

Abstract
Prion diseases are caused by conversion of a normal cell-surface glycoprotein (PrPC) into a conformationally altered isoform (PrPSc) that is infectious in the absence of nucleic acid. Although a great deal has been learned about PrPSc and its role in prion propagation, much less is known about the physiological function of PrPC. In this review, we will summarize some of the major proposed functions for PrPC, including protection against apoptotic and oxidative stress, cellular uptake or binding of copper ions, transmembrane signaling, formation and maintenance of synapses, and adhesion to the extracellular matrix. We will also outline how loss or subversion of the cytoprotective or neuronal survival activities of PrPC might contribute to the pathogenesis of prion diseases, and how similar mechanisms are probably operative in other neurodegenerative disorders.
Keywords: prion, neurodegeneration, apoptosis, oxidative stress, copper, Bax
1. Introduction
Prion diseases result from conversion of a normal, cell-surface glycoprotein (PrPC) into a conformationally altered isoform (PrPSc) that is infectious in the absence of nucleic acids. Although we know a great deal about the role of PrPSc in the disease process, the normal, physiological function of PrPC has remained enigmatic. Attempts to deduce the function of PrPC from the phenotypes of PrP-null mice have been uninformative, since lines of these mice in which the adjacent Doppel (Dpl) gene is not artifactually up-regulated display no major anatomical or developmental deficits [1, 2]. Even mouse lines in which the PrP gene is deleted postnatally using a conditional Cre-Lox system are phenotypically relatively normal, arguing against the existence of other proteins that compensate for essential PrP functions in the adult [3]. Parts of the PrPC sequence have been highly conserved in evolution [4], suggesting important biological roles for the protein.
Investigating the biological activity of PrPC is likely to be crucial for understanding the pathogenesis of prion diseases, since alteration of this function could play a role in the disease process. In this review, we will summarize some of the major proposed functions for PrPC, with a particular emphasis on a role for the protein in protection from cellular stress. We will also outline how gain, loss, or subversion of PrPC function can play a role in the pathogenesis of prion diseases, and how similar mechanisms may be operative in other neurodegenerative disorders.
2. Cellular biology of PrPC
PrPC is expressed beginning early in embryogenesis, and in the adult it is present at highest levels in neurons of the brain and spinal cord [5, 6]. PrPC is also found at lower levels in glial cells of the CNS as well as in a number of peripheral cell types [7, 8]. Most PrPC molecules are normally localized on the cell surface, where they are attached to the lipid bilayer via a C-terminal, glycosyl-phosphatidylinositol (GPI) anchor [9]. The biosynthetic pathway followed by PrPC is similar to that of other membrane and secreted proteins, involving synthesis on ER-attached ribosomes, transit to the Golgi, followed by delivery to the cell surface [reviewed in 10]. PrPC is a glycoprotein, with two N-linked oligosaccharide chains of the complex type. Although most cell-surface PrPC is found in lipid rafts, some of the protein is transferred to clathrin-coated pits where is subject to constitutive endocytosis and recycling [11-14].
3. Identification of proteins that interact with PrPC
A powerful strategy for elucidating the physiological function of PrPC would be to identify other cellular proteins with which PrPC interacts. At least some of these interactors are likely to be components of the physiological pathways in which PrP plays a role. Over the years, a number of candidates have been identified as potential PrP-binding partners using conventional yeast two-hybrid screens, co-immunoprecipitation, cross-linking and other methods (Table 1) [reviewed in 15]. Several of the candidates listed in Table 1 (STI-1, N-CAM, Bcl-2, caveolin) will be discussed below, in conjunction with the cellular function of PrP in which they are purported to play a role. In almost every case, however, the physiological relevance of the proposed interactions remains uncertain. Some of the putative interactors are primarily or exclusively cytoplasmic proteins, and so would be unlikely to associate directly with PrP, which is localized to the outer face of the plasma membrane and to the lumen of organelles in the secretory pathway. One example of such a candidate is the laminin receptor precursor (LRP), which was originally identified in a yeast two-hybrid screen and has been claimed to serve as an endocytic receptor for cellular uptake of both PrPC and PrPSc [16, 17]. However, LRP lacks a signal peptide or transmembrane anchor that could target it to the secretory pathway, and the majority of the molecules are likely to be cytoplasmic.
TABLE 1. Putative PrP interactors.
| Candidate Interactor | Candidate Function | Identification Method | Localization | Reference |
|---|---|---|---|---|
| Grb2 | Signal transduction (adaptor protein) | Yeast two-hybrid; co-immunoprecipitation | Cytoplasm | [101] |
| Pint1 | Unknown | Yeast two-hybrid; co-immunoprecipitation | Cytoplasm | [101] |
| Synapsin 1b | Synaptic vesicle trafficking | Yeast two-hybrid; co-immunoprecipitation | Cytoplasm (synaptic vesicles) | [101] |
| TREK-1 | Two-pore K+ channel | Yeast two-hybrid; co-immunoprecipitation | Plasma membrane (transmembrane) | [169] |
| Tubulin | Microtubule subunit | Cross-linking | Cytoplasm (cytoskeleton) | [170] |
| NRAGE (Neurotrophin receptor-interacting MAGE homologue) | Activator of apoptosis | Yeast two-hybrid; co-immunoprecipitation | Cytoplasm | [171] |
| Laminin receptor precursor (LRP) | Extracellular matrix interactions | Yeast two-hybrid | Cytoplasm; Plasma membrane? | [16] |
| STI-1 (stress-inducible protein 1) | Heat shock protein | Complementary hydropathy; co-immunoprecipitation | Cytoplasm; Plasma membrane? | [107] |
| Hsp60 | Chaperone | Yeast two-hybrid | Cytoplasm | [172] |
| N-CAM | Cell adhesion | Cross-linking | Plasma membrane (transmembrane and GPI-anchored forms) | [144] |
| Bcl-2 | Multi-domain anti-apoptotic regulator | Yeast two-hybrid | Cytoplasm | [173] |
| Caveolin-1 | Caveolar coat | Co-immunoprecipitation | Plasma membrane (hairpin loop) | [103] |
Since PrP is a GPI-anchored protein, the entire polypeptide chain is located on the extracytoplasmic face of the lipid bilayer. Thus, direct PrP binding partners are most likely to be transmembrane and secreted proteins. Although PrP potentially exists as two transmembrane variants (NtmPrP and CtmPrP) that could theoretically interact with cytoplasmic partners [18, 19], these forms are normally present in minute amounts in the absence of predisposing mutations in the PrP molecule [18]. PrP molecules that lie entirely in the cytoplasm have also been described in cultured cells [20], but our own data indicate that these forms are likely to be artifacts of over-expression or treatment with proteasome inhibitors [21]. Of course, PrP could associate with cytoplasmic proteins indirectly, via involvement of intermediary proteins that serve as transmembrane linkers.
4. Anti-apoptotic activity of PrP
Several intriguing lines of evidence have emerged recently suggesting that PrPC may exert a cytoprotective activity, particularly against internal or environmental stresses that initiate an apoptotic program [reviewed in 22, 23]. This activity has been demonstrated in a variety of experimental systems, including cultured mammalian cells, yeast, and mice.
4.1. Cultured cells
One of the clearest examples of a cytoprotective activity of PrPC is the protein's ability to protect human fetal neurons in culture against apoptosis induced by Bax. Bax is a pro-apoptotic member of the Bcl-2 family that plays a major role in postmitotic neurons of the central nervous system [24, 25]. When human fetal neurons in culture were microinjected with a plasmid encoding Bax, ∼90% of the neurons underwent apoptosis; but when the neurons were co-injected with both Bax- and PrP-encoding plasmids, the percentage of apoptotic cells was reduced to ∼10% [26, 27]. This cytoprotective effect of PrP appeared to be specific for Bax, since PrP did not prevent neuronal apoptosis induced by Bak, t-Bid, staurosporine, or thapsigargin [28].
It has also been reported that expression of PrPC rescued immortalized hippocampal neurons (HpL3-4 cells) derived from Prn-p0/0 mice from apoptosis induced by serum deprivation [29, 30]. This effect appeared to involve a Bax-mediated apoptotic pathway.
Another system involves MCF-7 cells, which are derived from a human mammary adenocarcinoma and undergo apoptosis in response to treatment with the cytokine, tumor necrosis factor (TNF-α). Diarra-Mehrpour et al. [31] isolated a variant sub-clone of MCF-7 cells that was resistant to TNF-α-induced apoptosis, and found that this clone displayed a dramatic up-regulation of endogenous PrP gene expression. Moreover, over-expression of PrPC in the parent MCF-7 line was sufficient to render it resistant to TNF-α-induced cell death. At least part of the protective effect of PrPC involved suppression of a mitochondria-dependent death pathway.
PrP has also been found to rescue cultured cerebellar granule neurons [32] and N2a neuroblastoma cells [33] from apoptosis induced by Dpl. Dpl is a PrP paralog which causes a neurodegenerative phenotype when ectopically expressed in the CNS of Prn-p0/0 transgenic mice (see Section 4.3). In N2a cells, it was claimed that the protective effect involved a physical interaction between PrP and Dpl on the cell membrane.
In a recently published study, it was reported that PrPC promoted self-renewal of hematopoietic stem cells during serial transplantation [34]. This phenomenon could also relate to the cytoprotective activities of PrPC, since serial transplantation is likely to subject cells to apoptotic stress. In what could be a related effect, it was recently reported that PrPC positively regulates proliferation and differentiation of neural precursor cells in vitro and in vivo [35].
While these diverse model systems suggest a common mechanism for PrP cytoprotection, the specific cellular and molecular pathways involved remain largely unknown. Since Bax-dependent apoptosis is a common theme in several of these systems, one possibility is that PrP acts by inhibiting Bax-mediated cell death. Fig. 1 illustrates several possible mechanisms by which this might occur, based on known pathways for Bax action [24, 36]. First, GPI-anchored, PrP on the cell surface might interact with a putative transmembrane receptor, thereby initiating a signal transduction cascade. This cascade might then alter Bax activity directly, for example by inhibiting of its mitochondrial translocation, conformational change, or oligomerization (Fig. 1A). These same effects might also result from a physical interaction between Bax and cytoplasmic forms of PrP (Fig. 1B). Alternatively, a PrP-dependent signal might act upstream of Bax, causing either inhibition of pro-apoptotic, BH3-only activators of Bax (Fig. 1C), or enhanced association of Bax with anti-apoptotic regulators such as Bcl-2 or Bcl-XL (Fig. 1D). It is also possible that PrP suppresses downstream effects of Bax, such as release of cytochrome c, or activation of Apaf-1 and caspases (Fig. 1E). Finally, since Bax is known to have effects on calcium release and the unfolded protein response (UPR) in the ER, [37, 38], PrP traversing the secretory pathway could also affect Bax activity associated with this organelle, possibly via a transmembrane receptor (Fig. 1F).
FIGURE 1. Possible mechanisms for PrP suppression of Bax-induced apoptosis.
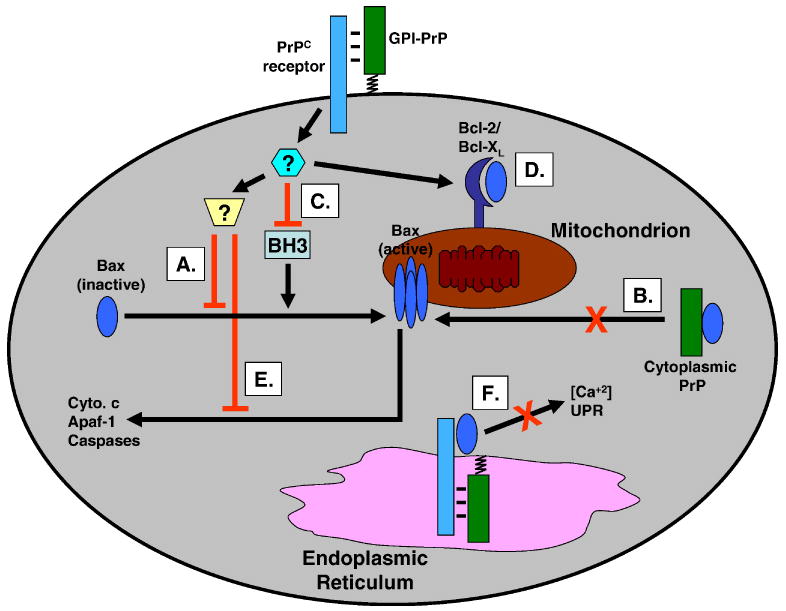
PrPC may inhibit Bax-mediated apoptotic pathways at several different points, either by a direct interaction between the two proteins or by involvement of additional, intermediary proteins. PrPC on the cell surface (GPI-PrP) may bind to a putative transmembrane receptor, initiating a signal transduction cascade that culminates in inhibition of Bax mitochondrial translocation, conformational change, or oligomerization (A). Cytoplasmic forms of PrP may produce similar effects via a direct interaction with Bax (B). PrP may inhibit pro-apoptotic, BH3-only proteins (C), or enhance an interaction between Bax and anti-apoptotic, multi-domain proteins such as Bcl-2 and Bcl-XL (D). PrP may suppress downstream events in the Bax pathway, such as cytochrome c (cyto. c) release, or activation of Apaf-1 and caspases (E). Finally, PrP in the ER may alter Bax function in this organelle, via effects on intracellular calcium and the unfolded protein response (UPR) (F).
At this point, there is a dearth of experimental data to distinguish among the different mechanisms outlined in Fig. 1. A recent study of the Bax suppressive effects of PrPC utilizing several different cell types (fetal neurons, HpL3-4 cells, MCF-7 cells) concluded that PrP inhibits the conformational change that occurs when Bax is activated and targeted to the outer mitochondrial membrane [28]. This result would be consistent with mechanisms A and B in Fig. 1. Mechanism B is also suggested by a study showing that an engineered form of cytoplasmic PrP protects fetal neurons against Bax [27]. However, other studies have shown that cytoplasmic PrP produces a toxic rather than protective effect by binding to and sequestering Bcl-2 [39]. In any event, the physiological significance of cytoplasmic PrP is uncertain at this point, since very little of this form appears to be generated in vivo from the wild-type molecule [40] Clearly, further progress in elucidating the mechanism underlying PrP cytoprotection will require identification of other proteins that interact physically and functionally with PrP and connect it to cell death and survival pathways.
4.2. Yeast
In order to investigate the anti-apoptotic effect of PrP in a genetically tractable experimental system, we have recently turned to baker's yeast, Saccharomyces cerevisiae. Extensive work by other laboratories has shown that heterologous expression of mammalian Bax in yeast is lethal [41]. Although S. cerevisiae does not contain endogenous Bcl-2 family members or caspases, the initial events underlying Bax activity in yeast and mammalian cells are similar, including translocation of the protein to mitochondria, release of cytochrome c, and alterations in mitochondrial function [41, 42].
We have found that expression of mammalian PrP in yeast efficiently suppresses cell death induced by Bax [43]. Our assay system utilizes yeast expressing mammalian Bax from a galactose-inducible promoter (GAL10). The yeast also constitutively express a form of PrP containing a modified signal peptide that we have shown to be targeted to the secretory pathway [44]. The assay involves comparing growth of the yeast on glucose vs. galactose medium. The basic effect is shown in Fig. 2A, B, which demonstrates that Bax-expressing yeast transformed with the PrP plasmid grow well on galactose medium, while Bax-expressing yeast transformed with the vector plasmid fail to grow on galactose. In contrast, both stains of yeast grow well on glucose medium, which does not induce synthesis of Bax. Fig. 2C demonstrates that PrP does not alter expression levels of Bax, ruling out a trivial explanation for PrP rescue activity. We have carried out a detailed structure-function analysis to define which domains of the PrP molecule are necessary for the Bax protective effect (Fig. 2D). We are currently using this yeast system to perform screens aimed at identifying genes that are essential for PrP rescue activity, and to characterize the cellular mechanisms involved.
FIGURE 2. PrP targeted to the secretory pathway protects yeast against Bax-induced cell death.
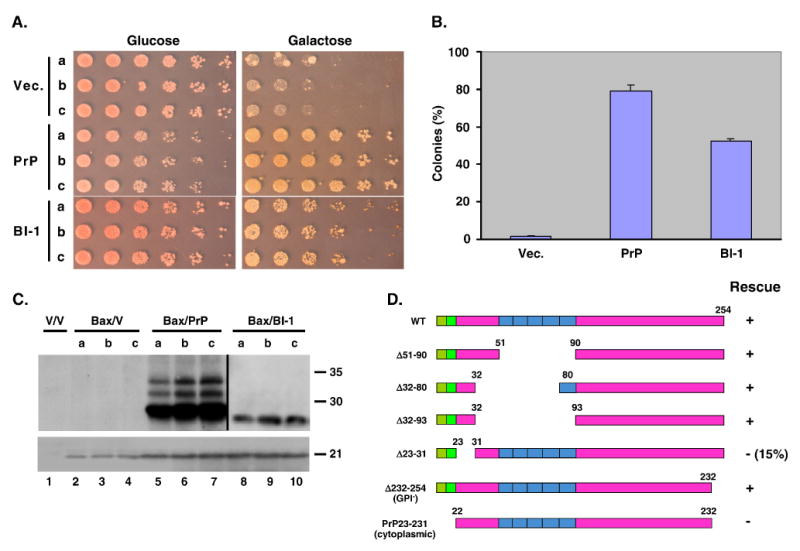
(A) S. cerevisiae expressing Bax from a galactose-inducible promoter were transformed with empty vector, or with vector constitutively expressing either PrP or human Bax-inhibitor 1 (BI-1) (a positive control protein). The PrP construct was engineered to allow expression in the secretory pathway [44]. Three independent transformants (a-c) were spotted in serial 5-fold dilutions (left to right) on glucose or galactose plates and allowed to grow for 3 or 6 days, respectively. Co-expression of PrP allows yeast to grow on galactose medium (i.e., under conditions where Bax synthesis is induced). (B) Quantitation of the protective effects of PrP and BI-1. Yeast transformed as in (A) were plated onto glucose or galactose plates, and the number of colonies counted. Results are expressed as the number of colonies on the galactose plates as a % of those on the glucose plates. PrP is even more potent than BI-1 in restoring growth in the presence of Bax. (C) Lysates prepared from three independent yeast transformants (as described in A) were subjected to Western blotting using anti-PrP (upper panel, lanes 1-7), anti-HA to detect BI-1 (upper panel, lanes 8-10), or anti-Bax (lower panel, lanes 1-10). Lane 1 shows yeast that carry the empty vectors used for Bax and PrP/BI-1 expression. Neither PrP nor BI-1 affect expression levels of Bax. (D) Deletion analysis to determine which domains of PrP are required for its ability to protect yeast from Bax-induced cell death. Yeast expressing Bax from a galactose-inducible promoter were transformed with plasmids encoding wild-type (WT) PrP, or the indicated deletion constructs. Growth on glucose and galactose plates was assessed as in panels A and B in order to score PrP rescue activity. The green boxes indicate the hybrid signal sequence used to target PrP to the secretory pathway [44], and the blue boxes indicate the octapeptide repeats. Data are from Li and Harris [43].
The ability of PrP to rescue yeast from Bax-induced death has been confirmed by Bounhar et al. [45] using dye exclusion and colony-forming ability as measures of cell viability. In contrast to us, however, these authors found that non-secreted (cytoplasmic) forms of PrP possessed rescue activity; neither of the two PrP constructs they analyzed entered the secretory pathway to any significant extent. They also reported that PrP failed to prevent growth inhibition caused by Bax, although PrP expression did have effects on the yeast cell cycle as monitored by flow cytometry.
4.3. Mice
Two kinds of mouse models dramatically exemplify a cytoprotective activity of PrPC in vivo. In one model, Shmerling et al. [46] expressed a series of N-terminally deleted forms of PrP in transgenic mice. Deletions encompassing residues 32 through 106 did not cause any abnormalities, but two larger deletions (Δ32-121 or Δ32-134, collectively referred to PrPΔN) caused a progressive neurodegenerative illness in mice lacking both copies of the endogenous PrP gene (Prn-p). Strikingly, a single Prn-p allele was sufficient to completely abrogate the phenotype of these animals. A similar phenomenon was described in certain Prn-p0/0 mouse lines in which the prion-like protein Dpl was ectopically expressed in the brain as a result of intergenic splicing events between the adjacent PrP and Dpl genes [47-50]. Dpl is structurally similar to PrPΔN, since it lacks regions homologous to the flexible, N-terminal tail of PrP. Reintroduction of wild-type PrP also rescued Dpl-expressing mice from neurodegenerative illness [49, 51-53].
We have recently documented an analogous rescue effect in Tg(Δ105-125) mice that express PrP harboring a deletion of residues 105-125 [54]. These mice spontaneously develop a severe neurodegenerative illness that is lethal within one week of birth in the absence of endogenous PrP. This phenotype is reversed in a dose-dependent fashion by co-expression of wild-type PrP, with 5-fold over-expression delaying death beyond 1 year. Thus, the phenotype of Tg(PrPΔ105-125) mice is reminiscent of, but much more severe than, those described in mice that express PrPΔN and Dpl. The fact that PrP suppresses the phenotypes induced by PrPΔN, PrPΔ105-125 and Dpl suggests that a common mechanism facilitates the protective effect of PrP against each of these toxic proteins. We have speculated that the greatly enhanced toxicity of PrPΔ105-125, coupled with the requirement for supraphysiological levels of wild-type PrP to rescue the neurodegenerative phenotype, is due to increased affinity of PrPΔ105-125 for a hypothetical receptor that transduces the toxic signal [54].
4.4. Structure-activity analysis of PrP cytoprotection in yeast, mammalian cells and mice
It is instructive to compare the sequence domains of PrP that are essential for its cytoprotective activity in yeast, cultured mammalian cells, and mice. Such comparisons suggest a number of similarities between these three systems, as well as a few differences (Table 2). Thus, this cytoprotective function of PrPC appears to be partially but not completely conserved in evolution.
TABLE 2. Structure-activity relationships for PrP cytoprotection in yeast, mammalian cells, and mice.
| PrP MOLECULE | YEAST | MAMMALIAN CELLS | MICE | ||
|---|---|---|---|---|---|
| Rescue from Bax | Rescue | Toxic Stimulus / Cell Type | Rescue | Toxic Stimulus | |
| Wild-type PrP | + [43, 45] | + | Bax / human fetal neurons [26] | + | PrPΔN [46] |
| + | Serum deprivation / immortalized hippocampal neurons [29] | + | Doppel [49, 52, 53] | ||
| + | Doppel / cerebellar granule neurons [32] | + | PrPΔ105-125 [54] | ||
|
| |||||
| Δ23-31 | − (Δ23-31) [43] | − (Δ23-28) | Doppel / cerebellar granule neurons [32] | − (Δ23-88) | Doppel [57] |
| + (Δ32-80/93) | PrP N [46] | ||||
|
| |||||
| Δ octarepeats | + [43] | − | Bax / human fetal neurons [26] | + (Δ32-93) | PrPΔN [46] |
| − | Serum deprivation / immortalized hippocampal neurons [60] | ||||
| − | Doppel / cerebellar granule neurons [32] | ||||
|
| |||||
| GPI- | + [43] | + | Bax / human fetal neurons [26] | ND | |
| + | Doppel / cerebellar granule neurons [32] | ||||
|
| |||||
| Cytoplasmic PrP (23−231) | − [43] | + | Bax / human fetal neurons [27] | ND | |
|
| |||||
| D178N | + [43] | − | Bax / human fetal neurons [26] | ND | |
|
| |||||
| E199K | + [43] | ND | + | Doppel [57] | |
|
| |||||
| PG14 | − [43] | ND | +/− | PrPΔN (Li et al., ms. in prep.) | |
ND, not determined
The first region of importance (residues 23-31; KKRPKPGGW in mouse) lies at the extreme N-terminus of PrP and includes four positively charged residues. This segment is known to play a role in endocytic trafficking of PrP [14, 55] and in localization of the protein to lipid rafts [56]. This region has been found to be essential for PrP protection against Bax in yeast [43] and Dpl in cerebellar granule neurons [32]. In transgenic mice, deletions beginning at residue 32 (PrPΔ32-80 and PrPΔ32-93) do not affect the ability of PrP to suppress neurodegeneration induced by PrPΔN [46] (Li et al., manuscript in preparation). In contrast, deletion of residues 23-88 obliterates the ability of PrP to rescue mice from Dpl-induced toxicity [57]. The latter two results together implicate residues 23-31 in protection against PrPΔN and Dpl, although generation of Tg(PrPΔ23-31) mice will be necessary to confirm this inference. The requirement for residues 23-31 suggests that PrP cytoprotective activity may depend on localization to the cell surface, lipid rafts, or endosomal compartments.
Another domain that has been tested is the C-terminal, GPI addition signal. In both yeast [43] and human neurons [26], deletion of this region does not impair the ability of PrP to suppress Bax-mediated apoptosis, implying that PrP cytoprotective activity does not require tethering to the plasma membrane. Similarly, PrP rescue of granule neurons from Dpl-induced apoptosis does not require the GPI anchor [32]. To determine whether these observations in yeast and cultured neurons hold true in vivo, transgenic mice expressing GPI-anchorless PrP [58] could be crossed with mice expressing PrPΔN or Doppel. The lack of requirement for the GPI anchor suggests that the cytoprotective activity of PrP does not involve signal transduction through the PrP molecule itself, but perhaps through a transmembrane protein to which PrP binds on the extracellular surface.
A related issue concerns the necessity for expression of PrP in the secretory pathway. In our experiments, deletion of the N- and C-terminal signal peptides (PrP23-231) completely abolishes the ability of PrP to rescue yeast from Bax-induced cell death [43]. This result indicates that PrP must be targeted to the secretory pathway to be active in yeast, and also makes it unlikely that PrP interacts directly with cytosolic Bax (mechanism B in Fig. 1). In contrast, PrP23-231 retains full rescue activity against Bax in cultured human neurons [27]. Whether this discrepancy implies a fundamental difference in the cytoprotective pathways operative in yeast and mammalian cells remains to be determined. The protective activity of cytoplasmic PrP in human neurons is surprising, since in other cultured cell types and in transgenic mice cytoplasmic PrP is toxic [39, 59].
The role of the PrP octapeptide repeats has also been analyzed in several systems. The repeats are known to bind copper ions, which have been postulated to play a role in the function of PrPC (see Section 6). Deletion of the octapeptide repeats abolishes the ability of PrP to protect cortical neurons from Bax [26], granule neurons from Doppel [32], and immortalized hippocampal neurons from serum deprivation [60]. In contrast, removal of the repeats does not affect the ability of PrP to protect yeast from Bax [43] or transgenic mice from PrPΔN [46]. These observations are difficult to reconcile, perhaps because the octapeptide repeats perform distinct functions in different cell types or in the context of different toxic insults.
Another interesting point of comparison concerns the effect of disease-causing mutations on the rescuing activity of PrP. D178N and E199K mutants retain the ability to rescue yeast from Bax-mediated cell death [43]. In contrast, PG14, a mutant harboring an insertion of nine additional octapeptide repeats, lacks rescue activity [43]. Biochemical analysis of these mutant proteins suggests that the loss of protective activity of PG14 may be due to its aggregated state, whereas D178N and E199K are completely soluble in yeast. This observation is consistent with studies in transgenic mice, since PG14 PrP is aggregated when expressed in the brain [61] and is also partially deficient in its ability to suppress the neurotoxicity of PrPΔ32-134 (Li et al., manuscript in preparation). PrP carrying the E199K mutation, which has been shown to be completely soluble in mouse models [62], maintains the ability to suppress Doppel-induced neurodegeneration [57]. Although soluble in yeast, D178N PrP is partially detergent resistant in cultured neurons [63], which may explain the inability of this mutant to rescue neurons from Bax [26]. Taken together, these results indicate that the cytoprotective activity of PrP requires a soluble form of the protein, and that mutations inducing aggregation impair activity.
5. PrPC and oxidative stress
A frequently discussed hypothesis to explain the pathogenesis of several neurodegenerative disorders involves chronic oxidative stress. Dysfunction of any of several interconnected cellular pathways is sufficient to cause oxidative stress in the brain, including impaired mitochondrial function, increased oxidative damage, defects in the ubiquitin–proteasome system, the presence of aggregated proteins, changes in iron metabolism, excitotoxicity, and inflammation [reviewed in 64].
Several lines of evidence suggest that PrPC may play a role in protecting cells from oxidative stress [reviewed in 65]. Perhaps the most compelling observation is that neurons (cerebellar granular and neocortical) cultured from Prn-p0/0 mice are more susceptible than neurons from wild-type mice to treatments with agents that induce oxidative stress, including hydrogen peroxide, xanthine oxidase and copper ions [66, 67]. Consistent with these cell culture results, brain tissue from Prn-p0/0 mice exhibits biochemical changes indicative of oxidative stress, such as increased levels of protein carbonyls and lipid peroxidation products [68]. In addition, brain lesions induced by hypoxia and ischemia are larger in Prn-p0/0 compared to Prn-p+/+ mice [69-71]. Since hypoxia and ischemia probably cause neuronal death via oxidative damage, these observations also tie PrPC to protection from oxidative stress.
How might PrPC protect cells from oxidative stress? One possibility is that PrPC itself acts directly to detoxify reactive oxygen species (ROS). Consistent with this idea, it has been claimed that PrPC (both recombinant, and immunoprecipitated from brain tissue or cultured cells) displays a copper-dependent superoxide dismutase (SOD) activity [72, 73]. However, the biological significance of these results is questionable for several reasons. First, the SOD activity measured for recombinant PrP depended on refolding the protein from a denatured state in the presence of supra-physiological concentrations of copper. Second, even small organic molecules like amino acids can bind copper and exhibit weak dismutase activity. Finally, copper binds much more weakly to PrPC than to known cuproenzymes like Cu-Zn SOD [74], arguing against the possibility that the copper plays a specific catalytic role in PrPC. In a recent report, Jones et al. [75] failed to detect SOD activity above background levels in recombinant PrP refolded from a denatured state in the presence of excess copper (as in the original studies of Brown and colleagues), or in PrP loaded with copper after folding.
A second hypothesis is that PrPC acts indirectly to protect cells from oxidative stress by up-regulating the activities of other proteins, such as Cu-Zn SOD, that detoxify ROS. It has been reported that the enzymatic activity and the 64Cu loading of Cu-Zn SOD from the brains of Prn-p0/0 mice is 10-50% of normal [66, 76, 77]. Conversely, it has been claimed that the activity and copper loading of Cu-Zn SOD are increased in PrP-over-expressing mice. However, we [78] and others [79] have been unable to replicate these results. The activities of other anti-oxidant enzymes such as catalase and glutathione reductase have been reported to be decreased in Prn-p0/0 mice [77, 80], but whether PrPC plays a direct role in regulating these molecules remains to be determined.
It is also possible that PrPC acts either upstream or downstream of ROS to protect cells from oxidative stress. In some situations, for example, oxidative stress may activate apoptotic pathways [64]. In such cases, the anti-apoptotic effects of PrPC described in Section 4 may account for the protein's ability to protect cells against oxidative stress.
6. PrPC and copper
Copper is an essential cofactor for a number of enzymes that catalyze redox reactions. Because ionic copper is highly reactive, however, cells have evolved specialized mechanisms for its uptake and transport [81]. Defects in copper metabolism have been linked to a number of human diseases, including several neurodegenerative disorders [82]. An extensive body of evidence has accumulated suggesting a connection between PrPC and copper ions [reviewed in 83, 84].
The most widely agreed upon observation is that PrPC is a copper-binding protein [85-88]. The histidine-containing octapeptide repeats specifically bind up to four Cu2+ ions copper in a pH-dependent and negatively cooperative manner, with an affinity that may be as high as 0.1 nanomolar (depending on binding site occupancy) [89]. Binding involves coordination with nitrogen atoms in the imidazole side chains of histidine residues, as well as with nitrogen and oxygen atoms in main-chain amide linkages involving glycine residues. Two additional copper binding sites exist at residues 96 and 111 [90]. Copper binding has been shown to cause conformational changes in the flexible, N-terminal tail of PrP [91, 92].
Copper ions also alter the biochemical and cell biological properties of PrPC. Copper causes PrPC in brain homogenates to assume an aggregated and protease-resistant form that is distinct from PrPSc [93]. In addition, micromolar concentrations of copper stimulate endocytosis of cell-surface PrP via clathrin-coated pits [94-96]. This effect requires binding of copper to the octapeptide repeats, and exit of PrP from lipid raft domains [55]. Based on the effects of copper on PrP trafficking, we have hypothesized that the protein serves as a receptor for cellular uptake or efflux of copper ions [94]. It is also possible that PrPC acts as a sink for binding of copper ions at the cell surface without actually undergoing endocytosis.
One piece of evidence that would strongly support a role for PrPC in copper uptake or efflux would be a correlation between PrPC expression levels and the copper content of cells or tissues. An initial report indicated that the content of copper, but not of several other transition metals, is only 10% of normal in crude membranes, synaptosomes, and endosomes derived from the brains of Prn-p0/0 mice [85]. A subsequent study from the same authors [97] reported that synaptosomes from Prn-p0/0 mice had a copper content that was 50% of the wild-type level, a considerably smaller difference than in the original report. Based on these results, the authors proposed that PrPC may play a role in regulating copper release at the synapse [98].
We have re-examined this subject by using mass spectrometry to measure the concentrations of several transition metals in brain tissue from wild-type and Prn-p0/0 mice, as well as in Tga20 mice that over-express PrP by 10-fold. We were unable to find any differences in metal content in either whole brain or of several subcellular fractions among mice of these three genotypes [78], and we believe that the results of Brown and colleagues are likely to be in error.
We have also utilized yeast as an experimental system to test the role of PrPC in copper metabolism [44]. Much of what we know about cellular utilization of copper ions in eukaryotes is derived from studies of S. cerevisiae [81]. We therefore tested the effect of PrP expression on the growth phenotypes of yeast strains harboring deletions of genes that encode key components of copper utilization pathways, including transporters, chaperones, pumps, reductases, and cuproenzymes. We failed to find an effect of PrP expression on the growth deficiency of any of the yeast strains tested.
Taken together, these studies in yeast and mammalian systems suggest that PrPC is unlikely to be part of a major pathway for copper uptake or efflux by cells and tissues. Of course, the results do not rule out involvement of PrPC in more specialized processes for metabolism or trafficking of this metal ion. Additionally, copper binding may play a regulatory role by modulating the physiological activity of PrPC.
7. PrPC and transmembrane signaling
Since most PrPC is localized to the cell surface, it is reasonable to hypothesize that the protein could participate in transmembrane signaling processes. Like other GPI-anchored proteins, PrPC resides in lipid raft domains on the plasma membrane, which are known to serve as molecular scaffolds for signal transduction [99, 100]. Since its polypeptide chain is entirely extracellular, PrPC would presumably need to interact with transmembrane adaptor proteins in order to transmit signals into the cytoplasm. Yeast two-hybrid screens have identified several signaling molecules that bind to PrP, including Grb2 and synapsin 1b [101]. However, because these molecules are cytoplasmic, they are unlikely to directly associate with membrane-bound PrPC in a cellular environment. Clearly, a crucial challenge in the field is to identify the molecular components of putative PrPC-mediated signal transduction pathways.
There are now a number of studies suggesting that PrPC can activate transmembrane signaling pathways involved in several different phenomena, including neuronal survival, neurite outgrowth, and neurotoxicity. In some of these cases, signal transduction is initiated by interaction of PrPC with specific protein or peptide ligands. In other cases, PrPC appears to act constitutively.
An artificial situation in which PrPC-mediated signaling has been observed involves antibody-induced cross-linking of cell-surface PrPC. Presumably, the antibodies in this type of experiment are mimicking the action of naturally occurring PrPC ligands. Antibody-induced cross-linking of GPI-anchored proteins on the cell surface is a technique commonly used to activate phosphorylation-dependent signaling cascades in lymphocytes [102]. Similarly, antibody-mediated cross-linking of PrPC on a neuroectodermal cell line (1C11) was found to stimulate the activity of the non-receptor tyrosine kinase, fyn [103]. This effect was reported to require interaction of PrPC with the raft protein caveolin. In a subsequent study, it was shown that antibody-induced fyn activation in 1C11 cells led to downstream stimulation of NADPH oxidase and extracellular-regulated kinases (ERKs), as well as production of reactive oxygen species [104]. The activities of several G protein-coupled serotonin receptors found on the surface of these cells were also altered by PrP cross-linking [105]. The signaling pathways engaged by anti-PrP antibodies in 1C11 cells are postulated to have pro-survival effects [104].
Another kind of pro-survival signaling pathway that has been characterized involves an interaction between PrPC and stress-inducible protein 1 (STI-1). STI-1 was originally described as a co-chaperone found in macromolecular complexes with heat shock proteins of the Hsp70 and Hsp90 families [106]. Although STI-1 lacks a signal sequence and is primarily localized to the cytoplasm and nucleus, some molecules have been reported to reside on the plasma membrane and to co-immunoprecipitate with PrPC [107]. Interaction with cell surface STI-1 has been proposed to mediate PrPC-dependent protection of retinal explants from anisomycin-mediated cell death [107]. This effect was found to depend on activation of a cAMP/protein kinase A pathway [108]. A recent study also demonstrated that incubation of cultured hippocampal neurons with recombinant STI-1 stimulated neurite outgrowth in a PrP-dependent manner, an effect requiring signaling through a mitogen-activated protein kinase (MAPK) pathway [109].
Two recent studies have suggested a potential role for the PI3 kinase/Akt signaling pathway in the neuroprotective effects of PrPC. In one study [110], infarct volumes were measured in Prn-p+/+ and Prn-p0/0 mice that had been subjected to focal cerebral ischemia. It was found that Prn-p0/0 mice displayed significantly larger infarct volumes, demonstrating a protective role for PrPC in response to brain injury. Additionally, the level of phosphorylated Akt was diminished in Prn-p0/0 mice, indicating that PrPC may enhance Akt-dependent cell survival pathways to prevent damage inflicted by ischemic brain injury. In a second study [111], Akt activity was found to be diminished in neurons and brain tissue from Prn-p0/0 mice compared to Prn-p+/+ mice. Moreover, it was reported that pharmacological inhibition of Akt reduced the ability of PrPC to protect cells against oxidative damage.
Another context in which PrPC-mediated signal transduction events have been observed involves neurite outgrowth. Cell surface PrPC facilitates axonal outgrowth via cis and trans interactions with N-CAM, a process that involves recruitment of N-CAM to lipid rafts and activation of fyn kinase [112]. Treatment of cultured neurons with recombinant PrP also enhances neurite outgrowth and neuronal survival, concomitant with activation of several kinases, including fyn, PKC, PKA, PI-3 kinase/Akt, and ERK [113, 114].
The signaling processes described thus far involve positive effects of PrPC on neuronal survival or differentiation. However, there is also evidence that PrPC can mediate neurotoxic effects via activation of specific signaling cascades. One example involves the synthetic peptide PrP106-126, which displays certain biochemical properties of PrPSc and has been used to mimic the effects of PrPSc on cultured cells [115]. PrP106-126 is toxic to cultured neurons and neuronal cell lines, but only those that express PrPC, suggesting that the toxic action is mediated by a PrPC-dependent signaling pathway [116, 117]. Consistent with this idea, PrP106-126 has been reported to stimulate a number of intracellular kinase cascades, including those involving p38, ERK1/2, and JNK1/2 [118-120]. In a second example of PrPC-mediated neurotoxicity, PrPC expression has been reported to sensitize neurons and neuronal cell lines to the apoptotic action of the kinase inhibitor, staurosporine [121-123]. This effect required endocytosis of PrPC, and was mediated by increased activity of the key transcriptional regulator p53.
8. A role for PrPC at synapses
Several experimental observations suggest that PrPC could play a role in synaptic structure, function or maintenance. This hypothesis is consistent with the fact that synaptic pathology is often a prominent feature of prion diseases [124]. Light and electron microscopic immunocytochemical studies, as well as localization of a PrP-EGFP fusion protein, indicate that PrPC is preferentially concentrated along axons and in pre-synaptic terminals [125-130]. In addition, PrPC is subject to anterograde and retrograde axonal transport [131, 132], and PrP-EGFP fusion proteins can be visualized in what appear to be axonally transported synaptic vesicles (Medrano and Harris, unpublished observations). Incubation of cultured hippocampal neurons with recombinant PrP induces rapid elaboration of axons and dendrites, and increases the number of synaptic contacts [113]. This result suggests that PrPC could play a regulatory role in synapse formation. There is evidence that PrPC could serve a function at peripheral as well as central synapses. It was reported that PrPC is concentrated at the neuromuscular junction where it is localized in the sub-synaptic sarcoplasm, possibly associated with endosomal structures [133]. In addition, nanomolar concentrations of recombinant PrP have been found to potentiate acetylcholine release at the neuromuscular junction [134].
Electrophysiological recordings from brain slices of Prn-p0/0 mice also support a functional role for PrP in synaptic transmission. In hippocampal slices from Prn-p0/0 mice, it was initially reported that long term potentiation was impaired and receptor-mediated fast inhibition involving GABA-A receptors was decreased [135, 136]. However, this result was subsequently disputed [137]. More recent studies have demonstrated a positive correlation between the expression level of PrPC and the overall strength of glutamatergic transmission in the hippocampus, with PrP-over-expressing mice exhibiting supra-physiological responses [138]. At least part of this effect seemed to result from more efficient recruitment of pre-synaptic fibers as the level of PrPC increased. Correlating with reduced after-hyperpolarization seen in hippocampal CA1 neurons of Prn-p0/0 mice [139], cerebellar Purkinje cells from these animals have been reported to show decreased Ca2+-activated K+ currents [140]. Reduced after-hyperpolarization was observed in pyramidal neurons regardless of whether the PrP gene was deleted pre- or postnatally [3].
Prn-p0/0 mice have been reported to display several other neurobiological abnormalities that may also relate to the participation of PrPC in synapse formation and function. These include alterations in nerve fiber organization [141], circadian rhythm [142], and spatial learning [143].
9. PrPC and cell adhesion
PrPC has been found to interact with several proteins involved in cell adhesion. For example, cross-linking experiments have identified a binding interaction between PrP and neural cell adhesion molecule (N-CAM) [144]. In cultured hippocampal neurons, this interaction results in redistribution of N-CAM to lipid rafts, activation of fyn kinase, and enhancement of neurite outgrowth [112]. Laminin, a major structural component of basement membranes, has also been shown to be a binding partner of PrPC. In neurons, laminin plays a significant role in cell proliferation, supports neurite outgrowth, and aids in cellular migration. Graner et al. [145] have shown that PrP binding to laminin promotes neurite outgrowth in PC12 cells and hippocampal neurons. Laser ablation of cell-surface PrP caused retraction of neurites [146]. Finally, expression of PrP has been found to enhance aggregation of neuroblastoma cells, although the molecular mechanisms underlying this phenomenon are uncertain [147].
10. Prion pathogenesis may involve alterations in the physiological function of PrPC
A great deal of effort in the prion field has been devoted to understanding of the chemical nature of the infectious agent, and testing the validity of the protein-only mechanism of infectivity. In contrast, much less attention has been paid to the question of how PrPSc or other abnormal forms of PrP cause CNS pathology. It is now becoming clear, however, that prions may kill nerve cells by virtue of their ability to perturb the normal, physiological activities of PrPC. In this section, we will outline three different hypotheses explaining how changes in PrPC function can cause neurodegeneration [reviewed in 148, 149].
10.1. Gain of function
The most widely discussed hypothesis is that prion pathology is attributable to a toxic gain of function mechanism (Fig. 3A). In this view, PrPSc possesses novel toxic properties that are not related to the normal, physiological function of PrPC. For example, aggregates of PrPSc may block axonal transport, interfere with synaptic function, or trigger apoptotic pathways.
FIGURE 3. Models for the cellular toxicity of PrPSc.
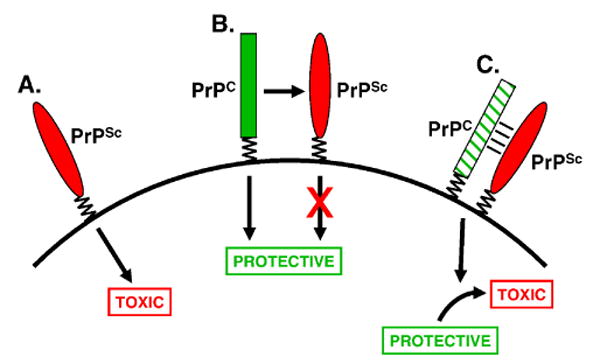
(A) Toxic gain-of-function mechanism. PrPSc (or PrPtoxic, a pathogenic intermediate) possesses a novel neurotoxic activity that is independent of the normal function of PrPC. (B) Loss-of-function mechanism. PrPC possesses a normal, physiological activity, in this case neuroprotection, that is lost upon conversion to PrPSc. (C) Subversion-of-function mechanism. The normal, neuroprotective activity of PrPC is subverted by binding to PrPSc (or PrPtoxic). Cross-hatching of the rectangle representing PrPC indicates a change in its signaling properties such that a neurotoxic rather than a neuroprotective signal is delivered. Taken from Harris and True [148].
10.2. Loss of function
An alternative hypothesis for prion toxicity postulates that PrPC possesses a biological activity that is lost upon conversion to or contact with PrPSc (Fig. 3B). Loss of this putative PrPC function would then cause neurodegeneration. In principle, loss of any of several of the putative functions of PrPC discussed above could produce pathogenic consequences. However, the anti-apoptotic activity of PrPC (Section 4) is most easily accommodated in such a mechanism, since loss of this activity could lead directly to neuronal death.
Two considerations would seem to argue against a loss of function mechanism in prion diseases. First, genetic ablation of PrP expression, either prenatally [1, 2] or postnatally [3], has relatively little phenotypic effect, and does not produce any features of a prion disease. Thus, loss of PrPC function cannot, by itself, account for prion-induced neurodegeneration. However, it is possible that a loss of function mechanism exacerbates pathology caused by toxic gain of function or other mechanisms. For example, a cytoprotective activity of PrPC that is dispensable under normal conditions may become essential in the disease state due to cellular or organismal stress. Loss of PrPC function as a pathogenic mechanism also appears to be incompatible with the dominant mode of inheritance of familial prion diseases. In this regard, however, PrPSc or mutant PrP may sequester wild-type PrPC into aggregates that lack functional activity, thereby producing a dominant-negative effect.
A consideration which argues for a loss rather than a gain of function in some familial prion diseases relates to the effect of PrP mutations on the thermodynamic stability and biochemical properties of PrP. Although some mutations markedly destabilize the protein, favoring misfolding and formation of PrPSc-like aggregates, others do not [150, 151]. It is difficult to explain the pathogenicity of the latter mutants on the basis of the accumulation of toxic protein aggregates. Rather, it seems more likely that these proteins are deficient in some functional property normally displayed by wild-type PrP. Consistent with this proposal, we have shown that an octapeptide insertional mutation in PrP partially impairs the cytoprotective activity of the protein in both a yeast model [43] and a transgenic mouse model (Li et al., manuscript in preparation).
10.3. Subversion of function
A third possible hypothesis for prion pathogenesis involves a subversion of the normal neuroprotective function of PrPC (Fig. 3C). In this mechanism, interaction with PrPSc converts PrPC from a transducer of neuroprotective signals into a transducer of neurotoxic signals. Consistent with this model, there are now a number of experimental situations in which expression of PrPC in neurons, rather than being neuroprotective, appears to be essential for conferring sensitivity to PrP-related neurotoxic insults. First, Prn-p0/0 neurons are resistant to the toxic effects of PrPSc supplied from grafted brain tissue [152] or from nearby astrocytes [153]. Second, scrapie-inoculated mice expressing a GPI-negative version of PrP develop minimal brain pathology and neurological dysfunction despite the accumulation of numerous PrPSc-containing amyloid plaques [58]. This result implies that PrP must be membrane-anchored to efficiently transduce a toxic signal. Third, Prn-p0/0 neurons in culture have been found to be resistant to apoptosis induced by exposure to the synthetic peptide PrP106-126, which has been used as a model for PrPSc [117].
How might the neuroprotective activity of PrPC be subverted to produce a neurotoxic effect? Presumably, a physical interaction of PrPSc with PrPC is required. One possibility is that PrPSc induces aggregation of cell surface PrPC, thereby generating a neurotoxic rather than a neuroprotective signal. Consistent with this model, cross-linking of PrPC with anti-PrP antibodies induces apoptosis of CNS neurons in vivo [154]. Alternatively, PrPSc may bind to and block specific regions of PrPC, thereby altering the signaling properties of the latter.
Support for the latter mechanism comes from Tg(PrPΔ105-125) mice we have made that express PrP containing a deletion of a highly conserved block of 21 amino acids in the unstructured, N-terminal tail of the protein [54] (see Section 4.3). The neurotoxicity of PrPΔ105-125 observed in these animals suggests a model in which residues 105-125 constitute a critical binding site for interaction between PrPC and a hypothetical, cell-surface receptor (designated Tr) that can transduce either neuroprotective or neurotoxic signals. PrPC binding to Tr normally elicits a neuroprotective signal, but deletion of the 105-125 domain of PrPC subverts this interaction in such a way that a neurotoxic signal is generated. We speculate that exogenous PrPSc perturbs binding interactions between Tr and the 105-125 domain of PrPC, thereby producing a neurotoxic effect equivalent to deletion of residues 105-125 [54]. A similar phenomenon might occur with the synthetic peptide PrP106-126, which is toxic to cultured neurons in PrPC-dependent fashion [117]. In this scenario, the neurotoxic pathways activated by PrPSc and PrP106-126 would be the same as those activated by PrPΔ105-125, PrPΔN and Dpl. However, wild-type PrPC would have opposite effects in the two situations: it would suppress the toxicity of PrPΔ105-125, PrPΔN and Dpl, but it would be required for the toxicity of PrPSc and PrP106-126.
10.4. Loss of function as a pathogenic mechanism in other neurodegenerative diseases
Toxic gain of function is commonly invoked to explain the phenotypes of other dominantly inherited neurodegenerative disorders including Alzheimer's disease, Huntington's and other polyglutamine expansion diseases, Parkinson's disease, frontotemporal dementia, and amyotrophic lateral sclerosis. In these disorders, it is postulated that intracellular or extracellular aggregates of the relevant misfolded protein (Aβ, huntingtin, α-synuclein, tau, or SOD1) possess a neurotoxic activity that is not directly related to the normal, physiological function of the parent protein [155]. However, in at least some of these disorders, a loss of function mechanism similar to what we have proposed for prion diseases (Section 10.2) may also be operative. Huntington's and Parkinson's diseases provide especially clear examples of this idea.
Huntington (Htt), like PrP, has been found to possess physiological activities that promote neuronal survival, and loss of these activities may contribute to disease pathogenesis [156]. For example mice lacking Htt show extensive cell death in the embryonic ectoderm [157]. Moreover, it has been reported that wild-type Htt up-regulates transcription of BDNF, and that mutant Htt lacks this activity, leading to compromised neuronal differentiation and survival [158]. Wild-type Htt may also facilitate vesicular transport of BDNF along axons [159]. Consistent with a neuroprotective effect of Htt, over-expression of wild type Htt in transgenic mice significantly reduced the cellular toxicity elicited by various Htt mutants [160, 161].
Similarly, there is evidence that α-synuclein, which comprises the Lewy bodies of Parkinson's disease, may have anti-apoptotic activities. Expression of wild type α-synuclein in primary neurons and neuronal cell lines protects the cells from death induced by serum deprivation, H2O2-induced oxidative stress, and glutamate-induced toxicity [162-164]. This protective effect was not observed with the two disease-associated mutants of α-synuclein (A30P and A53T) [162].
These results suggest that some of the pathogenic mechanisms operative in prion diseases may be closely related to those underlying other, more common neurodegenerative disorders. This idea is consistent with evidence that prion neurotoxicity is attributable to small, non-infectious PrP oligomers that are distinct from PrPSc, and that are similar to the pathogenic aggregates seen in other protein misfolding disorders [148, 165].
11. Conclusions
For many years the PrPSc isoform of the prion protein has consumed the attention of investigators because of its relationship to infectivity, and its specific accumulation during the disease process. PrPC, the cellular isoform, was of interest primarily because it served as a necessary precursor to PrPSc, but its intrinsic physiological function seemed irrelevant to understanding the disease process. It is now clear, however, that alterations in the normal function of PrPC may play a crucial role in causing or contributing to the disease phenotype. For this reason, elucidating the physiological activity of PrPC has become a major priority in the prion field. In this review, we have discussed each of the major functions that have been proposed for PrPC. Although the evidence is not conclusive for any them, we have emphasized the anti-apoptotic activity of PrPC as being of particular interest, because it has been demonstrated in a variety of experimental systems, from yeast to mice. In addition, there is evidence that loss or subversion of this activity may be related to neuronal death in prion diseases.
Understanding the normal function of PrPC has important implications for the therapy of prion disorders. At present, most therapeutic strategies are directed at inhibiting the formation of PrPSc [166]. If alterations in PrPC function play an important role in prion-induced pathology, then an alternative approach is to target the cellular pathways mediating the biological actions of PrPC. In this regard, it might be possible to use the physiological activity of PrPC to develop in vitro assays to screen for drugs that have therapeutic potential. In addition, if the toxicity of PrPSc is partly attributable to a loss of PrP function, then over-expression of wild-type PrP may represent a strategy for suppressing the disease phenotype. Conversely, reduction of PrP expression, a strategy that has been proposed for preventing or treating prion diseases [153, 167], may have detrimental consequences due to loss of the neuroprotective activity of PrPC.
Future progress in understanding the normal function of PrPC will require identification of physiologically relevant PrP-interacting partners, and elucidation of the cellular pathways in which they participate. Accomplishing these objectives is likely to be facilitated by the use of model systems such as yeast [43] and Drosophila [168] that are amenable to genetic analysis, as well as by the development of additional cultured cell and transgenic mouse models. Given the rapid pace of work in the prion field, new discoveries will certainly be forthcoming.
Acknowledgments
Work in the Harris laboratory is supported by grants from the NIH (NS052526 and NS040975) and the Hope Center for Neurological Disorders at Washington University. H.M.C. was supported by a pre-doctoral fellowship (NS04691003) from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Büeler H, Fischer M, Lang Y, Fluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 2.Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 3.Mallucci GR, Ratte S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002;21:202–210. doi: 10.1093/emboj/21.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rivera-Milla E, Oidtmann B, Panagiotidis CH, Baier M, Sklaviadis T, Hoffmann R, Zhou Y, Solis GP, Stuermer CA, Malaga-Trillo E. Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. FASEB J. 2006;20:317–319. doi: 10.1096/fj.05-4279fje. [DOI] [PubMed] [Google Scholar]
- 5.Manson J, West JD, Thomson V, McBride P, Kaufman MH, Hope J. The prion protein gene: a role in mouse embryogenesis? Development. 1992;115:117–122. doi: 10.1242/dev.115.1.117. [DOI] [PubMed] [Google Scholar]
- 6.Harris DA, Lele P, Snider WD. Localization of the mRNA for a chicken prion protein by in situ hybridization. Proc Natl Acad Sci USA. 1993;90:4309–4313. doi: 10.1073/pnas.90.9.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moser M, Colello RJ, Pott U, Oesch B. Developmental expression of the prion protein gene in glial cells. Neuron. 1995;14:509–517. doi: 10.1016/0896-6273(95)90307-0. [DOI] [PubMed] [Google Scholar]
- 8.Ford MJ, Burton LJ, Morris RJ, Hall SM. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience. 2002;113:177–192. doi: 10.1016/s0306-4522(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 9.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–249. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 10.Harris DA, Peters PJ, Taraboulos A, Lingappa VR, DeArmond SJ, Prusiner SB. Cell biology of prions. In: Prusiner SB, editor. Prion Biology and Diseases. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2004. pp. 483–544. [Google Scholar]
- 11.Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem. 1996;272:6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- 12.Gorodinsky A, Harris DA. Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J Cell Biol. 1995;129:619–627. doi: 10.1083/jcb.129.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shyng SL, Heuser JE, Harris DA. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J Cell Biol. 1994;125:1239–1250. doi: 10.1083/jcb.125.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003;22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KS, Linden R, Prado MA, Brentani RR, Martins VR. Towards cellular receptors for prions. Rev Med Virol. 2003;13:399–408. doi: 10.1002/rmv.408. [DOI] [PubMed] [Google Scholar]
- 16.Gauczynski S, Peyrin JM, Haik S, Leucht C, Hundt C, Rieger R, Krasemann S, Deslys JP, Dormont D, Lasmezas CI, Weiss S. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001;20:5863–5875. doi: 10.1093/emboj/20.21.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gauczynski S, Nikles D, El-Gogo S, Papy-Garcia D, Rey C, Alban S, Barritault D, Lasmezas CI, Weiss S. The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J Infect Dis. 2006;194:702–709. doi: 10.1086/505914. [DOI] [PubMed] [Google Scholar]
- 18.Stewart RS, Harris DA. Most pathogenic mutations do not alter the membrane topology of the prion protein. J Biol Chem. 2001;276:2212–2220. doi: 10.1074/jbc.M006763200. [DOI] [PubMed] [Google Scholar]
- 19.Hegde RS, Mastrianni JA, Scott MR, Defea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- 20.Ma J, Lindquist S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science. 2002;298:1785–1788. doi: 10.1126/science.1073619. [DOI] [PubMed] [Google Scholar]
- 21.Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, Fioriti L, Chiesa R, Harris DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–21743. doi: 10.1074/jbc.M213247200. [DOI] [PubMed] [Google Scholar]
- 22.Roucou X, LeBlanc AC. Cellular prion protein neuroprotective function: implications in prion diseases. J Mol Med. 2005;83:3–11. doi: 10.1007/s00109-004-0605-5. [DOI] [PubMed] [Google Scholar]
- 23.Roucou X, Gains M, LeBlanc AC. Neuroprotective functions of prion protein. J Neurosci Res. 2004;75:153–161. doi: 10.1002/jnr.10864. [DOI] [PubMed] [Google Scholar]
- 24.van Delft MF, Huang DC. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 2006;16:203–213. doi: 10.1038/sj.cr.7310028. [DOI] [PubMed] [Google Scholar]
- 25.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 26.Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- 27.Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- 28.Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005;12:783–795. doi: 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- 29.Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Yokoyama T, Itohara S, Onodera T. Prions prevent neuronal cell-line death. Nature. 1999;400:225–226. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- 30.Sakudo A, Lee DC, Saeki K, Nakamura Y, Inoue K, Matsumoto Y, Itohara S, Onodera T. Impairment of superoxide dismutase activation by N-terminally truncated prion protein (PrP) in PrP-deficient neuronal cell line. Biochem Biophys Res Commun. 2003;308:660–667. doi: 10.1016/s0006-291x(03)01459-1. [DOI] [PubMed] [Google Scholar]
- 31.Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Pietu G, Pitaval A, Ripoche H, Eloit M, Dormont D, Chouaib S. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004;64:719–727. doi: 10.1158/0008-5472.can-03-1735. [DOI] [PubMed] [Google Scholar]
- 32.Drisaldi B, Coomaraswamy J, Mastrangelo P, Strome B, Yang J, Watts JC, Chishti MA, Marvi M, Windl O, Ahrens R, Major F, Sy MS, Kretzschmar H, Fraser PE, Mount HT, Westaway D. Genetic mapping of activity determinants within cellular prion proteins: N-terminal modules in PrPC offset pro-apoptotic activity of the Doppel helix B/B' region. J Biol Chem. 2004;279:55443–55454. doi: 10.1074/jbc.M404794200. [DOI] [PubMed] [Google Scholar]
- 33.Qin K, Zhao L, Tang Y, Bhatta S, Simard JM, Zhao RY. Doppel-induced apoptosis and counteraction by cellular prion protein in neuroblastoma and astrocytes. Neuroscience. 2006;141:1375–1388. doi: 10.1016/j.neuroscience.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 34.Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc Natl Acad Sci USA. 2006;103:2184–2189. doi: 10.1073/pnas.0510577103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPC) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci USA. 2006;103:3416–3421. doi: 10.1073/pnas.0511290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 37.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, Korsmeyer SJ. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1a. Science. 2006;312:572–576. [Google Scholar]
- 38.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 39.Rambold AS, Miesbauer M, Rapaport D, Bartke T, Baier M, Winklhofer KF, Tatzelt J. Association of Bcl-2 with misfolded prion protein is linked to the toxic potential of cytosolic PrP. Mol Biol Cell. 2006;17:3356–3368. doi: 10.1091/mbc.E06-01-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stewart RS, Harris DA. Mutational analysis of topological determinants in prion protein (PrP) and measurement of transmembrane and cytosolic PrP during prion infection. J Biol Chem. 2003;278:45960–45968. doi: 10.1074/jbc.M307833200. [DOI] [PubMed] [Google Scholar]
- 41.Zha H, Fisk HA, Yaffe MP, Mahajan N, Herman B, Reed JC. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol Cell Biol. 1996;16:6494–6508. doi: 10.1128/mcb.16.11.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin C, Reed JC. Yeast and apoptosis. Nat Rev Mol Cell Biol. 2002;3:453–459. doi: 10.1038/nrm832. [DOI] [PubMed] [Google Scholar]
- 43.Li A, Harris DA. Mammalian prion protein suppresses Bax-induced cell death in yeast. J Biol Chem. 2005;280:17430–17434. doi: 10.1074/jbc.C500058200. [DOI] [PubMed] [Google Scholar]
- 44.Li A, Dong J, Harris DA. Cell surface expression of the prion protein in yeast does not alter copper utilization phenotypes. J Biol Chem. 2004;279:29469–29477. doi: 10.1074/jbc.M402517200. [DOI] [PubMed] [Google Scholar]
- 45.Bounhar Y, Mann KK, Roucou X, LeBlanc AC. Prion protein prevents Bax-mediated cell death in the absence of other Bcl-2 family members in Saccharomyces cerevisiae. FEMS Yeast Res. 2006;6:1204–1212. doi: 10.1111/j.1567-1364.2006.00122.x. [DOI] [PubMed] [Google Scholar]
- 46.Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- 47.Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, Westaway D. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol. 1999;292:797–817. doi: 10.1006/jmbi.1999.3108. [DOI] [PubMed] [Google Scholar]
- 48.Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y, Houtani T, Shirabe S, Okada H, Hasegawa S, Miyamoto T, Noda T. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature. 1996;380:528–531. doi: 10.1038/380528a0. [DOI] [PubMed] [Google Scholar]
- 49.Rossi D, Cozzio A, Flechsig E, Klein MA, Rülicke T, Aguzzi A, Weissmann C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001;20:694–702. doi: 10.1093/emboj/20.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li A, Sakaguchi S, Atarashi R, Roy BC, Nakaoke R, Arima K, Okimura N, Kopacek J, Shigematsu K. Identification of a novel gene encoding a PrP-like protein expressed as chimeric transcripts fused to PrP exon 1/2 in ataxic mouse line with a disrupted PrP gene. Cell Mol Neurobiol. 2000;20:553–567. doi: 10.1023/a:1007059827541. [DOI] [PubMed] [Google Scholar]
- 51.Anderson L, Rossi D, Linehan J, Brandner S, Weissmann C. Transgene-driven expression of the Doppel protein in Purkinje cells causes Purkinje cell degeneration and motor impairment. Proc Natl Acad Sci USA. 2004;101:3644–3649. doi: 10.1073/pnas.0308681101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D, DeArmond SJ, Tremblay P. Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci USA. 2001;98:15288–15293. doi: 10.1073/pnas.251550798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel SP, Nakaoke R, Atarashi R, Houtani T, Torchia M, Sakaguchi S, DeArmond SJ, Prusiner SB, Katamine S. A mouse prion protein transgene rescues mice deficient for the prion protein gene from Purkinje cell degeneration and demyelination. Lab Invest. 1999;79:689–697. [PubMed] [Google Scholar]
- 54.Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J. 2007;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor DR, Watt NT, Perera WS, Hooper NM. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J Cell Sci. 2005;118:5141–5153. doi: 10.1242/jcs.02627. [DOI] [PubMed] [Google Scholar]
- 56.Walmsley AR, Zeng F, Hooper NM. The N-terminal region of the prion protein ectodomain contains a lipid raft targeting determinant. J Biol Chem. 2003;278:37241–37248. doi: 10.1074/jbc.M302036200. [DOI] [PubMed] [Google Scholar]
- 57.Atarashi R, Nishida N, Shigematsu K, Goto S, Kondo T, Sakaguchi S, Katamine S. Deletion of N-terminal residues 23-88 from prion protein (PrP) abrogates the potential to rescue PrP-deficient mice from PrP-like protein/doppel-induced Neurodegeneration. J Biol Chem. 2003;278:28944–28949. doi: 10.1074/jbc.M303655200. [DOI] [PubMed] [Google Scholar]
- 58.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 59.Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- 60.Sakudo A, Lee DC, Nishimura T, Li S, Tsuji S, Nakamura T, Matsumoto Y, Saeki K, Itohara S, Ikuta K, Onodera T. Octapeptide repeat region and N-terminal half of hydrophobic region of prion protein (PrP) mediate PrP-dependent activation of superoxide dismutase. Biochem Biophys Res Commun. 2005;326:600–606. doi: 10.1016/j.bbrc.2004.11.092. [DOI] [PubMed] [Google Scholar]
- 61.Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- 62.Rosenmann H, Talmor G, Halimi M, Yanai A, Gabizon R, Meiner Z. Prion protein with an E200K mutation displays properties similar to those of the cellular isoform PrPC. J Neurochem. 2001;76:1654–1662. doi: 10.1046/j.1471-4159.2001.00195.x. [DOI] [PubMed] [Google Scholar]
- 63.Fioriti L, Dossena S, Stewart LR, Stewart RS, Harris DA, Forloni G, Chiesa R. Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J Biol Chem. 2005;280:11320–11328. doi: 10.1074/jbc.M412441200. [DOI] [PubMed] [Google Scholar]
- 64.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 65.Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Rev. 2002;38:328–339. doi: 10.1016/s0165-0173(01)00150-3. [DOI] [PubMed] [Google Scholar]
- 66.Brown DR, Schulzschaeffer WJ, Schmidt B, Kretzschmar HA. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol. 1997;146:104–112. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- 67.Brown DR, Nicholas RS, Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res. 2002;67:211–224. doi: 10.1002/jnr.10118. [DOI] [PubMed] [Google Scholar]
- 68.Wong BS, Liu T, Li R, Pan T, Petersen RB, Smith MA, Gambetti P, Perry G, Manson JC, Brown DR, Sy MS. Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J Neurochem. 2001;76:565–572. doi: 10.1046/j.1471-4159.2001.00028.x. [DOI] [PubMed] [Google Scholar]
- 69.McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol. 2004;165:227–235. doi: 10.1016/S0002-9440(10)63291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakurai-Yamashita Y, Sakaguchi S, Yoshikawa D, Okimura N, Masuda Y, Katamine S, Niwa M. Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience. 2005;136:281–287. doi: 10.1016/j.neuroscience.2005.06.095. [DOI] [PubMed] [Google Scholar]
- 71.Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol Dis. 2005;20:442–449. doi: 10.1016/j.nbd.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 72.Brown DR, Clive C, Haswell SJ. Antioxidant activity related to copper binding of native prion protein. J Neurochem. 2001;76:69–76. doi: 10.1046/j.1471-4159.2001.00009.x. [DOI] [PubMed] [Google Scholar]
- 73.Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, Jones IM. Normal prion protein has an activity like that of superoxide dismutase. Biochem J. 1999;344:1–5. [PMC free article] [PubMed] [Google Scholar]
- 74.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 75.Jones S, Batchelor M, Bhelt D, Clarke AR, Collinge J, Jackson GS. Recombinant prion protein does not possess SOD-1 activity. Biochem J. 2005;392:309–312. doi: 10.1042/BJ20051236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brown DR, Besinger A. Prion protein expression and superoxide dismutase activity. Biochem J. 1998;334:423–429. doi: 10.1042/bj3340423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klamt F, Dal-Pizzol F, Conte da Frota ML, Walz R, Andrades ME, da Silva EG, Brentani RR, Izquierdo I, Fonseca Moreira JC. Imbalance of antioxidant defense in mice lacking cellular prion protein. Free Radic Biol Med. 2001;30:1137–1144. doi: 10.1016/s0891-5849(01)00512-3. [DOI] [PubMed] [Google Scholar]
- 78.Waggoner DJ, Drisaldi B, Bartnikas TB, Casareno RLB, Prohaska JR, Gitlin JD, Harris DA. Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J Biol Chem. 2000;275:7455–7458. doi: 10.1074/jbc.275.11.7455. [DOI] [PubMed] [Google Scholar]
- 79.Hutter G, Heppner FL, Aguzzi A. No superoxide dismutase activity of cellular prion protein in vivo. Biol Chem. 2003;384:1279–1285. doi: 10.1515/BC.2003.142. [DOI] [PubMed] [Google Scholar]
- 80.White AR, Collins SJ, Maher F, Jobling MF, Stewart LR, Thyer JM, Beyreuther K, Masters CL, Cappai R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am J Pathol. 1999;155:1723–1730. doi: 10.1016/S0002-9440(10)65487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Puig S, Thiele DJ. Molecular mechanisms of copper uptake and distribution. Curr Opin Chem Biol. 2002;6:171–180. doi: 10.1016/s1367-5931(02)00298-3. [DOI] [PubMed] [Google Scholar]
- 82.Waggoner DJ, Bartnikas TB, Gitlin JD. The role of copper in neurodegenerative disease. Neurobiol Dis. 1999;6:221–230. doi: 10.1006/nbdi.1999.0250. [DOI] [PubMed] [Google Scholar]
- 83.Vassallo N, Herms J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J Neurochem. 2003;86:538–544. doi: 10.1046/j.1471-4159.2003.01882.x. [DOI] [PubMed] [Google Scholar]
- 84.Millhauser GL. Copper and the prion protein: methods, structures, function, and disease. Annu Rev Phys Chem. 2007;58 doi: 10.1146/annurev.physchem.1158.032806.104657. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brown DR, Qin KF, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, Vonbohlen A, Schulzschaeffer W, Giese A, Westaway D, Kretzschmar H. The cellular prion protein binds copper in vivo. Nature. 1997;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- 86.Jackson GS, Murray I, Hosszu LL, Gibbs N, Waltho JP, Clarke AR, Collinge J. Location and properties of metal-binding sites on the human prion protein. Proc Natl Acad Sci USA. 2001;98:8531–8535. doi: 10.1073/pnas.151038498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kramer ML, Kratzin HD, Schmidt B, Romer A, Windl O, Liemann S, Hornemann S, Kretzschmar H. Prion protein binds copper within the physiological concentration range. J Biol Chem. 2001;276:16711–16719. doi: 10.1074/jbc.M006554200. [DOI] [PubMed] [Google Scholar]
- 88.Stöckel J, Safar J, Wallace AC, Cohen FE, Prusiner SB. Prion protein selectively binds copper(II) ions. Biochemistry. 1998;37:7185–7193. doi: 10.1021/bi972827k. [DOI] [PubMed] [Google Scholar]
- 89.Walter ED, Chattopadhyay M, Millhauser GL. The affinity of copper binding to the prion protein octarepeat domain: evidence for negative cooperativity. Biochemistry. 2006;45:13083–13092. doi: 10.1021/bi060948r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jones CE, Klewpatinond M, Abdelraheim SR, Brown DR, Viles JH. Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: the unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations. J Mol Biol. 2005;346:1393–1407. doi: 10.1016/j.jmb.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 91.Jones CE, Abdelraheim SR, Brown DR, Viles JH. Preferential Cu2+ coordination by His96 and His111 induces beta-sheet formation in the unstructured amyloidogenic region of the prion protein. J Biol Chem. 2004;279:32018–32027. doi: 10.1074/jbc.M403467200. [DOI] [PubMed] [Google Scholar]
- 92.Leclerc E, Serban H, Prusiner SB, Burton DR, Williamson RA. Copper induces conformational changes in the N-terminal part of cell-surface PrP(C) Arch Virol. 2006;151:2103–2109. doi: 10.1007/s00705-006-0804-1. [DOI] [PubMed] [Google Scholar]
- 93.Quaglio E, Chiesa R, Harris DA. Copper converts the cellular prion protein into a protease-resistant species that is distinct from the scrapie isoform. J Biol Chem. 2001;276:11432–11438. doi: 10.1074/jbc.M009666200. [DOI] [PubMed] [Google Scholar]
- 94.Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998;273:33107–33110. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- 95.Brown LR, Harris DA. Copper and zinc cause delivery of the prion protein from the plasma membrane to a subset of early endosomes and the Golgi. J Neurochem. 2003;87:353–363. doi: 10.1046/j.1471-4159.2003.01996.x. [DOI] [PubMed] [Google Scholar]
- 96.Perera WS, Hooper NM. Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol. 2001;11:519–523. doi: 10.1016/s0960-9822(01)00147-6. [DOI] [PubMed] [Google Scholar]
- 97.Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schurmann P, Windl O, Brose N, Kretzschmar H. Evidence of presynaptic location and function of the prion protein. J Neurosci. 1999;19:8866–8875. doi: 10.1523/JNEUROSCI.19-20-08866.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brown DR. Prion and prejudice: normal protein and the synapse. Trends Neurosci. 2001;24:85–90. doi: 10.1016/s0166-2236(00)01689-1. [DOI] [PubMed] [Google Scholar]
- 99.Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM. Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002;25:412–417. doi: 10.1016/s0166-2236(02)02215-4. [DOI] [PubMed] [Google Scholar]
- 100.Taylor DR, Hooper NM. The prion protein and lipid rafts. Mol Membr Biol. 2006;23:89–99. doi: 10.1080/09687860500449994. [DOI] [PubMed] [Google Scholar]
- 101.Spielhaupter C, Schatzl HM. PrPC directly interacts with proteins involved in signaling pathways. J Biol Chem. 2001;276:44604–44612. doi: 10.1074/jbc.M103289200. [DOI] [PubMed] [Google Scholar]
- 102.Loertscher R, Lavery P. The role of glycosyl phosphatidyl inositol (GPI)-anchored cell surface proteins in T-cell activation. Transpl Immunol. 2002;9:93–96. doi: 10.1016/s0966-3274(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 103.Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 104.Schneider B, Mutel V, Pietri M, Ermonval M, Mouillet-Richard S, Kellermann O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc Natl Acad Sci USA. 2003;100:13326–13331. doi: 10.1073/pnas.2235648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mouillet-Richard S, Pietri M, Schneider B, Vidal C, Mutel V, Launay JM, Kellermann O. Modulation of serotonergic receptor signaling and cross-talk by prion protein. J Biol Chem. 2005;280:4592–4601. doi: 10.1074/jbc.M406199200. [DOI] [PubMed] [Google Scholar]
- 106.Lässle M, Blatch GL, Kundra V, Takatori T, Zetter BR. Stress-inducible, murine protein mSTI1. Characterization of binding domains for heat shock proteins and in vitro phosphorylation by different kinases. J Biol Chem. 1997;272:1876–1884. doi: 10.1074/jbc.272.3.1876. [DOI] [PubMed] [Google Scholar]
- 107.Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21:3307–3316. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21:3317–3326. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, Brentani RR, Linden R, Martins VR. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci. 2005;25:11330–11339. doi: 10.1523/JNEUROSCI.2313-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I, Bahr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke. 2006;37:1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4. [DOI] [PubMed] [Google Scholar]
- 111.Vassallo N, Herms J, Behrens C, Krebs B, Saeki K, Onodera T, Windl O, Kretzschmar HA. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem Biophys Res Commun. 2005;332:75–82. doi: 10.1016/j.bbrc.2005.04.099. [DOI] [PubMed] [Google Scholar]
- 112.Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005;169:341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem. 2005;95:1373–1386. doi: 10.1111/j.1471-4159.2005.03469.x. [DOI] [PubMed] [Google Scholar]
- 114.Chen S, Mange A, Dong L, Lehmann S, Schachner M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol Cell Neurosci. 2003;22:227–233. doi: 10.1016/s1044-7431(02)00014-3. [DOI] [PubMed] [Google Scholar]
- 115.Selvaggini C, De Gioia L, Cantu L, Ghibaudi E, Diomede L, Passerini F, Forloni G, Bugiani O, Tagliavini F, Salmona M. Molecular characteristics of a protease-resistant, amyloidogenic and neurotoxic peptide homologous to residues 106-126 of the prion protein. Biochem Biophys Res Commun. 1993;194:1380–1386. doi: 10.1006/bbrc.1993.1977. [DOI] [PubMed] [Google Scholar]
- 116.Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- 117.Brown DR, Herms J, Kretzschmar HA. Mouse cortical cells lacking cellular PrP survive in culture with a neurotoxic PrP fragment. Neuroreport. 1994;5:2057–2060. doi: 10.1097/00001756-199410270-00017. [DOI] [PubMed] [Google Scholar]
- 118.Thellung S, Villa V, Corsaro A, Arena S, Millo E, Damonte G, Benatti U, Tagliavini F, Florio T, Schettini G. p38 MAP kinase mediates the cell death induced by PrP106-126 in the SH-SY5Y neuroblastoma cells. Neurobiol Dis. 2002;9:69–81. doi: 10.1006/nbdi.2001.0461. [DOI] [PubMed] [Google Scholar]
- 119.Pietri M, Caprini A, Mouillet-Richard S, Pradines E, Ermonval M, Grassi J, Kellermann O, Schneider B. Overstimulation of PrPC signaling pathways by prion peptide 106-126 causes oxidative injury of bioaminergic neuronal cells. J Biol Chem. 2006;281:28470–28479. doi: 10.1074/jbc.M602774200. [DOI] [PubMed] [Google Scholar]
- 120.Carimalo J, Cronier S, Petit G, Peyrin JM, Boukhtouche F, Arbez N, Lemaigre-Dubreuil Y, Brugg B, Miquel MC. Activation of the JNK-c-Jun pathway during the early phase of neuronal apoptosis induced by PrP106-126 and prion infection. Eur J Neurosci. 2005;21:2311–2319. doi: 10.1111/j.1460-9568.2005.04080.x. [DOI] [PubMed] [Google Scholar]
- 121.Paitel E, Sunyach C, Alves da Costa C, Bourdon JC, Vincent B, Checler F. Primary cultured neurons devoid of cellular prion display lower responsiveness to staurosporine through the control of p53 at both transcriptional and posttranscriptional levels. J Biol Chem. 2004;279:612–618. doi: 10.1074/jbc.M310453200. [DOI] [PubMed] [Google Scholar]
- 122.Sunyach C, Alfa Cisse M, Alves da Costa C, Vincent B, Checler F. The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent staurosporine-induced caspase-3 activation. J Biol Chem. 2006 doi: 10.1074/jbc M609663200. In Press. [DOI] [PubMed] [Google Scholar]
- 123.Sunyach C, Checler F. Combined pharmacological, mutational and cell biology approaches indicate that p53-dependent caspase 3 activation triggered by cellular prion is dependent on its endocytosis. J Neurochem. 2005;92:1399–1407. doi: 10.1111/j.1471-4159.2004.02989.x. [DOI] [PubMed] [Google Scholar]
- 124.Jeffrey M, Halliday WG, Bell J, Johnston AR, MacLeod NK, Ingham C, Sayers AR, Brown DA, Fraser JR. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol Appl Neurobiol. 2000;26:41–54. doi: 10.1046/j.1365-2990.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- 125.Lainé J, Marc ME, Sy MS, Axelrad H. Cellular and subcellular morphological localization of normal prion protein in rodent cerebellum. Eur J Neurosci. 2001;14:47–56. doi: 10.1046/j.0953-816x.2001.01621.x. [DOI] [PubMed] [Google Scholar]
- 126.Moya KL, Sales N, Hassig R, Creminon C, Grassi J, Di Giamberardino L. Immunolocalization of the cellular prion protein in normal brain. Microsc Res Tech. 2000;50:58–65. doi: 10.1002/1097-0029(20000701)50:1<58::AID-JEMT9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 127.Salès N, Hassig R, Rodolfo K, Di Giamberardino L, Traiffort E, Ruat M, Fretier P, Moya KL. Developmental expression of the cellular prion protein in elongating axons. Eur J Neurosci. 2002;15:1163–1177. doi: 10.1046/j.1460-9568.2002.01953.x. [DOI] [PubMed] [Google Scholar]
- 128.Mironov A, Jr, Latawiec D, Wille H, Bouzamondo-Bernstein E, Legname G, Williamson RA, Burton D, DeArmond SJ, Prusiner SB, Peters PJ. Cytosolic prion protein in neurons. J Neurosci. 2003;23:7183–7193. doi: 10.1523/JNEUROSCI.23-18-07183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ford MJ, Burton LJ, Li H, Graham CH, Frobert Y, Grassi J, Hall SM, Morris RJ. A marked disparity between the expression of prion protein and its message by neurones of the CNS. Neuroscience. 2002;111:533–551. doi: 10.1016/s0306-4522(01)00603-0. [DOI] [PubMed] [Google Scholar]
- 130.Barmada S, Piccardo P, Yamaguchi K, Ghetti B, Harris DA. GFP-tagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol Dis. 2004;16:527–537. doi: 10.1016/j.nbd.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 131.Moya KL, Hassig R, Creminon C, Laffont I, Di Giamberardino L. Enhanced detection and retrograde axonal transport of PrPC in peripheral nerve. J Neurochem. 2004;88:155–160. doi: 10.1046/j.1471-4159.2003.02150.x. [DOI] [PubMed] [Google Scholar]
- 132.Borchelt DR, Koliatsos VE, Guarnieri M, Pardo CA, Sisodia SS, Price DL. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J Biol Chem. 1994;269:14711–14714. [PubMed] [Google Scholar]
- 133.Gohel C, Grigoriev V, Escaig-Haye F, Lasmezas CI, Deslys JP, Langeveld J, Akaaboune M, Hantai D, Fournier JG. Ultrastructural localization of cellular prion protein (PrPc) at the neuromuscular junction. J Neurosci Res. 1999;55:261–267. doi: 10.1002/(SICI)1097-4547(19990115)55:2<261::AID-JNR14>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 134.Re L, Rossini F, Re F, Bordicchia M, Mercanti A, Fernandez OS, Barocci S. Prion protein potentiates acetylcholine release at the neuromuscular junction. Pharmacol Res. 2006;53:62–68. doi: 10.1016/j.phrs.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 135.Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–297. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- 136.Manson JC, Hope J, Clarke AR, Johnston A, Black C, MacLeod N. PrP gene dosage and long term potentiation. Neurodegeneration. 1995;4:113–114. doi: 10.1006/neur.1995.0014. [DOI] [PubMed] [Google Scholar]
- 137.Lledo PM, Tremblay P, Dearmond SJ, Prusiner SB, Nicoll RA. Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc Natl Acad Sci USA. 1996;93:2403–2407. doi: 10.1073/pnas.93.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Carleton A, Tremblay P, Vincent JD, Lledo PM. Dose-dependent, prion protein (PrP)-mediated facilitation of excitatory synaptic transmission in the mouse hippocampus. Pflugers Arch. 2001;442:223–229. doi: 10.1007/s004240100523. [DOI] [PubMed] [Google Scholar]
- 139.Colling SB, Collinge J, Jefferys JGR. Hippocampal slices from prion protein null mice: disrupted Ca2+-activated K+ currents. Neurosci Lett. 1996;209:49–52. doi: 10.1016/0304-3940(96)12596-9. [DOI] [PubMed] [Google Scholar]
- 140.Herms JW, Tings T, Dunker S, Kretzschmar HA. Prion protein affects Ca2+-activated K+ currents in cerebellar Purkinje cells. Neurobiol Dis. 2001;8:324–330. doi: 10.1006/nbdi.2000.0369. [DOI] [PubMed] [Google Scholar]
- 141.Colling SB, Khana M, Collinge J, Jefferys JGR. Mossy fibre reorganization in the hippocampus of prion protein null mice. Brain Res. 1997;755:28–35. doi: 10.1016/s0006-8993(97)00087-5. [DOI] [PubMed] [Google Scholar]
- 142.Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rulicke T, Moser M, Oesch B, McBride PA, Manson JC. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380:639–642. doi: 10.1038/380639a0. [DOI] [PubMed] [Google Scholar]
- 143.Criado JR, Sanchez-Alavez M, Conti B, Giacchino JL, Wills DN, Henriksen SJ, Race R, Manson JC, Chesebro B, Oldstone MB. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol Dis. 2005;19:255–265. doi: 10.1016/j.nbd.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 144.Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol. 2001;314:1209–1225. doi: 10.1006/jmbi.2000.5183. [DOI] [PubMed] [Google Scholar]
- 145.Graner E, Mercadante AF, Zanata SM, Forlenza OV, Cabral AL, Veiga SS, Juliano MA, Roesler R, Walz R, Minetti A, Izquierdo I, Martins VR, Brentani RR. Cellular prion protein binds laminin and mediates neuritogenesis. Mol Brain Res. 2000;76:85–92. doi: 10.1016/s0169-328x(99)00334-4. [DOI] [PubMed] [Google Scholar]
- 146.Graner E, Mercadante AF, Zanata SM, Martins VR, Jay DG, Brentani RR. Laminin-induced PC-12 cell differentiation is inhibited following laser inactivation of cellular prion protein. FEBS Lett. 2000;482:257–260. doi: 10.1016/s0014-5793(00)02070-6. [DOI] [PubMed] [Google Scholar]
- 147.Mange A, Milhavet O, Umlauf D, Harris D, Lehmann S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett. 2002;514:159–162. doi: 10.1016/s0014-5793(02)02338-4. [DOI] [PubMed] [Google Scholar]
- 148.Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–357. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 149.Hetz C, Maundrell K, Soto C. Is loss of function of the prion protein the cause of prion disorders? Trends Mol Med. 2003;9:237–243. doi: 10.1016/s1471-4914(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 150.Liemann S, Glockshuber R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry. 1999;38:3258–3267. doi: 10.1021/bi982714g. [DOI] [PubMed] [Google Scholar]
- 151.Swietnicki W, Petersen RB, Gambetti P, Surewicz WK. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J Biol Chem. 1998;273:31048–31052. doi: 10.1074/jbc.273.47.31048. [DOI] [PubMed] [Google Scholar]
- 152.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 153.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 154.Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303:1514–1516. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- 155.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 156.Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S. Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci. 2001;24:182–188. doi: 10.1016/s0166-2236(00)01721-5. [DOI] [PubMed] [Google Scholar]
- 157.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 158.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 159.Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 160.Rubinsztein DC. Lessons from animal models of Huntington's disease. Trends Genet. 2002;18:202–209. doi: 10.1016/s0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- 161.Sipione S, Cattaneo E. Modeling Huntington's disease in cells, flies, and mice. Mol Neurobiol. 2001;23:21–51. doi: 10.1385/MN:23:1:21. [DOI] [PubMed] [Google Scholar]
- 162.da Costa CA, Ancolio K, Checler F. Wild-type but not Parkinson's disease-related ala-53→Thr mutant α-synuclein protects neuronal cells from apoptotic stimuli. J Biol Chem. 2000;275:24065–24069. doi: 10.1074/jbc.M002413200. [DOI] [PubMed] [Google Scholar]
- 163.Lee M, Hyun D, Halliwell B, Jenner P. Effect of the overexpression of wild-type or mutant α-synuclein on cell susceptibility to insult. J Neurochem. 2001;76:998–1009. doi: 10.1046/j.1471-4159.2001.00149.x. [DOI] [PubMed] [Google Scholar]
- 164.Seo JH, Rah JC, Choi SH, Shin JK, Min K, Kim HS, Park CH, Kim S, Kim EM, Lee SH, Lee S, Suh SW, Suh YH. α-synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J. 2002;16:1826–1828. doi: 10.1096/fj.02-0041fje. [DOI] [PubMed] [Google Scholar]
- 165.Chiesa R, Harris DA. Prion diseases: what is the neurotoxic molecule? Neurobiol Dis. 2001;8:743–763. doi: 10.1006/nbdi.2001.0433. [DOI] [PubMed] [Google Scholar]
- 166.Cashman NR, Caughey B. Prion diseases--close to effective therapy? Nat Rev Drug Discov. 2004;3:874–884. doi: 10.1038/nrd1525. [DOI] [PubMed] [Google Scholar]
- 167.Pfeifer A, Eigenbrod S, Al-Khadra S, Hofmann A, Mitteregger G, Moser M, Bertsch U, Kretzschmar H. Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J Clin Invest. 2006;116:3204–3210. doi: 10.1172/JCI29236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gavin BA, Dolph MJ, Deleault NR, Geoghegan JC, Khurana V, Feany MB, Dolph PJ, Supattapone S. Accelerated accumulation of misfolded prion protein and spongiform degeneration in a Drosophila model of Gerstmann-Sträussler-Scheinker syndrome. J Neurosci. 2006;26:12408–12414. doi: 10.1523/JNEUROSCI.3372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Azzalin A, Ferrara V, Arias A, Cerri S, Avella D, Pisu MB, Nano R, Bernocchi G, Ferretti L, Comincini S. Interaction between the cellular prion (PrPC) and the 2P domain K+ channel TREK-1 protein. Biochem Biophys Res Commun. 2006;346:108–115. doi: 10.1016/j.bbrc.2006.05.097. [DOI] [PubMed] [Google Scholar]
- 170.Nieznanski K, Nieznanska H, Skowronek KJ, Osiecka KM, Stepkowski D. Direct interaction between prion protein and tubulin. Biochem Biophys Res Commun. 2005;334:403–411. doi: 10.1016/j.bbrc.2005.06.092. [DOI] [PubMed] [Google Scholar]
- 171.Bragason BT, Palsdottir A. Interaction of PrP with NRAGE, a protein involved in neuronal apoptosis. Mol Cell Neurosci. 2005;29:232–244. doi: 10.1016/j.mcn.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 172.Edenhofer F, Rieger R, Famulok M, Wendler W, Weiss S, Winnacker EL. Prion protein PrPC interacts with molecular chaperones of the Hsp60 family. J Virol. 1996;70:4724–4728. doi: 10.1128/jvi.70.7.4724-4728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kurschner C, Morgan JI. The cellular prion protein (PrP) selectively binds to Bcl-2 in the yeast two-hybrid system. Mol Brain Res. 1995;30:165–168. doi: 10.1016/0169-328x(95)00013-i. [DOI] [PubMed] [Google Scholar]