

Summary
In antiviral and cancer therapy, deoxyribonucleoside kinases (dNKs) are often the rate-limiting step in activating nucleoside analog (NA) prodrugs into their cytotoxic, phosphorylated forms. We have constructed libraries of hybrid enzymes by non-homologous recombination of the pyrimidine-specific human thymidine kinase 2 and the broad-specificity dNK from Drosophila melanogaster; their low sequence identity has precluded engineering by conventional, homology-dependent shuffling techniques. From these libraries, we identified chimeras that phosphorylate nucleoside analogs with higher activity than either parental enzyme, and that possess new activity towards the anti-HIV prodrug 2′,3′-didehydro-3′-deoxythymidine (d4T). These results demonstrate the potential of non-homologous recombination within the dNK family for creating enzymes with new and improved activities towards nucleoside analogs. In addition, our results exposed a previously unknown role for the C-terminal regions of these dNKs in determining substrate selectivity.
Keywords: enzyme engineering, non-homologous recombination, deoxyribonucleoside kinase, human thymidine kinase 2, nucleoside analog, substrate specificity
Introduction
This article describes the use of directed evolution to reprogram the catalytic activity and selectivity of human thymidine kinase 2 (hTK2), through non-homologous recombination with the deoxyribonucleoside kinase from Drosophila melanogaster (DmdNK). Deoxyribonucleoside kinases (dNKs) phosphorylate deoxyribonucleosides to their monophosphorylated forms, and are often the rate-limiting enzymes in the activation of anticancer and antiviral nucleoside analog (NA) prodrugs. In tumor cell lines, the administration of exogenous dNKs via gene therapy has been shown to potentiate the biological activity of several NA prodrugs.1–4 However, the kinetic properties of dNKs constitute a limiting factor in the efficacy of these therapies, and there is a need to identify enzymes with higher efficiency and improved specificity towards NAs for further development of this strategy.
Based on sequence similarity, hTK2 and DmdNK belong to a large subfamily of dNKs (type-1) that includes other human enzymes (deoxycytidine kinase, hdCK; and deoxyguanosine kinase, hdGK) and dNKs from insects.5 Type-1 dNKs share the same fold, but vary widely in their substrate specificities and overall catalytic efficiencies. DmdNK possesses the highest catalytic rates of any type-1 dNK, and is a multisubstrate enzyme that phosphorylates pyrimidine and purine deoxyribonucleosides, as well as several NAs. DmdNK has been the subject of both rational and random mutagenesis approaches to probe its substrate specificity.6;7 In contrast with DmdNK, the less studied hTK2 is ∼30 to 80-fold less efficient at phosphorylating pyrimidine nucleosides, and has no activity towards purines. Despite their functional differences, the active site residues of these enzymes are highly conserved. However, our previous research has shown that the narrow specificity of hTK2 is not a result of the primary shell differences with DmdNK.8 Furthermore, other studies suggest that non-active site features such as flexible loops9 and C-terminal extensions10 have subtle effects on the specificity of dNKs.
Given the conservation of active site residues, the lack of structural information for hTK2, and the limitations of rational design in investigating non-local interactions, we have utilized a directed evolution approach in this study. Recombination can be an effective tool for sampling sequence space and generating enzymes with new catalytic activities, and recent studies has also emphasized the utility of recombination techniques in identifying regions of functional importance in protein frameworks.11 One of the most successful examples of engineering dNKs utilized DNA family shuffling to recombine the closely related herpesvirus thymidine kinases (HSV TK-1 and HSV TK-2, 78% DNA identity).12 In theory this approach is a good strategy for other suicide gene/prodrug combinations; however, the sequence similarity between type-1 dNKs is below the threshold at which DNA shuffling techniques are effective. Homologous recombination-based methods can only shuffle sequences of >70% identity13, and preferentially generate crossovers in high-homology regions. Because DmdNK and hTK2 are only 48% identical at the DNA level, we have employed the non-homologous recombination techniques of ITCHY14–16 and enhanced crossover SCRATCHY17;18 as a route to their combinatorial diversification. From single- and triple-crossover libraries, we have isolated hybrids with novel activities, including the introduction of activity towards the anti-HIV prodrug d4T (2′,3′-didehydro-3′-deoxythymidine), and we have imparted purine activity on a predominantly hTK2 scaffold. These results provide insight into the structure/function relationships of hTK2 and DmdNK, and demonstrate the utility of non-homologous recombination within the dNK family for creating enzymes with new and improved activities towards nucleoside analogs.
Results
Library construction and analysis
Two single-crossover ITCHY libraries of hTK2 and DmdNK were generated to cover both possible orientations, consisting of 4 × 106 transformants (DmdNK-hTK2; DH) and 5 × 106 transformants (hTK2-DmdNK; HD). The theoretical diversity is 4.9 × 105 (648 bp × 750 bp); therefore, these libraries provide essentially full coverage of available sequence space. The distribution of crossover locations was checked by sequencing randomly-selected clones from each library; a broad distribution of crossover locations was observed (Supplementary Figure S1).
Because crossovers can occur at any base, two-thirds of all ITCHY library members will contain frameshifts which result in premature termination or non-functional, frame-shifted progeny. Before recombining the ITCHY libraries to generate multiple-crossover chimeras, the libraries were subcloned into pInSALect19 and plated on carbenicillin for selection of clones in the correct reading frame. As expected, approximately one-third of each library (DH: 33%; HD: 29%) survived this selection. Post-selection sequence analysis of 24 clones confirmed that all hybrid genes possessed in-frame crossovers.
For the creation of multiple-crossover chimeras, we divided the in-frame ITCHY libraries into three approximately equal sections (∼250 bp) with short overlaps between each section, then amplified the sections by PCR using skewed primers. The resulting fragments, each containing a single crossover, were assembled by overlap extension to generate two complementary triple-crossover libraries, each with >106 transformants: hTK2-DmdNK-hTK2-DmdNK (HDHD) and DmdNK-hTK2-DmdNK-hTK2 (DHDH). This strategy is a variation on the enhanced SCRATCHY protocol18; however, we assembled the multiple-crossover chimeras by overlap extension PCR, instead of by DNA shuffling, in order to control the number of crossovers, and to minimize the frequency of single-crossover variants and/or revertant wild-type sequences. With the previous protocol, 58% of clones had 1 crossover.18 Similar levels of background would have overwhelmed the genetic selection used herein, making it difficult to isolate functional, multi-crossover enzymes. We chose to design the libraries as triple-crossovers to increase the sequence diversity while limiting the structural disruption that >3 crossovers may introduce. Sequencing of 30 clones (14 from DHDH; 16 from HDHD) showed that each contained three crossovers, implying that any wild-type or single-crossover background was present at <3%. Each analyzed clone represented a unique sequence, and a broad diversity of crossover locations was observed.
Selection for thymidine kinase activity
Chimeras that retained thymidine kinase activity were selected from the HD, DH, HDHD and DHDH libraries by functional complementation of the tdk- E. coli strain KY895.20 Less than 0.1% of each ITCHY library survived this functional selection. DNA sequencing of these chimeras yielded a majority of clones that were DmdNK-like, though a small number of clones were found which more closely resembled hTK2. All functionally selected single-crossover clones examined by DNA sequencing showed a similar pattern, with the recombination site near the N- or C-terminus.
The two triple-crossover libraries were also subjected to functional selection in E. coli KY895. Eighteen HDHD clones survived the selection (<0.01%). DNA sequencing revealed that six of these clones contained three crossovers; however, the other 12 possessed only a single crossover. In the complementary DHDH library, only one of 24 chimeras was identified with three crossovers; the remaining 23 were single-crossover hybrids. These single-crossover clones were similar to the functionally selected ITCHY clones, with all crossovers occurring near the termini. No wild-type parental sequences were observed. From these results, it appears that despite the library design, single-crossover products were present at very low levels in each library. Furthermore, these single-crossover clones were more likely to retain function and survive the functional selection than the multiple crossover clones.
Modeling of selected chimeras
Homology models of selected chimeras were constructed, based on the crystal structures of DmdNK21;22 (Figure 1), to elucidate the effects of crossovers on key structural features. Full sequence details for each clone are available in Supplementary Data 1. Four representative clones from the single-crossover libraries were chosen for further study (Figure 2). DH-03 and HD-15 represent the predominantly DmdNK chimeras, with crossovers occurring near the C-terminus and N-terminus, respectively. DH-03 contains a crossover in a non-homologous region of α8, and based on alignment of the parental sequences, possesses a net insertion of 10 aa. HD-15 has only the first 11 aa from hTK2, followed by a crossover leading to a 1 aa insertion in the section of the P-loop (SGKTT) that is identical in the two parents. Clones HD-16 and DH-02 represent the chimeras with predominantly hTK2 sequence. HD-16 consists of the first 193 residues from hTK2, and residues 190–250 from DmdNK. A mid-codon crossover introduces a mutation (W190R, DmdNK numbering) at the beginning of the DmdNK segment. This crossover also yields a 6 aa insertion. Like HD-15, DH-02 has a crossover located in the P-loop.
Figure 1.
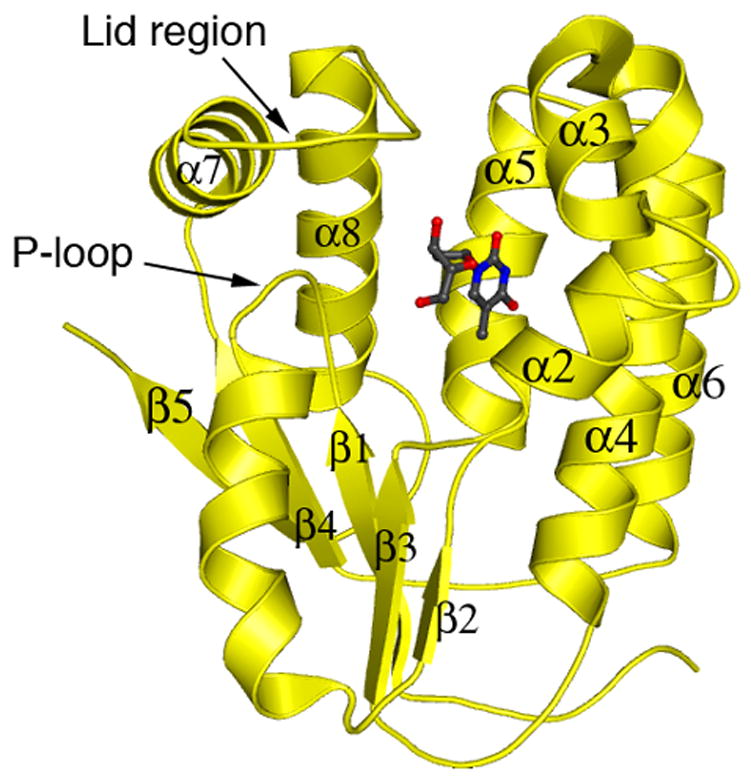
Conserved features of type-1 dNKs. The monomeric subunit of DmdNK22 is used as an archetypal example. The overall structure is a three-layer αβα Rossmann fold. Two conserved features are present in all type 1-dNKs: the phosphate binding P-loop (GX4GKS/T) located after strand β1; and the Lid region, a connecting loop between α7 and β8 which contains conserved arginine residues at the phosphate donor site.
Figure 2.
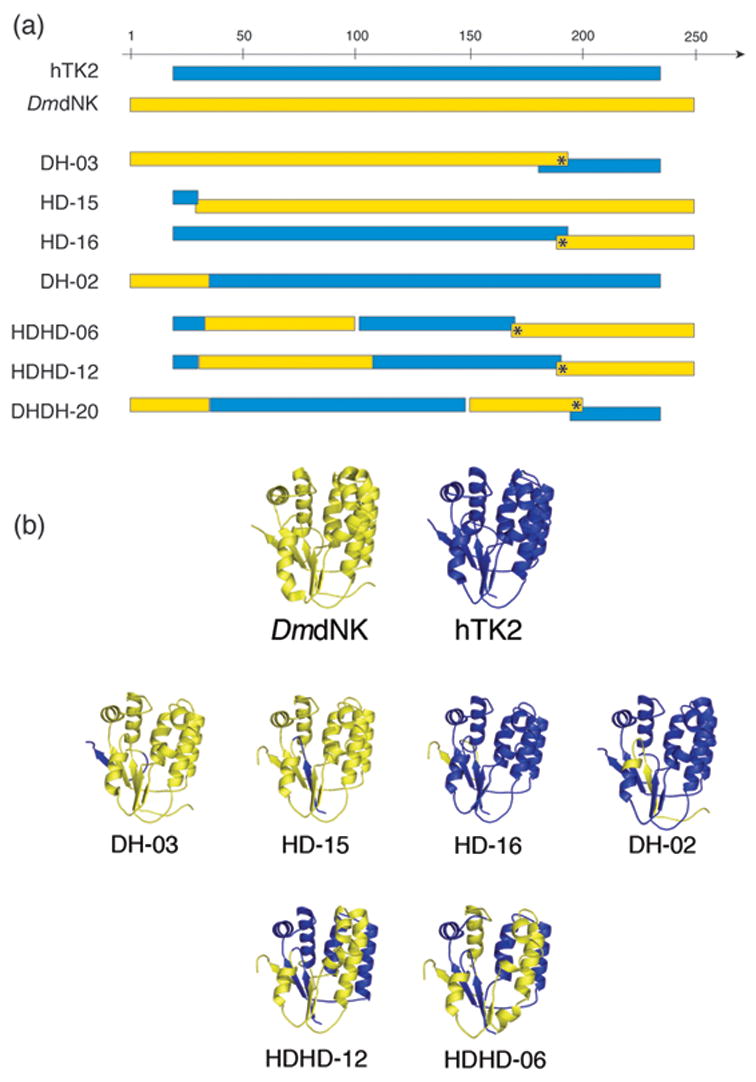
Comparison of parental enzymes and selected chimeras. (a) Schematic representation of selected chimeric protein sequences based on a CLUSTHALW sequence alignment of the parental enzymes. Segments from hTK2 and DmdNK are represented as blue and yellow bars, respectively. Amino acid substitutions (point mutations) are marked by asterisks. (b) Structural models of the parental enzymes and selected chimeras. The structure of DmdNK [PDB entry 1OT322] is shown in yellow. All homology models were generated using SWISS-MODEL.34 The homology model of hTK2 is colored blue. The homology models of the single- and multiple-crossover chimeras (DH-02, DH-03, HD-15, HD-16, HDHD-06 and HDHD-12) are colored as the parental enzymes, with segments from hTK2 and DmdNK shown in blue and yellow, respectively.
Two of the six functionally-selected HDHD triple-crossover chimeras were analyzed further: HDHD-06 and HDHD-12 (Figure 2). In each case, the first crossover is located in the P-loop. In HDHD-06, the second crossover creates a 2 aa deletion, which in the homology model appears to disrupt α4 and β3. The third crossover introduces a point mutation (S170A, DmdNK numbering) and extends the Lid by insertion of 2 aa between α7 and β8. In HDHD-12 there is no insertion or deletion at the second crossover. The third crossover occurs after the Lid region, at the end of α8, and results in a 2 aa insertion as well as the same point mutation (W190R) observed in HD-16.
Only one triple-crossover chimera from the DHDH library, DHDH-20, survived functional selection (Figure 2a). As observed in the other libraries, the first crossover was in the P-loop. The second crossover occurs after α6 and results in a single-residue deletion, while the third occurs at the end of α8 and introduces both a Phe to Ile mutation, and also a 5 aa insertion.
Enzyme purification and activity with natural deoxyribonucleosides
The seven selected chimeras were subcloned into pET14b to incorporate an N-terminal (His)6-tag for purification via metal affinity chromatography. Yields varied from 0.1–4 mg protein per liter of culture, with DH clones having lower yields than HD clones. DHDH-20 proved to be insoluble, and therefore could not be characterized further.
The catalytic activities of the selected hybrids and parental enzymes towards the four natural deoxyribonucleosides were measured spectrophotometrically (Table 1). Of these substrates, the pyrimidines are substrates for both parental enzymes, while the purines are substrates for DmdNK only. These differences allow the chimeras to be classified as “DmdNK-like” or “hTK2-like,” based on their substrate specificities.
Table 1a.
Kinetic parameters of wild-type and chimeric enzymes with pyrimidine nucleosides.
| dT | dC | |||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | |
| hTK2a | 1.8 ± 0.2 | 6.7 ± 1.1 | 2.7 × 105 | 2.0 ± 0.2 | 16.2 ± 1.1 | 1.2 × 105 |
| DmdNKa | 17.5 ± 0.6 | 1.9 ± 0.3 | 9.1 × 106 | 19.3 ± 0.5 | 2.2± 0.3 | 9.0 × 106 |
| DH-03 | 0.54 ± 0.01 | 10.4 ± 1.3 | 5.2 × 104 | 0.48 ± 0.01 | 23.5 ± 1.7 | 2.0 × 104 |
| HD-15 | 3.6 ± 0.1 | 6.2 ± 0.9 | 5.9 × 105 | 4.3 ± 0.1 | 8.2 ± 0.6 | 5.2 × 105 |
| HD-16 | 2.5 ± 0.1 | 1.9 ± 0.3 | 1.3 × 106 | 3.2 ± 0.1 | 14.1 ± 2.2 | 2.3 × 105 |
| DH-02 | 0.69 ± 0.02 | 9.5 ± 1.6 | 7.3 × 104 | 0.98 ± 0.01 | 74.0 ± 3.1 | 1.3 × 104 |
| HDHD-06 | 0.43 ± 0.01 | 5.4 ± 0.4 | 8.0 × 104 | 0.72 ± 0.02 | 19.5 ± 1.8 | 3.7 × 104 |
| HDHD-12 | 2.6 ± 0.1 | 1.8 ± 0.3 | 1.4 × 106 | 3.5 ± 0.1 | 3.8 ± 0.3 | 9.1 × 105 |
Three of the four single-crossover chimeras have substrate-activity profiles that mirror the predominant parental enzyme (Table 1). The two chimeras with the majority of their sequence derived from DmdNK (DH-03 and HD-15) exhibited DmdNK-like substrate profiles, demonstrating activity towards pyrimidines and purines. For HD-15, the introduction of 11 hTK2 amino acids at the N-terminus had little effect on purine activity, but reduced the kcat/KM for dT and dC ∼16-fold. The other chimera, DH-02, had 77% of its amino acids derived from hTK2 and exhibited hTK2-like, pyrimidine-specific activity. All three chimeras had reduced catalytic activity towards pyrimidine substrates, compared with DmdNK. DH-02 and DH-03 had kcat/KM values at least two orders of magnitude worse than DmdNK for every substrate.
In contrast, HD-16 shares 78% sequence identity with hTK2 and yet has a DmdNK-like substrate profile (Table 1). Surprisingly, this chimera gained the ability to phosphorylate the purines dA and dG, an activity not detectable in hTK2. Furthermore, HD-16 has 2–5 fold higher catalytic efficiency than hTK2 for pyrimidines.
Two triple-crossover hybrids, HDHD-06 and HDHD-12, were also profiled. Both have approximately equal contributions from each parent (HDHD-06: 44% hTK2; HDHD-12: 40% hTK2), but differ in their substrate specificities (Table 1). HDHD-06 is a pyrimidine-specific, hTK2-like enzyme. It possesses catalytic efficiencies for dT and dC that are three-fold lower than hTK2, and >100-fold lower than DmdNK, due to decreases in kcat for both substrates. The DmdNK-like hybrid, HDHD-12, has improved kcat /KM values relative to hTK2 for all four substrates. The Michaelis constants with pyrimidine substrates are comparable to DmdNK, though the kcat values mirror the lower values of hTK2. Relative to DmdNK, the catalytic efficiency of HDHD-12 is reduced by 38-fold and 40-fold towards dA and dG, respectively.
Chimeras HD-16 and HDHD-12 had DmdNK-like substrate profiles, despite having large segments from hTK2 (74% and 40% of total sequence, respectively). Interestingly, both enzymes possessed the W190R mutation (DmdNK numbering), introduced at a crossover. To control for the effect of this mutation on substrate specificity, we constructed the revertant tryptophan variants of each clone. These mutants were purified and characterized with all four natural deoxyribonucleosides. The catalytic activities of the revertant enzymes were the same as the W190R-containing chimeras (data not shown).
Specific activities with nucleoside analogs
Based on their broadened specificities for the natural nucleosides, the specific activities of HD-16 and HDHD-12 were determined with purine and pyrimidine NAs (Figure 3). As with the natural nucleosides, the parental enzymes have distinct NA substrate profiles. Of the nine analogs tested, DmdNK showed activity towards FAraA, dFdC, 3TC, ddC and ddT. hTK2 was able to phosphorylate dFdC and FAraA, albeit poorly. Neither DmdNK nor hTK2 showed activity against d4T, and no phosphorylation of GCV, ACV or ddI was detected for any parental or chimeric enzyme.
Figure 3.
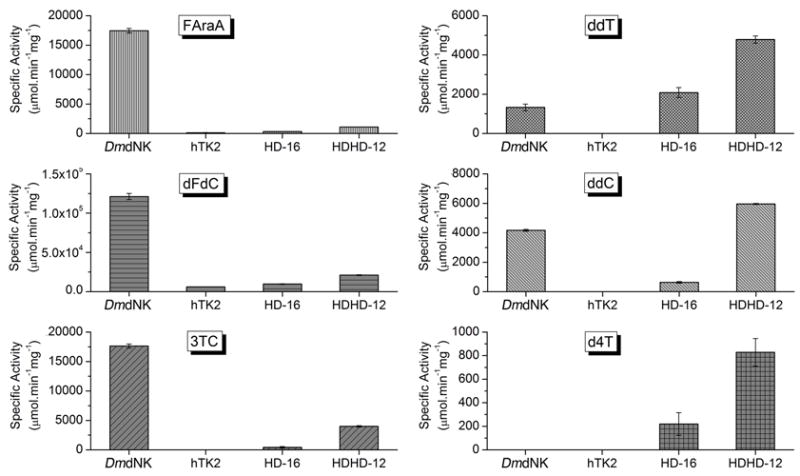
Specific activities of DmdNK, hTK2, HD-16 and HDHD-12 for phosphorylation of the following nucleoside analogs: FAraA (2-fluoro-9β-D-arabinofuranosyladenine), dFdC (2′,2′-difluorodeoxycytidine), 3TC (2′-deoxy-3′-thiacytidine), ddT (2′,3′-dideoxythymidine), ddC (2′,3′-dideoxycytidine) and d4T (2′,3′-didehydro-3′-deoxythymidine). No catalytic activity towards the NAs GCV (ganciclovir), ACV (acyclovir) and ddI (2′,3′-dideoxyinosine) was detected.
Both chimeras could phosphoylate FAraA, dFdC and 3TC, though in each case the specific activity was reduced compared to DmdNK (Figure 3). Such reductions in activity, relative to the most active parent, appear common for enzymes derived from homology-independent recombination.23 HD-16 retained 2–8% of DmdNK’s activity for these three substrates; for HDHD-12, the residual activities were 6–23%. The activity of each chimera was substantially higher than hTK2 (Figure 3).
The chimeras displayed their highest specific activities with the dideoxynucleosides ddT and ddC (Figure 3). Both HDHD-12 and HD-16 showed greater activity towards ddT (by 3.6-fold and 1.6-fold, respectively) than DmdNK. HDHD-12 also possessed 1.5-fold greater activity towards ddC than DmdNK, although HD-16 only retained 15% specific activity for this substrate. Most strikingly, both hybrid enzymes were able to phosphorylate d4T, an activity that neither DmdNK nor hTK2 possesses.24
ddC and d4T are both important components of the therapy regimens used for the treatment of AIDS. Therefore, the kinetic parameters of HD-16 and HDHD-12 with these NAs were analyzed in detail (Table 2). The catalytic efficiency of HD-16 for ddC phosphorylation was 11-fold lower than DmdNK, due to kcat and KM both worsening. For HDHD-12, a reduction in KM led to a 2.8-fold improvement in kcat/KM for ddC. Furthermore, the catalytic efficiency of HDHD-12 for ddC phosphorylation is 28-fold greater than that of hdCK25, the enzyme responsible for phosphorylating ddC in vivo.26 The new d4T activity is interesting because d4T is a poor substrate for dNKs; the catalytic efficiencies of our evolved enzymes are two orders of magnitude higher than those previously reported for natural or engineered dNKs27;28. The activities of HD-16 and HDHD-12 with d4T were similar (kcat/KM≈M−1s−1). Substrate turnover was slow (kcat <1 s−1), but the hybrids had surprisingly low KM values of 66.3 and 148 μespectively. Remarkably, these values indicate that the active sites of the chimeras are more optimized for d4T binding than DmdNK is for purines (KM(dA) = 315 μIT>KM(dG) = 710 μM; Table 1b).
Table 2.
Kinetic parameters of wild-type and selected chimeras with nucleoside analogs.
| ddC | d4T | |||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | |
| hTK2 | nd | nd | nd | nd | nd | nd |
| DmdNK | 1.5 ± 0.1 | 380 ± 20 | 3.9 × 103 | nd | nd | nd |
| HD-16 | 0.42 ± 0.01 | 1240 ± 110 | 3.4 × 102 | 0.07 ± 0.01 | 66.3 ± 10.3 | 1.1 × 103 |
| HDHD-12 | 1.5 ± 0.1 | 135 ± 11 | 1.1 × 104 | 0.29 ± 0.01 | 148 ± 17 | 1.9 × 103 |
Table 1b.
Kinetic parameters of wild-type and chimeric enzymes with purine nucleosides.
| dA | dG | |||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | |
| hTK2a | nd | nd | nd | nd | nd | nd |
| DmdNKa | 26.2 ± 0.5 | 315 ± 23 | 8.3 × 104 | 12.5 ± 0.4 | 710 ± 70 | 1.8 × 104 |
| DH-03 | 0.45 ± 0.01 | 1300 ± 190 | 1.7 × 102 | 0.23 ± 0.01 | 610 ± 70 | 7.4 × 102 |
| HD-15 | 4.6 ± 0.1 | 200 ± 14 | 2.2 × 104 | 5.6 ± 0.1 | 310 ± 20 | 1.8 × 104 |
| HD-16 | 1.2 ± 0.1 | 1240 ± 170 | 9.3 × 102 | 0.23 ± 0.1 | 1530 ± 110 | 1.5 × 102 |
| DH-02 | nd | nd | nd | nd | nd | nd |
| HDHD-06 | nd | nd | nd | nd | nd | nd |
| HDHD-12 | 2.5 ± 0.1 | 1100 ± 60 | 2.2 × 103 | 1.1 ± 0.1 | 2340 ± 200 | 4.5 × 102 |
Data from Gerth et al.6
Discussion
Human dNKs are important targets for engineering, due to their role in activating NA prodrugs and also their potential utility for suicide gene therapy. We have used non-homologous recombination to identify regions accounting for functional differences between hTK2 and DmdNK, and to improve the activity of hTK2 towards clinically-relevant NAs.
Selection for thymidine kinase activity from the DH and HD libraries yielded a majority of clones that were predominantly comprised of DmdNK sequence. This appears to reflect the impaired ability of wild-type hTK2 to fold in E. coli.19 Every functional single-crossover clone had its crossover situated near the N- or C-terminus. Single crossovers in the interior of these dNKs are clearly too disruptive for maintaining function, a bias previously observed for other enzymes.13 Crossovers near the C-terminus occurred in or beyond α8, and typically fell into non-homologous regions. In contrast, N-terminal crossovers always occurred in the conserved P-loop. This loop is located between β1 and α1 (Figure 1) and is critical for binding and positioning the phosphoryl donor, therefore the introduction of crossovers into this key structural and catalytic feature was unexpected. The crossover in the P-loop may be more tolerated than in the neighboring α helix or β sheet, due to the greater flexibility of loop secondary structure elements. Furthermore, crossovers in the P-loop region may also be favorable as many of the residues in the adjacent β1 and α1 are conserved between the parental enzymes, thus minimizing functional disruption.
Most of the single-crossover hybrids characterized kinetically had specificity profiles that mirrored the predominant parent; the exception was HD-16, which has an hTK2-like sequence but displayed a DmdNK-like substrate/activity profile. The crossover in HD-16 is located in the loop region connecting α8 and β5 near the C-terminus of the enzyme. Beyond β5, this portion of the protein sequence shows low overall sequence homology among type-1 dNKs and none of the critical active site residues are located in this region. Further complicating the evaluation of its functional relevance, no structures of DmdNK are available which include this region; the structures have been determined using a recombinant, C-terminal truncation mutant lacking the last twenty residues and in these structures, the last 14–16 residues have no ordered electron density.18,19 Crystal structures of full-length sequences from other type-1 dNKs,18,22 on the other hand, show that this portion of the protein folds into a helical segment (α9) but otherwise has no apparent critical contacts with the rest of the protein. In a separate study, it has been shown that C-terminal truncations of DmdNK affect its substrate specificity; the C-terminal 20 amino acids were not required for phosphorylation of deoxyribonucleosides but were necessary for full activity with dideoxy- and purine-ribonucleosides.8 The deletion of 10 additional residues reduced the activity to 1% that of wild-type. A similar C-terminal truncation of hdGK also resulted in a virtually inactive enzyme.29 Enzyme inactivation upon deletion of the C-terminus could be the result of misfolding or a lack of necessary conformational constraints. Based on our substitution experiments, we suggest that subtle structural changes in protein topology might be responsible for the observed changes in catalytic activity and broadened substrate specificity. Moreover, comparison of DH-03 and HD-16 (Figure 3, Table 1) suggests that the C-terminal subdomain of DmdNK activates hTK2, but not that the C-terminal region of hTK2 is incompatible with purine activity, a priori. Further biochemical and biophysical studies will be required to clarify the role of this subdomain.
HDHD-12 was the most active chimera isolated from the triple-crossover libraries. In addition to the first crossover in the N-terminal P-loop region, HDHD-12, like HD-16, possesses the 52 aa C-terminal subdomain derived from DmdNK and a DmdNK-like substrate profile. The second crossover occurs at the beginning of α5 that forms part of the deoxyribonucleoside binding pocket. As the region shows high amino acid sequence conservation between the two parental enzymes, we predict only small local-structure perturbations resulting from the crossover. On the other hand, the substitution with the 78-amino acid segment of DmdNK (Ser 31 – I107) divides the kinase scaffold, including the active site binding pocket, into two equal segments. Given the extensive interface between these segments and the low overall sequence homology between DmdNK and hTK2, protein crystallography might be one way for obtaining more detailed structural information and for identifying potential conformational changes that translate into altered enzyme function.
Upon testing for NA activity, HDHD-12 and HD-16 both displayed improved activity (relative to hTK2) for six of the nine compounds screened. Quite unexpectedly, the specific activities of HDHD-12 towards ddC and ddT, as well as of HD-16 towards ddC, were actually greater than either parental enzyme. Even more remarkably, the two chimeras are able to phosphorlyate d4T, an activity that both parents lack. Moreover, this activity is greater than that exhibited by any known natural or engineered dNK. While it is not surprising that the chimeras have activity towards ddC and ddT, given the known role of the DmdNK C-terminus in dideoxyribonucleoside phosphorylation, it is interesting that the catalytic efficiency of hybrid enzymes surpasses that of DmdNK. The reason for the improved activity for dideoxyribonucleosides, as well as the new activity towards d4T, is not clear from available structural information. In the case of ddC, the increase in the kcat/KM for HDHD-12 results from improvement in the apparent binding constant, possibly caused by small conformational changes as a result of chimeragenesis. A similar analysis for d4T is not possible as the parental enzymes have no detectable activity for the NA.
Though it is an oft-repeated rule in directed evolution that “you get what you select for”, our results suggest that even when a direct selection for novel or improved activity is not available, enzymes with these properties can be found with minimal screening. Furthermore, the chimeras obtained in this study contain successful combinations that would have been impossible to predict from the available structural and phylogenetic information. Future developments, such as the implementation of direct screens or selections for NA phosphorylation and recombination with other type-1 dNKs, should enable further improvement of the chimeras described. Moreover, as the percentage of active chimeras isolated was very low, creation of alternative crossover libraries is also a potential subject of future work. In a general sense, alternate crossovers could sample a greater portion of the sequence-function diversity. More specifically, it would be interesting to explore crossover combinations that result in N- and C-termini from the same parent. It has been observed in recombination experiments of other enzyme families, that functional chimeras tend to have the N- and C- termini from the same parent.30 The ultimate goal of our experiments is to identify humanized enzymes with higher activities towards NAs than towards natural nucleosides, making them even more promising candidates for suicide gene therapy. The results described here constitute a major step towards this goal.
Materials and Methods
Materials
Pfu Turbo from Strategene (La Jolla, CA), was used for all cloning, unless otherwise indicated. The α-phosphorothioate dNTP mix and Exonuclease III were purchased from Promega (Madison, WI). Pyruvate kinase and lactate dehydrogenase were from Roche Biochemicals (Indianapolis, IN). Plasmid DNA was isolated using the Qiaprep Spin Miniprep kit and PCR products were purified with the Qiaquick PCR Purification kit (Qiagen, Valencia, CA). Enzymes were purchased from New England Biolabs (Beverly, MA), and all other reagents were from Sigma-Aldrich (St. Louis, MO), unless noted. Constructs were confirmed by DNA sequencing.
ITCHY library construction
The single-crossover DH and HD libraries were generated using full-length DmdNK (DNA encoding amino acids 1–250) and a truncated variant of hTK2 (DNA encoding amino acids 20–234).8 The libraries were prepared using the thio-ITCHY protocol described previously14, though instead of using DmdNK and hTK2 cloned in tandem, each gene resided on separate pDIM plasmids which were linearized, mixed and assembled via PCR as described below. Taq DNA polymerase (Promega) was used for the PCR amplification. The template and primer combinations used in constructing the DH ITCHY library were: pDIM-hTK2 linearized with NdeI; pDIM-DmdNK linearized with SpeI; forward primer 5′-CGC CAT ATG AGT GGG AAG ACG ACA TGC-3′; and reverse primer 5’-GCG ACT AGT TCA CTT GCA CGA CTG CGG TCG TCT CTG-3′. For the HD library the templates and primers used were: pDIM-DmdNK linearized with NdeI; pDIM-hTK2 linearized with SpeI; forward primer 5′-CGC CAT ATG AGC GGG AAG ACC ACG TAT TTG AAC-3′; and reverse primer 5′-CGC ACT AGT TCA GGC TGC CAT GGG GAA AAG G-3′. Thermocycling conditions were: 95°C for 3 min; 2 cycles of 94°C for 10 sec and 68°C for 60 sec; add primers and α-dNTPs; 30 cycles of 94°C for 10 sec, 55°C (DH) or 58°C (HD) for 60 sec, 68°C for 210 sec; 1 cycle 68°C for 4.5 min. The PCR products were used to generate the DH and HD ITCHY libraries as described previously.16 The resulting ligation mixture was used to transform E. coli KY895. Libraries were plated on LB-agar containing carbenicillin (Carb; 100 μg/ml). After overnight growth, the colonies were recovered by scraping the plates with 2YT medium, supplemented with glucose (2% w/v) and glycerol (15% v/v). The recovered libraries were divided into aliquots; one portion was saved at −80°C as a frozen stock, and another portion was isolated in plasmid form.
Reading frame selection
The single-crossover libraries were amplified by PCR to replace the stop codon from pDIM-DH and pDIM-HD with a codon for glycine (shown underlined), using the forward primer 5′-ATT AAC CCT CAC TAA AGG GA-3′ (T3), and the reverse primers 5′-AGC ACT AGT TCC TGG GCA ATG-3′ (DH library) and 5′-GGC ACT AGT TCC TCT GCG ACC CTC TG-3′ (HD library). The PCR products were purified, digested with NdeI and SpeI, and ligated into pInSALect19 that had been digested with the same enzymes. The ligation products were used to transform E. coli DH5α-E by electroporation. Cells were plated on LB-agar containing chloramphenicol (Cm; 50 μg/ml) and incubated at 37°C. Colonies were recovered in 2YT medium supplemented with glucose (2% v/v) and glycerol (15% w/v). Reading frame selection was carried out by replating the libraries on LB-agar containing carbenicillin (Carb; 100 μg/ml) and incubating at 21°C. Libraries were recovered as described above, and the plasmid pools (pInSALect-DH and pInSALect-HD) were isolated.
SCRATCHY library construction
Each ITCHY library was divided into three approximately equal and overlapping sections. The sections were amplified by PCR using skewed primers (i.e. a DmdNK-specific forward primer and an hTK2-specific reverse primer, or vice versa), to generate products that each contained one crossover. The pInSALect-DH library plasmids were used to generate the three DH segments, with the following primers: DH-frag1, DF1 (5′-AGT TGT CAG AGG TCA TAT GGC GGA GGC AGC-3′) and HR1 (5′-GCG TAA GAC CCC AGC GAG AGG C-3′); DH-frag2, DF2 (5′-CAC ACC GCC CCA ACC AAC AAG AAG-3′) and HF2 (5′-CTG GTA ACA AGT CTC AGG ATT G-3′); DH-frag3, DF3 (5′-CCG GCA GCG GGC TCG TTC TG-3′) and HR3 5′-(TGC GAA TAC CAC TAG TTC CTG GGC AAT G-3′). Using pInSALect-HD as the template, the following HD fragments were generated: HD-frag1, HF1 (5′-AGT TGT CAG AGG TCA TAT GTC AGT G-3′) and DR1 (5′-CTT CTT GTT GGT TGG GGC GGT GTG-3′); HD-frag2, HF2 (5′-GCC TCT CGC TGG GGT CTT ACG C-3′) and DR2 (5′-CAG AA CGA GCC CGC TGC CGG-3′); HD-frag3, HF3 (5′-CAA TCC TGA GAC TTG TTA CCA G-3′) and DR3 5′-GAA TAC CAC TAG TTC CTC TGG CGA C-3′). The amplifications were carried out using the following cycling conditions: 95°C for 2 min; 30 cycles of 95°C for 30 sec, 57°C for 30 sec, 72°C for 45 sec; 1 cycle of 72°C for 10 min.
Overlap assembly was used to generate the full-length, triple-crossover products. For each library, three gel-purified DNA segments from the previous step were mixed in equal amounts to a final concentration of 15 ng/μl. Fragments DH-frag1, HD-frag2 and DH-frag3 were mixed for assembly of the DHDH library. The HDHD library was assembled using fragments HD-frag1, DH-frag2 and HD-frag3. The primerless assembly reaction used the following conditions: 95°C for 2 min; 35 cycles of 95°C for 30 sec, 52°C for 30 sec, 72°C for 1 min; 1 cycle of 72°C for 10 min.
Next, the reassembled full-length templates were purified from the PCR mix and used as templates for PCR amplification using gene-specific, flanking primers which incorporate NdeI and SpeI restriction sites to facilitate cloning. For amplification of the DHDH library the following primers were used: a DmdNK-specific forward primer, 5′-CTA TCA TAT GGC GGA GGC AGC ATC CTG TGC-3′; and a hTK2-specific reverse primer, 5′-GCG ACT AGT TCA TGG GCA ATG CTT-3′. Similarly, amplification of the HDHD library was performed with an hTK2-specific forward primer, 5′-CAG CCA TAT GTC AGT GAT CTG TGT CG-3′, and a DmdNK-specific reverse primer, 5′-CTC ACT AGT TCA TCT GGC GAC CCT CTG-3′. The cycling conditions were: 95°C for 2 min; 15 cycles of 95°C for 30 sec, 54°C for 30 sec, 72°C for 1 min; 1 cycle of 72°C for 10 min. The resulting products were purified, digested with NdeI and SpeI, and ligated with the pDIM vector. The resulting plasmids were used to transform E. coli KY895.
Selection for thymidine kinase activity
E. coli strain KY89520 was transformed by plasmids and plated on LB-agar containing Carb (100 μg/mL). For library selections, the cells were recovered and replated on thymidine kinase selection plates31 containing 2% casamino acids, 1.5% noble agar (both Difco, BD Biosciences, Sparks MD), 0.5% NaCl, 0.2% glucose, 50 μg/ml carbenicillin, 10 μg/ml 5’-fluorodeoxyuridine (Acros Organics, Morris Plains, NJ), 12.5 μg/ml uridine and 2 μg/ml thymidine.
Site-directed mutagenesis
The R190W mutations were introduced by overlap extension PCR. For mutagenesis of HD-16, two overlapping PCR products were formed with the primer pairs (mutation sites are underlined): forward primer 5′-TAA TAC GAC TCA CTA TAG GG-3′ (T7F) and reverse primer 5′-GTG TAT CAG CCA GCT GCC TTT GAT GAG C-3; forward primer 5′-CAT CAA AGG CAG CTG GCT GAT ACA CC-3′ and reverse primer 5′-TAT GCT AGT TAT TGC TCA G-3′ (T7R). The purified products were assembled in a PCR reaction with the primers T7F and T7R. For mutagenesis of HDHD-12, the primer pairs were: T7F with reverse primer 5′-GGT GTA TCA GCC AGA GCC ACT CCT CAT GG-3′; and forward primer 5′-CAT GAG GAG TGG CTC TGG CTG ATA CAC C-3′ with T7R. The products were also assembled using T7F and T7R.
Enzyme purification
Genes of interest were subcloned into the NdeI and SpeI sites of pET14b (vector modified to include SpeI site). The resulting plasmids were used to transform E. coli BL21(DE3) pLysS (Novagen). Two liter LB cultures were used for IPTG-induced protein expression, as described previously.8 Enzymes were purified by batch/gravity flow column purification using Talon resin (Clontech, Mountain View, CA), also as described previously.8 The pH of each lysis and storage buffer was adjusted such that it was at least one unit away from the pI (estimated using the Expasy ProtParam tool32) of the (His)6-tagged protein being purified; this was done to maximize solubility. The buffers used were: pH 7.0 for HD-15, HD-16 and HDHD-06; pH 7.5 for DmdNK and HDHD-12; and pH 8.0 for hTK2, DH-02 and DH-03. Amicon Ultra centrifugal filter units (Amicon Bioseparations, Billercia, MA) were used to exchange the purified proteins into storage buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM β-mercaptoethanol, 10% glycerol). Yields were 0.1–4 mg per liter culture, depending on the enzyme, and preparations were typically >90% pure as analyzed by SDS-PAGE. Protein concentrations were quantified by measuring A280, using molar extinction coefficients calculated according to Pace et al.33 Aliquots of purified proteins were frozen in liquid nitrogen and stored at −80°C until use.
Steady-state kinetic assays
Spectrophotometric assays to measure nucleoside and NA phosphorylation were performed as described previously.8 All experiments were performed in triplicate. Data were fit to the Michaelis-Mention equation using Origin (OriginLab, Northhampton, MA). Specific activities towards nucleoside analogs were measured with 1mM each substrate, and 0.4 mu;g of each enzyme.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health (GM69958), as well as by a grant to the Emory Center for AIDS Research (AI050409) from the National Institutes of Health and by institutional funding from the Emory University Health Science Center. M.L.G. is a Teem Family Century ARCS Fellow. DNA sequencing was performed at the Center for Fundamental and Applied Molecular Evolution at Emory University (NSF-MRI 0320786). The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: dideoxyinosine (ddI) and Lamivudine (3TC).
Abbreviations
- dNK
deoxyribonucleoside kinase
- NA
nucleoside analog
- d4T
2′,3′-didehydro-3′-deoxythymidine
- hTK2
human thymidine kinase 2
- DmdNK
Drosophila melanogaster deoxyribonucleoside kinase
- hdCK
human deoxycytidine kinase
- hdGK
human deoxyguanosine kinase
- TK
thymidine kinase
- HSV TK
herpesvirus thymidine kinase
- dT
thymidine
- dC
deoxycytidine
- dA
deoxyadenosine
- dG
deoxyguanosine
- FAraA
2-fluoro-9β-D-arabinofuranosyladenine
- dFdC
2′,2′-difluorodeoxycytidine
- 3TC
2′-deoxy-3′-thiacytidine
- ddT
2′,3′-dideoxythymidine
- ddC
2′,3′-dideoxycytidine
- GCV
ganciclovir
- ACV
acyclovir
- ddI
2′,3′-dideoxyinosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550–2. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 2.Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46:5276–81. [PubMed] [Google Scholar]
- 3.Zheng X, Johansson M, Karlsson A. Retroviral transduction of cancer cell lines with the gene encoding Drosophila melanogaster multisubstrate deoxyribonucleoside kinase. J Biol Chem. 2000;275:39125–9. doi: 10.1074/jbc.M006212200. [DOI] [PubMed] [Google Scholar]
- 4.Jordheim LP, Galmarini CM, Dumontet C. Gemcitabine resistance due to deoxycytidine kinase deficiency can be reverted by fruitfly deoxynucleoside kinase, DmdNK, in human uterine sarcoma cells. Cancer Chemother Pharmacol. 2006;58:547–54. doi: 10.1007/s00280-006-0195-8. [DOI] [PubMed] [Google Scholar]
- 5.Eriksson S, Munch–Petersen B, Johansson K, Eklund H. Structure and function of cellular deoxyribonucleoside kinases. Cell Mol Life Sci. 2002;59:1327–46. doi: 10.1007/s00018-002-8511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knecht W, Sandrini MP, Johansson K, Eklund H, Munch-Petersen B, Piskur J. A few amino acid substitutions can convert deoxyribonucleoside kinase specificity from pyrimidines to purines. EMBO J. 2002;21:1873–80. doi: 10.1093/emboj/21.7.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knecht W, Munch-Petersen B, Piskur J. Identification of residues involved in the specificity and regulation of the highly efficient multisubstrate deoxyribonucleoside kinase from Drosophila melanogaster. J Mol Biol. 2000;301:827–37. doi: 10.1006/jmbi.2000.3990. [DOI] [PubMed] [Google Scholar]
- 8.Gerth ML, Lutz S. Mutagenesis of non-conserved active site residues improves the activity and narrows the specificity of human thymidine kinase 2. Biochem Biophys Res Commun. 2007;354:802–807. doi: 10.1016/j.bbrc.2007.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godsey MH, Ort S, Sabini E, Konrad M, Lavie A. Structural basis for the preference of UTP over ATP in human deoxycytidine kinase: illuminating the role of main-chain reorganization. Biochemistry. 2006;45:452–61. doi: 10.1021/bi0518646. [DOI] [PubMed] [Google Scholar]
- 10.Munch-Petersen B, Knecht W, Lenz C, Søndergaard L, Piskur J. Functional expression of a multisubstrate deoxyribonucleoside kinase from Drosophila melanogaster and its C-terminal deletion mutants. J Biol Chem. 2000;275:6673–9. doi: 10.1074/jbc.275.9.6673. [DOI] [PubMed] [Google Scholar]
- 11.Otey CR, Landwehr M, Endelman JB, Hiraga K, Bloom JD, Arnold FH. Structure-guided recombination creates an artificial family of cytochromes P450. PLoS Biol. 2006;4:e112. doi: 10.1371/journal.pbio.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christians FC, Scapozza L, Crameri A, Folkers G, Stemmer WP. Directed evolution of thymidine kinase for AZT phosphorylation using DNA family shuffling. Nat Biotechnol. 1999;17:259–64. doi: 10.1038/7003. [DOI] [PubMed] [Google Scholar]
- 13.Sieber V, Martinez CA, Arnold FH. Libraries of hybrid proteins from distantly related sequences. Nat Biotechnol. 2001;19:456–460. doi: 10.1038/88129. [DOI] [PubMed] [Google Scholar]
- 14.Lutz S, Ostermeier M, Benkovic SJ. Rapid generation of incremental truncation libraries for protein engineering using alpha-phosphothioate nucleotides. Nucleic Acids Res. 2001;29:E16. doi: 10.1093/nar/29.4.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ostermeier M, Shim JH, Benkovic SJ. A combinatorial approach to hybrid enzymes independent of DNA homology. Nat Biotechnol. 1999;17:1205–9. doi: 10.1038/70754. [DOI] [PubMed] [Google Scholar]
- 16.Ostermeier M, Lutz S. The creation of ITCHY hybrid protein libraries. Methods Mol Biol. 2003;231:129–41. doi: 10.1385/1-59259-395-X:129. [DOI] [PubMed] [Google Scholar]
- 17.Lutz S, Ostermeier M, Moore GL, Maranas CD, Benkovic SJ. Creating multiple-crossover DNA libraries independent of sequence identity. Proc Natl Acad Sci USA. 2001;98:11248–53. doi: 10.1073/pnas.201413698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawarasaki Y, Griswold KE, Stevenson JD, Selzer T, Benkovic SJ, Iverson BL, Georgiou G. Enhanced crossover SCRATCHY: construction and high-throughput screening of a combinatorial library containing multiple non-homologous crossovers. Nucleic Acids Res. 2003;31:e126. doi: 10.1093/nar/gng126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerth ML, Patrick WM, Lutz S. A second-generation system for unbiased reading frame selection. Protein Eng, Des Sel. 2004;17:595–602. doi: 10.1093/protein/gzh068. [DOI] [PubMed] [Google Scholar]
- 20.Igarashi K, Hiraga S, Yura T. A deoxythymidine kinase deficient mutant of Escherichia coli. II Mapping and transduction studies with phage phi 80. Genetics. 1967;57:643–54. doi: 10.1093/genetics/57.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansson K, Ramaswamy S, Ljungcrantz C, Knecht W, Piskur J, Munch-Petersen B, Eriksson S, Eklund H. Structural basis for substrate specificities of cellular deoxyribonucleoside kinases. Nat Struct Biol. 2001;8:616–20. doi: 10.1038/89661. [DOI] [PubMed] [Google Scholar]
- 22.Mikkelsen NE, Johansson K, Karlsson A, Knecht W, Andersen G, Piskur J, Munch-Petersen B, Eklund H. Structural basis for feedback inhibition of the deoxyribonucleoside salvage pathway: studies of the Drosophila deoxyribonucleoside kinase. Biochemistry. 2003;42:5706–12. doi: 10.1021/bi0340043. [DOI] [PubMed] [Google Scholar]
- 23.Griswold KE, Kawarasaki Y, Ghoneim N, Benkovic SJ, Iverson BL, Georgiou G. Evolution of highly active enzymes by homology-independent recombination. Proc Natl Acad Sci USA. 2005;102:10082–7. doi: 10.1073/pnas.0504556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Rompay AR, Johansson M, Karlsson A. Substrate specificity and phosphorylation of antiviral and anticancer nucleoside analogues by human deoxyribonucleoside kinases and ribonucleoside kinases. Pharmacol Ther. 2003;100:119–39. doi: 10.1016/j.pharmthera.2003.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabini E, Ort S, Monnerjahn C, Konrad M, Lavie A. Structure of human dCK suggests strategies to improve anticancer and antiviral therapy. Nat Struct Biol. 2003;10:513–9. doi: 10.1038/nsb942. [DOI] [PubMed] [Google Scholar]
- 26.Chen CH, Cheng YC. The role of cytoplasmic deoxycytidine kinase in the mitochondrial effects of the anti-human immunodeficiency virus compound, 2',3'–dideoxycytidine. J Biol Chem. 1992;267:2856–9. [PubMed] [Google Scholar]
- 27.Knecht W, Petersen GE, Sandrini MP, Søndergaard L, Munch-Petersen B, Piskur J. Mosquito has a single multisubstrate deoxyribonucleoside kinase characterized by unique substrate specificity. Nucleic Acids Res. 2003;31:1665–72. doi: 10.1093/nar/gkg257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munch-Petersen B, Cloos L, Tyrsted G, Eriksson S. Diverging substrate specificity of pure human thymidine kinases 1 and 2 against antiviral dideoxynucleosides. J Biol Chem. 1991;266:9032–8. [PubMed] [Google Scholar]
- 29.Mousson de Camaret B, Taanman JW, Padet S, Chassagne M, Mayencon M, Clerc-Renaud P, Mandon G, Zabot MT, Lachaux A, Bozon D. Kinetic properties of mutant deoxyguanosine kinase in a case of reversible hepatic mtDNA depletion. Biochem J. 2006 doi: 10.1042/BJ20060705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyer MM, Hochrein L, Arnold FH. Structure-guided SCHEMA recombination of distantly related beta-lactamases. Protein Eng Des Sel. 2006;19:563–70. doi: 10.1093/protein/gzl045. [DOI] [PubMed] [Google Scholar]
- 31.Black ME, Loeb LA. Identification of important residues within the putative nucleoside binding site of HSV-1 thymidine kinase by random sequence selection: analysis of selected mutants in vitro. Biochemistry. 1993;32:11618–26. doi: 10.1021/bi00094a019. [DOI] [PubMed] [Google Scholar]
- 32.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; USA: 2005. pp. 571–607. [Google Scholar]
- 33.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–23. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.