insult significantly increased the phosphorylation of Bad at 2, 3, and 6 h following a 4 h period of anoxia. In combination with anoxia, NIC (12.5 mM) 1 h pre-treatment further enhanced the expression of p-Bad over a 24 h period (Fig. 4B). Furthermore, application of wortmannin (W, 500 nM) and LY294002 (LY, 10 μM) to inhibit Akt activation (Fig. 3A) prevented Bad phosphorylation induced by NIC during anoxia (Fig. 4C).
Fig. (4). Nicotinamide (NIC) induces phosphorylation of Bad during anoxia exposure.
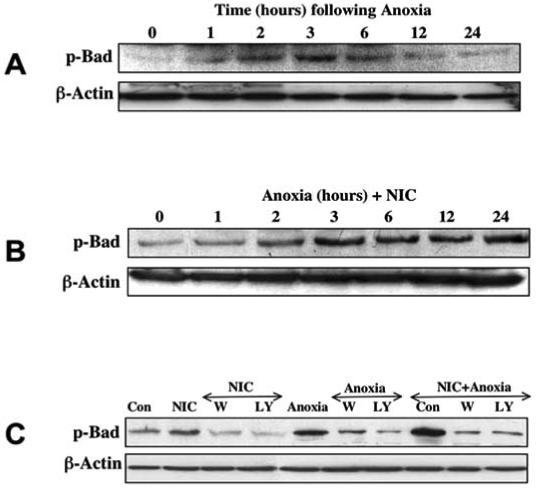
Equal amounts of neuronal protein extracts (50 μg/lane) were immunoblotted with anti - phospho-Bad (p-Bad, Ser136) antibody. The expression of p-Bad was increased 2-6 h following the exposure to a 4 h period of anoxia (A). NIC (12.5 mM) applied 1 h prior to anoxia further increased the phosphorylation of Bad during anoxia over a 24 h period (B). Application of the Akt1 phosphorylation inhibitor wortmannin (W, 500 nM) or LY294002 (LY, 10 μM) 1 h prior to anoxia was sufficient to block the expression of p-Bad in the presence of NIC 3 h later (C). Con=Control.
Exposure to anoxia produced a significant decrease in the red/green fluorescence intensity ratio when cells were labeled by a cationic membrane potential indicator JC-1 within 3 h when compared with untreated control cultures (Figs. 5A and Fig. 5B), suggesting that anoxia results in mitochondrial membrane depolarization. Application of NIC (12.5 mM) 1 h prior to anoxia significantly increased the red/green fluorescence intensity of the neurons, indicating that mitochondrial permeability transition pore membrane potential was restored to baseline (Figs. 5A and 5B). Administration of NIC (12.5 mM) 1 h prior to anoxia maintained mitochondrial permeability transition pore membrane function and prevented mitochondrial cytochrome c release as demonstrated by Western analysis (Fig. 5C).
Fig. (5). Nicotinamide (NIC) inhibits mitochondrial membrane depolarization and cytochrome c release during anoxia exposure.
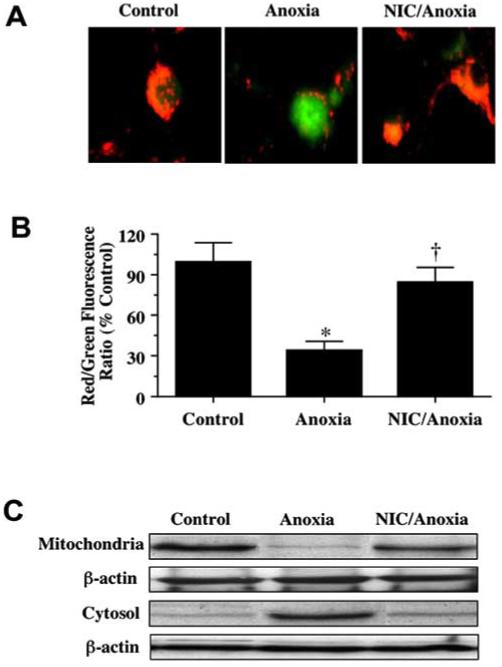
(A) Exposure to anoxia produced a significant decrease in the red/green fluorescence intensity ratio using a cationic membrane potential indicator JC-1 within 3 h when compared with untreated control cultures, suggesting that anoxia results in mitochondrial membrane depolarization. Application of NIC (12.5 mM) 1 h prior to anoxia exposure significantly increased the red/green fluorescence intensity of neurons, indicating that membrane potential was restored. (B) The relative ratio of red/green fluorescent intensity of mitochondrial staining in both untreated (control) neurons and neurons exposed to anoxia or NIC (12.5 mM) and anoxia 3 h following anoxia was measured in 4 independent experiments with analysis performed using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/) (Control vs. anoxia, *p<0.01; Anoxia vs. NIC/Anoxia, †p<0.01). (C) A representative Western blot with equal amounts of mitochondrial or cytosol protein extracts (50 μg/lane) were immunoblotted demonstrating that application of NIC (12.5 mM) significantly prevented cytochrome c release from mitochondria during anoxia.
NIC Protects Neurons Through the Inhibition of Caspases 8, 1, and 3-Like Activities During Anoxia
As shown in Fig. 6A, NIC (12.5 mM) was applied to neuronal cultures 1 h prior to a 4 h period of anoxia and data for caspase 8, caspase 1, and caspase 3 activities were obtained 12 h post injury since this time period represented the peak activities for these cysteine proteases (Chong, ZZ et al., 2003, Lin, S and Maiese, K, 2001). NIC pre-treatment significantly decreased caspase 8 - like activity from 0.25 ± 0.04 μmol/min/g of anoxia treated alone to 0.08 ± 0.04 μmol/min/g (P<0.01). Similarly, NIC pre-treatment significantly reduced the activity of caspase 1 - like activity (0.09 ± 0.02 μmol/min/g) and caspase 3 - like activity (0.11 ± 0.03 μmol/min/g) when compared to cultures treated with anoxia alone (0.18 ± 0.04 μmol/min/g and 0.28 ± 0.04 μmol min/g, respectively).
Fig. (6). Nicotinamide (NIC) prevents neuronal injury through the inhibition of anoxia-induced caspases 8, 1, and 3-like activities.
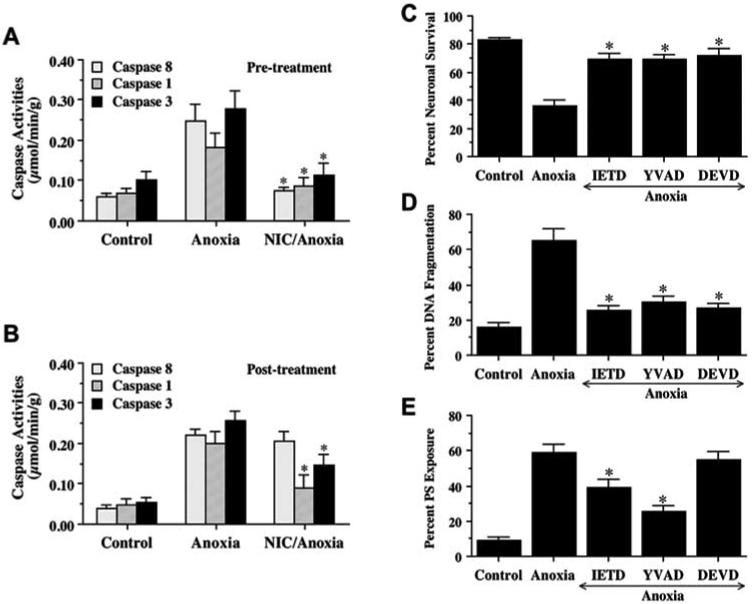
(A) NIC (12.5 mM) was administered directly into culture media 1 h prior to anoxia, and caspases activity were determined at 12 h following a 4 h period of anoxia by measuring the cleavage of substrate Ac-IETD-pNA (for caspase 8), Ac-YVAD-pNA (for caspase 1), and Ac-DEVD-pNA (for caspase 3). Caspases 8, 1, and 3-like activities increased significantly following anoxia when compared to untreated control groups. Pretreatment with NIC significantly prevented the induction of caspases 8, 1, and 3-like activities induced by anoxia (*p < 0.01 vs. anoxia treated only). (B) To examine the effects of post-treatment with NIC on caspase activity following anoxic exposure, NIC (12.5 mM) was added directly to culture media at 4 h after anoxic exposure. The caspases 8, 1, and 3-like activities were determined at 12 h after anoxia. Posttreatment with NIC significantly attenuated the increase in caspases 1, and 3, but not caspase 8-like activity during anoxia. (*p < 0.01 vs. anoxia treated only). (C, D, and E) Neurons were pretreated with an inhibitor of caspase 8 (IETD, 50 μM), caspase 1 (YVAD, 50 μM), or caspase 3 (DEVD, 50 μM) 1 h prior to anoxia and neuronal survival (C), DNA fragmentation (D), and membrane PS exposure (E) were determined 24 h following anoxia (*p<0.01 vs. anoxia treated alone). In all cases, data represent the mean and SEM from 4 experimental preparations, control = untreated neurons.
During a post-treatment paradigm, NIC (12.5 mM) was administered into cultures 4 h following a 4 h period of anoxia and caspase activity was determined at 12 h after anoxia (Fig. 6B). Post-treatment with NIC significantly decreased caspases 1, and 3-like activities. Yet, NIC post-treatment did not alter anoxia-induced caspase 8 activation.
We next examined whether the induction of caspase 8, caspase 1, and caspase 3 - like activities was required for anoxia induced cell injury. IETD, YVAD, and DEVD were applied to neuronal cultures to inhibit caspase 8, caspase 1, and caspase 3 - like activities, neuronal survival, DNA fragmentation, and membrane PS exposure were determined 24 h following anoxia. As shown in Fig. 6C, IETD (50 μM), YVAD (50 μM), and DEVD (50 μM) significantly increased neuronal survival 24 h following a 4 h period of anoxia. Pretreatment of neurons with 50 μM IETD, YVAD, and DEVD significantly decreased DNA fragmentation to 25 ± 3%, 30 ± 4%, and 27 ± 2%, respectively (Fig. 6D). In addition, inhibition of caspase 1 (YVAD) or caspase 8 (IETD), not of caspase 3 (DEVD), activity maintained cellular membrane asymmetry and significantly prevented PS externalization (Fig. 6E).
NIC Inhibits PARP Cleavage Induced by Anoxia
Since increased caspase activity such as caspase 3 leads to the specific cleavage of cellular protein substrates including PARP and preserving PARP integrity contributes to repair of damaged DNA during injury, we next characterized the ability of NIC to modulate neuronal PARP cleavage following anoxic exposure. Fig. 7 illustrates that the intact form of PARP (116 kDa) is detectable in neuronal extracts from untreated cultures (lane 3). Extracts from the human HL60 leukemia cells were analyzed as a positive control to compare migration of the intact form of PARP (lane 1) and its 85 kDa proteolytic fragment (lane 2) from cells treated with apoptotic inducer etoposide. Anoxic exposure (4 h) dramatically decreased the intact form of the PARP 12 h following anoxia (lane 4). However, this decrease in the amount of the 116 kDa of PARP protein was attenuated by pretreatment with NIC (12.5 mM) 1 h prior to anoxia) (lane 5). Although anoxia exposure significantly induced the degradation of intact form of PARP protein, the 85 kDa proteolytic fragment of PARP was not detected from extracts of hippocampal neurons.
Fig. (7). Nicotinamide (NIC) inhibits anoxia-induced poly(ADP-ribose) polymerase (PARP) cleavage.
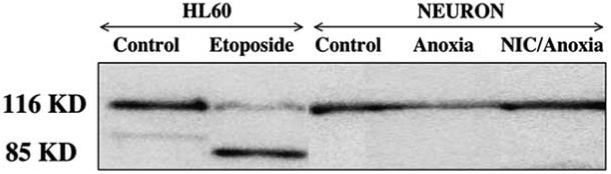
To examine the effect of NIC on PARP expression, total protein extracts were prepared from either untreated control cultures or from extracts prepared 12 h after a 4 h period of anoxia with or without pretreatment of NIC (12.5 mM). For a positive control, lanes 1 and 2 employed 20 μl of protein extract from human HL60 leukemia cells uninduced (lane 1) or induced (lane 2) to undergo apoptosis by the agent etoposide. In lanes 3, 4, and 5, equal amounts of neuronal protein extracts (25 μg/lane) were separated by 7.5% SDS-PAGE and were then immunoblotted with polyclonal anti-PARP antibody. Detection was by enhanced chemiluminescence. In lanes 1 and 2, extracts of the human HL60 cells were analyzed to compare migration of PARP and its 85 kDa proteolytic fragment. Lane 3 contained an extract from untreated rat hippocampal cultures. A decrease in the amount of 116 kDa PARP occurred within 12 h following anoxia exposure (lane 4). In lane 5, pretreatment with NIC (12.5 mM) 1 h prior to anoxic exposure inhibited PARP cleavage.
NIC does not Prevent p38 and JNK Phosphorylation During Anoxia
Since activation of p38 and JNK could be one of the mediators during anoxic insults, we monitored the effects of NIC on anoxia-induced activation of p38 and JNK. Cell extracts were analyzed at 1 h following a 4 h period anoxia. As shown in Fig. 8A, anoxic exposure resulted in a dramatic increase in the level of active form of p38. Pretreatment of neurons with NIC (12.5 mM) 1 h prior to anoxia did not prevent p38 activation induced by anoxia. The level of total p38 remained unchanged in each group. In a similar manner, anoxic exposure resulted in a significant increased expression of active form of JNK 1 h following anoxic exposure. Yet, pretreatment with NIC (12.5 mM) did not alter anoxia-induced phosphorylation of JNK in hippocampal neurons (Fig. 8B). The total level of cellular JNK remains unchanged in each group of untreated control, anoxia only, and NIC/anoxia. Treatment with NIC (12.5 mM) alone did not induce activation of p38 and JNK (data not shown).
Fig. (8). Nicotinamide (NIC) does not alter anoxia-induced p38 and JNK activation.
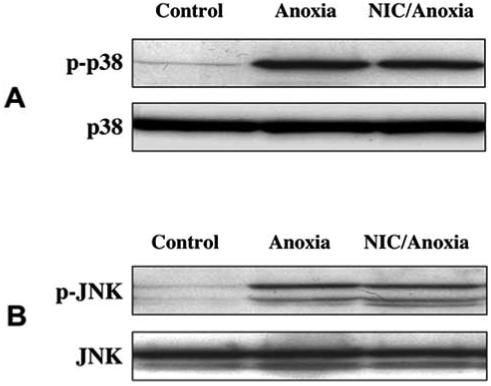
The effect of NIC on anoxia-induced phosphorylation of p38 (A) and JNK (B) was determined. Total protein extracts were prepared from either untreated control cultures or from cultures 1 h after a 4 h period of anoxia with or without pretreatment with NIC (12.5 mM). The cellular level of total p38 (A) and JNK (B) remained unchanged in each group. A significant expression of active form of p38 (p-p38) and JNK (p-JNK) was detected from cultures exposed to anoxia. Pretreatment with NIC 1 h prior to anoxia did not prevent anoxia-induced the activation of p38 (A) and JNK (B). Data presented are taken from one individual experiment of three giving similar results.
DISCUSSION
Dissecting the role and biological effects of NIC in different cell systems represents a significant challenge in several respects. Since PNC1 deaminates NIC to convert it into nicotinic acid and reduce intracellular NIC concentrations, Sir2 can become activated and PNC1 expression is increased to lead to lifespan extension during calorie extension, at least in experimental models of yeast (Li, F et al., 2004, Porcu, M et al., 2005). Furthermore, NIC can function as an inhibitor of sirtuins in concentrations that range from 50-100 μM (Porcu, M et al., 2005). On the converse side, NIC offers protection in millimole concentrations against NO (Lin, SH et al., 2000), anoxia (Lin, SH et al., 2001), and oxygen glucose deprivation (Chong, ZZ et al., 2004) in primary cultures rat hippocampal neurons. In cortical neurons, NIC antagonizes cell injury during free radical generating toxins such as tertiary butylhydroperoxide (Sonee, M et al., 2003). NIC also can protect both rod and cone photoreceptor cells against N-methyl-N-nitrosourea toxicity (Kiuchi, K et al., 2003, Kiuchi, K et al., 2002) as well as against glycation end products in all layers of the retina (Reber, F et al., 2003). In animal studies, NIC prevents neuronal degeneration against trauma (Wallis, RA et al., 1996), oxidative stress (Chong, ZZ et al., 2004, Chong, ZZ et al., 2002, Crowley, CL et al., 2000, Lin, SH et al., 2001, Lin, SH et al., 2000), cerebral ischemia (Gupta, S et al., 2004, Yang, J et al., 2002), and spinal cord injury (Brewer, KL and Hardin, JS, 2004, Isbir, CS et al., 2003). NIC can directly protect against both neuronal necrosis and apoptosis mechanisms of injury and prevent brain damage through reducing DNA fragmentation during ischemic reperfusion injury (Chong, ZZ et al., 2004, Chong, ZZ et al., 2002, Lin, SH et al., 2001, Lin, SH et al., 2000, Maiese, K et al., 2003, Maiese, K et al., 2001).
We demonstrate that NIC can reduce anoxia-induced neuronal injury and increase cell viability in a concentration-specific manner in concentration ranges similar to prior studies involving cell protection. Treatment with NIC in a range of 5.0 -12.5 mM significantly protected neurons during anoxia. This concentration range is similar to cellular protection with NIC in cerebral microvascular endothelial cells (ECs) and neurons during nitric oxide exposure (Chong, ZZ et al., 2002, Lin, SH et al., 2001, Lin, SH et al., 2000) and in neurons with oxygen glucose deprivation (Chong, ZZ et al., 2004). Post-treatment strategies with NIC also illustrate that neuronal injury is reversible by NIC. Consistent with the results in ECs during NO exposure, our current results suggest that application of NIC, within a 6 h period post the onset of anoxia, can increase neuronal survival. The results may also suggest that NIC can modulate critical cellular pathways prior to the induction of cellular mechanisms that can destine a cell to die. This fixed time frame for protection by NIC most likely coincides with the progressive induction of secondary cellular pathways during this 6 h time span, such as cysteine protease induction in neurons and ECs (Li, F et al., 2004, Li, F et al., 2004, Maiese, K et al., 2005, Maiese, K et al., 2000).
NIC also protects neurons against apoptotic injury through the prevention of DNA fragmentation and the maintenance of cellular membrane asymmetry. Apoptosis is an active process that can lead to a cell’s demise in a variety of tissues and has recently been identified in organisms as diverse as plants (Hatsugai, N et al., 2004). Apoptosis consists of two independent processes that involve membrane PS exposure and DNA fragmentation (Maiese, K et al., 2004). Apoptotic injury is believed to contribute significantly to a variety of disease states that especially involve the nervous system such as ischemic stroke, AD, PD, and spinal cord injury (Chong, ZZ and Maiese, K, 2004, Li, F et al., 2004). As an early event in the dynamics of cellular apoptosis, the biological role of membrane PS externalization tion can vary in different cell populations. In some cell systems, PS may be required for embryogenesis (Bose, J et al., 2004). Yet, in mature tissues, membrane PS externalization can become a signal for the phagocytosis of cells (Hong, JR et al., 2004). In addition, the externalization of membrane PS residues in ECs can promote the formation of a procoagulant surface (Chong, ZZ et al., 2004). In contrast to the early externalization of membrane PS residues, the cleavage of genomic DNA into fragments is considered to be a delayed event that occurs late during apoptosis (Dombroski, D et al., 2000, Jessel, R et al., 2002, Kang, JQ et al., 2003, Maiese, K et al., 2000). Administration of NIC during anoxia prevented not only DNA fragmentation but also the induction of membrane PS inversion over a 24 h period. Immediate protection by NIC is afforded through the maintenance of genomic DNA integrity. Long-term protection may result from the inhibition of membrane PS exposure, since membrane PS externalization functions as a cellular “marker” for removal by microglia (Chong, ZZ et al., 2003, Li, F et al., 2004).
To further elucidate the mechanisms of NIC neuroprotection, we next focused on Akt. Akt is a critical survival factor that can modulate cellular pathways in both the central and peripheral nervous systems (Chong, ZZ et al., 2005). Akt can be both necessary and sufficient for the survival of neurons, since expression of a dominant-negative Akt or inhibition of PI 3-K yields apoptotic cell death during trophic factor administration (Crowder, RJ and Freeman, RS, 1998) and precipitates cell death during oxidative stress (Kang, JQ et al., 2003, Kang, JQ et al., 2003). Increased Akt activity also can foster cell survival during free radical exposure (Chong, ZZ et al., 2003, Matsuzaki, H et al., 1999), matrix detachment (Rytomaa, M et al., 2000), neuronal axotomy (Namikawa, K et al., 2000), DNA damage (Chong, ZZ et al., 2002, Chong, ZZ et al., 2004, Henry, MK et al., 2001, Kang, JQ et al., 2003), anti-Fas antibody administration (Suhara, T et al., 2001), oxidative stress (Chong, ZZ et al., 2003, Kang, JQ et al., 2003, Kang, JQ et al., 2003, Yamaguchi, H and Wang, HG, 2001), Aß exposure (Martin, D et al., 2001), and transforming growth factor-β (TGF-β) application (Conery, AR et al., 2004). Application of NIC alone or NIC during anoxia increased Akt1 phosphorylation. Inhibition of Akt1 activation by using PI 3-K inhibitors not only reduced the expression of phosphorylated Akt1 (pAkt1), but also attenuated the efficacy of NIC on cellular survival during anoxia. Moreover, application of selective antagonists of Akt, SH-5 and SH-6 to neuronal cultures during anoxia also attenuated p-Akt1 expression during NIC treatment. Similar to the PI 3-K inhibitors, SH-5 and SH-6 also attenuated the efficacy of NIC on cell survival during anoxia. These results suggest that induction of Akt1 activation by NIC is required, at least to some extent, for NIC to promote neuroprotection.
This protection by NIC may be linked to the phosphorylation of Bad, a downstream substrate of Akt1. The Bcl-2 family member Bad promotes apoptosis through its interaction with Bcl-2/Bcl-xL. The phosphorylation of Bad at Ser136 by Akt triggers its interaction with 14-3-3 protein and prevents Bad from binding to the anti-apoptotic protein Bcl-2/Bcl-xL (Hsu, SY et al., 1997). In our studies, anoxia lead to the phosphorylation of Bad (p-Bad) and promoted p-Bad expression 2-6 h following anoxia. Application of NIC further increased the expression of p-Bad over a 24 h period following anoxia. The results suggest that NIC can precipitate the phosphorylation of Bad and therefore prevent its anti-apoptotic function. In addition, the phosphorylation of Bad by NIC was attenuated by PI 3-K inhibition, suggesting that NIC results in Bad phosphorylation through the PI 3-K/Akt pathway.
Phosphorylation of Bad by Akt can lead to the binding of Bad with the cytosolic protein 14-3-3 to release Bcl-xL and allows it to block apoptosis (Li, Y et al., 2001). Bcl-2 and Bcl-xL subsequently prevent Bax translocation to the mitochondria, maintain mitochondrial membrane potential, and prevent the release of cytochrome c from the mitochondria (Chong, ZZ et al., 2003, Putcha, GV et al., 1999). Therefore, we next evaluated the effects of NIC on mitochondria during anoxia. Exposure to anoxia resulted in an early loss of mitochondria membrane potential within 3 h. Administration of NIC attenuated the loss of membrane potential and prevented cytochrome c release into the cytosol, suggesting that NIC can preserve mitochondria membrane integrity in neurons during anoxia similar to other insults (Li, F et al., 2004, Maiese, K et al., 2003).
Cytochrome c normally resides exclusively in the intermembrane space of mitochondria, but once released can lead to caspase activation. Activation of caspases proceeds through extrinsic and intrinsic pathways. The extrinsic pathway is initiated by death receptor activation at the cell surface, resulting in the recruitment and activation of the initiator caspase 8 upon apoptotic stimuli (Ashkenazi, A and Dixit, VM, 1998). Caspase 8 can subsequently activate caspase 3. The intrinsic caspase pathway involves mitochondrial dysfunction and is more closely associated with the release of cytochrome c and the activation of caspase 9 (Liu, X et al., 1996). Caspase 9 can subsequently activate caspase 3 (Li, P et al., 1997) as well as caspase 1 through the intermediary caspase 8 (Takahashi, H et al., 1999). Together, caspase 1 and caspase 3 lead to both DNA fragmentation and membrane PS exposure (Chong, ZZ et al., 2002, Li, P et al., 1997, Maiese, K et al., 2000). We show that pre-treatment with NIC significantly prevents caspase 8, 1, and 3-like activities. The ability of NIC to modulate caspase 1, caspase 3, and caspase 8 - like activities appears to play a critical role in the neuroprotection of NIC. Our post-treatment paradigm demonstrated that NIC, applied 4 h post anoxia, could not prevent caspase 8 activation, also suggesting that caspase 8 act as an upstream caspase during injury. Interestingly, NIC prevents membrane PS exposure primarily through the inhibition of caspase 8 and caspase 1 -like activities but not through caspase 3 - like activity. Given that caspase 8 can result in the downstream activation of caspase 1, caspase 1 is believed to be principally responsible for the externalization of membrane PS residues in several cell systems (Chong, ZZ et al., 2003, Chong, ZZ et al., 2005, Chong, ZZ et al., 2005).
NIC also may provide cellular protection through the maintenance of PARP integrity and the preservation of cellular energy reserves. PARP is a nuclear protein that binds to DNA strand breaks and cleaves NAD+ into NIC and ADP-ribose (Southan, GJ and Szabo, C, 2003). During DNA repair, ADP-ribose is polymerized onto nuclear proteins that include histones and transcription factors at DNA strand breaks (Burkle, A, 2001). We show that NIC (12.5 mM) can prevent the degradation of PARP during anoxia. NIC concentrations of at least 1 mM have been shown to provide sufficient stores of NAD+ during PARP activation (Smets, LA et al., 1990) and prevent PARP degradation and allow for DNA repair through the direct inhibition of caspase 3 - like activity (Chong, ZZ et al., 2002, Lin, SH et al., 2000).
An alternate pathway that may mediate protection by NIC may involve the MAPKs that include the p38 kinases and the c-Jun N-terminal kinases (JNKs). These proteins are activated by phosphorylation and play a significant function during cell differentiation, growth, and death (Chong, ZZ et al., 2003). Significant activation of p38 and JNK is present in both neurons and ECs during oxidative stress (Lin, SH et al., 2001, Lin, SH et al., 2000, Maiese, K et al., 2003, Maiese, K et al., 2001). In addition, JNK can promote Bax translocation through phosphorylation of 14-3-3 proteins and lead to cytochrome c release (Tsuruta, F et al., 2004). Furthermore, during cellular injury such as with cyanideinduced apoptosis, p38 can modulate Bax translocation from the cytosol to the mitochondria and result in both cytochrome c release and caspase activation (Shou, Y et al., 2003). Yet, similar to prior work (Lin, SH et al., 2001, Lin, SH et al., 2000, Maiese, K et al., 2003, Maiese, K et al., 2001), NIC does not alter the activity of either p38 or JNK, suggesting that protection by NIC is independent of p38 and JNK.
ACKNOWLEDGEMENTS
This research was supported by the following grants (KM): American Heart Association (National), Bugher Foundation Award, Janssen Neuroscience Award, Johnson and Johnson Focused Investigator Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
REFERENCES
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Bose J, Gruber AD, Helming L, Schiebe S, Wegener I, Hafner M, Beales M, Kontgen F, Lengeling A. The phosphatidylserine receptor has essential functions during embryogenesis but not in apoptotic cell removal. J Biol. 2004;3(4):15. doi: 10.1186/jbiol10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer KL, Hardin JS. Neuroprotective effects of nicotinamide after experimental spinal cord injury. Acad Emerg Med. 2004;11(2):125–30. doi: 10.1111/j.1553-2712.2004.tb01421.x. [DOI] [PubMed] [Google Scholar]
- Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays. 2001;23(9):795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- Chen JH, Hsiao G, Lee AR, Wu CC, Yen MH. Androgra-pholide suppresses endothelial cell apoptosis via activation of phosphatidyl inositol-3-kinase/Akt pathway. Biochem Pharmacol. 2004;67(7):1337–45. doi: 10.1016/j.bcp.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106(23):2973–9. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, cytochrome c, and caspase 9 from the critical elements for cerebral vascular protection by erythropoietin. J Cereb Blood Flow Metab. 2003;23(3):320–330. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol. 2003;138(6):1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin: cytoprotection in vascular and neuronal cells. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3(2):141–54. doi: 10.2174/1568006033481483. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Metabotropic glutamate receptors promote neuronal and vascular plasticity through novel intracellular pathways. Histol Histopathol. 2003;18(1):173–89. doi: 10.14670/HH-18.173. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp Cell Res. 2004;296(2):196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol. 2005;20(1):299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75(3):207–46. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Li F, Maiese K. Stress in the brain: Novel cellular mechanisms of injury linked to Alzheimer’s disease. Brain Res Brain Res Rev. 2005;49(1):1–21. doi: 10.1016/j.brainresrev.2004.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li FQ, Maiese K. Employing new cellular therapeutic targets for Alzheimer’s disease: A change for the better? Curr Neurovasc Res. 2005;2(1):55–72. doi: 10.2174/1567202052773508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Lin S-H, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J Cereb Blood Flow Metab. 2004;24(7):728–743. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Kang JQ, Maiese K. The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell Mol Neurobiol. 2003;23(45):561–78. doi: 10.1023/A:1025158314016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Maiese K. Nicotinamide Modulates Mitochondrial Membrane Potential and Cysteine Protease Activity during Cerebral Vascular Endothelial Cell Injury. J Vasc Res. 2002;39(2):131–47. doi: 10.1159/000057762. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J Cereb Blood Flow Metab. 2004;24(7):728–43. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Maiese K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol Histopathol. 2004;19(2):495–504. doi: 10.14670/hh-19.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conery AR, Cao Y, Thompson EA, Townsend CM, Jr., Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol. 2004;6(4):366–72. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18(8):2933–43. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley CL, Payne CM, Bernstein H, Bernstein C, Roe D. The NAD+ precursors, nicotinic acid and nicotinamide protect cells against apoptosis induced by a multiple stress inducer, deoxycholate. Cell Death Differ. 2000;7(3):314–26. doi: 10.1038/sj.cdd.4400658. [DOI] [PubMed] [Google Scholar]
- Dombroski D, Balasubramanian K, Schroit AJ. Phosphatidylserine expression on cell surfaces promotes antibody- dependent aggregation and thrombosis in beta2-glycoprotein I-immune mice. J Autoimmun. 2000;14(3):221–9. doi: 10.1006/jaut.2000.0365. [DOI] [PubMed] [Google Scholar]
- Friguls B, Justicia C, Pallas M, Planas AM. Focal cerebral ischemia causes two temporal waves of Akt activation. Neuroreport. 2001;12(15):3381–4. doi: 10.1097/00001756-200110290-00046. [DOI] [PubMed] [Google Scholar]
- Gupta S, Kaul CL, Sharma SS. Neuroprotective effect of combination of poly (ADP-ribose) polymerase inhibitor and antioxidant in middle cerebral artery occlusion induced focal ischemia in rats. Neurol Res. 2004;26(1):103–7. doi: 10.1179/016164104773026624. [DOI] [PubMed] [Google Scholar]
- Hatsugai N, Kuroyanagi M, Yamada K, Meshi T, Tsuda S, Kondo M, Nishimura M, Hara-Nishimura I. A plant vacuolar protease, VPE, mediates virus-induced hypersensitive cell death. Science. 2004;305(5685):855–8. doi: 10.1126/science.1099859. [DOI] [PubMed] [Google Scholar]
- Henry MK, Lynch JT, Eapen AK, Quelle FW. DNA damage-induced cell-cycle arrest of hematopoietic cells is overridden by activation of the PI-3 kinase/Akt signaling pathway. Blood. 2001;98(3):834–41. doi: 10.1182/blood.v98.3.834. [DOI] [PubMed] [Google Scholar]
- Hong JR, Lin GH, Lin CJ, Wang WP, Lee CC, Lin TL, Wu JL. Phosphatidylserine receptor is required for the engulfment of dead apoptotic cells and for normal embryonic development in zebrafish. Development. 2004;131(21):5417–27. doi: 10.1242/dev.01409. [DOI] [PubMed] [Google Scholar]
- Hsu SY, Kaipia A, Zhu L, Hsueh AJ. Interference of BAD (Bcl-xL/Bcl-2-associated death promoter)-induced apoptosis in mammalian cells by 14-3-3 isoforms and P11. Mol Endocrinol. 1997;11(12):1858–67. doi: 10.1210/mend.11.12.0023. [DOI] [PubMed] [Google Scholar]
- Isbir CS, Ak K, Kurtkaya O, Zeybek U, Akgun S, Scheitauer BW, Sav A, Cobanoglu A. Ischemic preconditioning and nicotinamide in spinal cord protection in an experimental model of transient aortic occlusion. Eur J Cardiothorac Surg. 2003;23(6):1028–33. doi: 10.1016/s1010-7940(03)00110-6. [DOI] [PubMed] [Google Scholar]
- Ishaque A, Al-Rubeai M. Role of vitamins in determining apoptosis and extent of suppression by bcl-2 during hybridoma cell culture. Apoptosis. 2002;7(3):231–9. doi: 10.1023/a:1015343616059. [DOI] [PubMed] [Google Scholar]
- Jessel R, Haertel S, Socaciu C, Tykhonova S, Diehl HA. Kinetics of apoptotic markers in exogeneously induced apoptosis of EL4 cells. J Cell Mol Med. 2002;6(1):82–92. doi: 10.1111/j.1582-4934.2002.tb00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallmann B, Burkart V, Kroncke KD, Kolb-Bachofen V, Kolb H. Toxicity of chemically generated nitric oxide towards pancreatic islet cells can be prevented by nicotinamide. Life Sci. 1992;51(9):671–8. doi: 10.1016/0024-3205(92)90240-p. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J Neurosci Res. 2003;74(1):37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol Pharmacol. 2003;64(3):557–69. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19(8):5800–10. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuchi K, Kondo M, Ueno S, Moriguchi K, Yoshizawa K, Miyake Y, Matsumura M, Tsubura A. Functional rescue of N-methyl-N-nitrosourea-induced retinopathy by nicotinamide in Sprague-Dawley rats. Curr Eye Res. 2003;26(6):355–62. doi: 10.1076/ceyr.26.5.355.15435. [DOI] [PubMed] [Google Scholar]
- Kiuchi K, Yoshizawa K, Shikata N, Matsumura M, Tsubura A. Nicotinamide prevents N-methyl-N-nitrosourea-induced photoreceptor cell apoptosis in Sprague-Dawley rats and C57BL mice. Exp Eye Res. 2002;74(3):383–92. doi: 10.1006/exer.2001.1127. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Erythropoietin on a Tightrope: Balancing Neuronal and Vascular Protection between Intrinsic and Extrinsic Pathways. Neurosignals. 2004;13(6):265–89. doi: 10.1159/000081963. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Navigating novel mechanisms of cellular plasticity with the NAD+ precursor and nutrient nicotinamide. Front Biosci. 2004;9:2500–2520. doi: 10.2741/1412. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Tennekoon GI, Birnbaum M, Marchionni MA, Rutkowski JL. Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol Cell Neurosci. 2001;17(4):761–7. doi: 10.1006/mcne.2000.0967. [DOI] [PubMed] [Google Scholar]
- Lin S, Maiese K. The metabotropic glutamate receptor system protects agnist ischemic free radical programmed cell death in rat brain endothelial cells. J Cereb Blood Flow Metab. 2001;21(3):262–275. doi: 10.1097/00004647-200103000-00010. [DOI] [PubMed] [Google Scholar]
- Lin SH, Chong ZZ, Maiese K. Nicotinamide: A Nutritional Supplement that Provides Protection Against Neuronal and Vascular Injury. J Med Food. 2001;4(1):27–38. doi: 10.1089/10966200152053686. [DOI] [PubMed] [Google Scholar]
- Lin SH, Vincent A, Shaw T, Maynard KI, Maiese K. Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J Cereb Blood Flow Metab. 2000;20(9):1380–91. doi: 10.1097/00004647-200009000-00013. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–57. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol Sci. 2003;24(5):228–32. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. Erythropoietin in the brain: can the promise to protect be fulfilled? Trends Pharmacol Sci. 2004;25(11):577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005;293(1):90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Lin S, Chong ZZ. Elucidating neuronal and vascular injury through the cytoprotective agent nicotinamide. Curr Med ChemImm Endoc Metab. Agents. 2001;1(3):257–267. [Google Scholar]
- Maiese K, Vincent AM. Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J Neurosci Res. 2000;59(4):568–80. doi: 10.1002/(SICI)1097-4547(20000215)59:4<568::AID-JNR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Majamaa K, Rusanen H, Remes A, Hassinen IE. Metabolic interventions against complex I deficiency in MELAS syndrome. Mol Cell Biochem. 1997;174(12):291–6. [PubMed] [Google Scholar]
- Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Abeta(25-35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem. 2001;78(5):1000–8. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S, Tohyama M. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J Neurochem. 1999;73(5):2037–46. [PubMed] [Google Scholar]
- Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H. Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci. 2000;20(8):2875–86. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien BA, Harmon BV, Cameron DP, Allan DJ. Nicotinamide prevents the development of diabetes in the cyclophosphamideinduced NOD mouse model by reducing beta-cell Apoptosis. J Pathol. 2000;191(1):86–92. doi: 10.1002/(SICI)1096-9896(200005)191:1<86::AID-PATH573>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Porcu M, Chiarugi A. The emerging therapeutic potential of sirtuin-interacting drugs: from cell death to lifespan extension. Trends Pharmacol Sci. 2005;26(2):94–103. doi: 10.1016/j.tips.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19(17):7476–85. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reber F, Geffarth R, Kasper M, Reichenbach A, Schleicher ED, Siegner A, Funk RH. Graded sensitiveness of the various retinal neuron populations on the glyoxal-mediated formation of advanced glycation end products and ways of protection. Graefes Arch Clin Exp Ophthalmol. 2003;241(3):213–25. doi: 10.1007/s00417-002-0528-1. [DOI] [PubMed] [Google Scholar]
- Rytomaa M, Lehmann K, Downward J. Matrix detachment induces caspase-dependent cytochrome c release from mitochondria: inhibition by PKB/Akt but not Raf signalling. Oncogene. 2000;19(39):4461–8. doi: 10.1038/sj.onc.1203805. [DOI] [PubMed] [Google Scholar]
- Shou Y, Li L, Prabhakaran K, Borowitz JL, Isom GE. p38 Mitogen-activated protein kinase regulates Bax translocation in cyanide-induced Apoptosis. Toxicol Sci. 2003;75(1):99–107. doi: 10.1093/toxsci/kfg157. [DOI] [PubMed] [Google Scholar]
- Simakajornboon N, Szerlip NJ, Gozal E, Anonetapipat JW, Gozal D. In vivo PDGF beta receptor activation in the dorsocaudal brain-stem of the rat prevents hypoxia-induced apoptosis via activation of Akt and BAD. Brain Res. 2001;895(12):111–8. doi: 10.1016/s0006-8993(01)02054-6. [DOI] [PubMed] [Google Scholar]
- Smeitink J, van den Heuvel L, Koopman W, Nijtmans L, Ugalde C, Willems P. Cell Biological Consequences of Mitochondrial NADH: Ubiquinone Oxidoreductase Deficiency. Curr Neurovasc Res. 2004;1(1):29–40. doi: 10.2174/1567202043480224. [DOI] [PubMed] [Google Scholar]
- Smets LA, Loesberg C, Janssen M, Van Rooij H. Intracellular inhibition of mono(ADP-ribosylation) by meta- iodobenzylguanidine: specificity, intracellular concentration and effects on glucocorticoid-mediated cell lysis. Biochim Biophys Acta. 1990;1054(1):49–55. doi: 10.1016/0167-4889(90)90204-q. [DOI] [PubMed] [Google Scholar]
- Sonee M, Martens JR, Evers MR, Mukherjee SK. The effect of tertiary butylhydroperoxide and nicotinamide on human cortical neurons. Neurotoxicology. 2003;24(3):443–8. doi: 10.1016/S0161-813X(03)00019-6. [DOI] [PubMed] [Google Scholar]
- Southan GJ, Szabo C. Poly(ADP-Ribose) Polymerase Inhibitors. Curr Med Chem. 2003;10(4):321–40. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- Suhara T, Mano T, Oliveira BE, Walsh K. Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE- inhibitory protein (FLIP) Circ Res. 2001;89(1):13–9. doi: 10.1161/hh1301.092506. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Nakamura S, Asano K, Kinouchi M, Ishida-Yamamoto A, Iizuka H. Fas antigen modulates ultraviolet B-induced apoptosis of SVHK cells: sequential activation of caspases 8, 3, and 1 in the apoptotic process. Exp Cell Res. 1999;249(2):291–8. doi: 10.1006/excr.1999.4476. [DOI] [PubMed] [Google Scholar]
- Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N, Gotoh Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004;23(8):1889–99. doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. Direct temporal analysis of apoptosis induction in living adherent neurons. J Histochem Cytochem. 1999;47(5):661–72. doi: 10.1177/002215549904700508. [DOI] [PubMed] [Google Scholar]
- Wallis RA, Panizzon KL, Girard JM. Traumatic neuroprotection with inhibitors of nitric oxide and ADP- ribosylation. Brain Res. 1996;710(12):169–77. doi: 10.1016/0006-8993(95)01278-8. [DOI] [PubMed] [Google Scholar]
- Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22(15):6401–7. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene. 2001;20(53):7779–86. doi: 10.1038/sj.onc.1204984. [DOI] [PubMed] [Google Scholar]
- Yang J, Klaidman L, Chang M, Kem S, Sugawara T, Chan P, Adams J. Nicotinamide therapy protects against both necrosis and apoptosis in a stroke model. Pharmacol Biochem Behav. 2002;73(4):901–910. doi: 10.1016/s0091-3057(02)00939-5. [DOI] [PubMed] [Google Scholar]
- Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, Hamada J, Miyamoto E, Ushio Y. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab. 2001;21(4):351–60. doi: 10.1097/00004647-200104000-00004. [DOI] [PubMed] [Google Scholar]
- Zhuang S, Demirs JT, Kochevar IE. p38 mitogen-activated protein kinase mediates bid cleavage, mitochondrial dysfunction, and caspase-3 activation during apoptosis induced by singlet oxygen but not by hydrogen peroxide. J Biol Chem. 2000;275(34):25939–48. doi: 10.1074/jbc.M001185200. [DOI] [PubMed] [Google Scholar]