

Abstract
Cerebellar granule neurons receive inhibitory input from Golgi cells in the form of phasic and tonic currents that are mediated by postsynaptic and extrasynaptic GABAA receptors, respectively. Extrasynaptic receptors are thought to contain α6βxδ subunits. Here we review studies on ethanol (EtOH) modulation of these receptors, which have yielded contradictory results. Although studies with recombinant receptors expressed in Xenopus oocytes indicate that α6β3δ receptors are potently enhanced by acute exposure to low (≥3 mM) EtOH concentrations, this effect was not observed when these receptors were expressed in Chinese hamster ovary cells. Slice recordings of cerebellar granule neurons have consistently shown that EtOH increases the frequency of phasic spontaneous inhibitory postsynaptic currents (sIPSCs), as well as the tonic current amplitude and noise. However, there is a lack of consensus as to whether EtOH directly acts on extrasynaptic receptors or modulates them indirectly; i.e. via an increase in spillover of synaptically released GABA. It was recently demonstrated that an R to Q mutation of amino acid 100 of the α6 subunit increases the effect of EtOH on both sIPSCs and tonic current. These electrophysiological findings have not been reproducible in our hands. Moreover, it was shown the α6-R100Q mutation enhances sensitivity to the motor-impairing effects of EtOH in outbred Sprague-Dawley rats, but this was not observed in a line of rats selectively bred for high sensitivity to EtOH-induced motor alterations (Alcohol Non-Tolerant rats). We conclude that currently there is insufficient evidence conclusively supporting a direct potentiation of extrasynaptic GABAA receptors following acute EtOH exposure in cerebellar granule neurons.
Keywords: GABA, ethanol, extrasynaptic, presynaptic, granule
The Cerebellum as a Target of Ethanol
The cerebellum is involved in the control of balance as well as the coordination, planning, and fine regulation of voluntary movement. In addition, this brain region has been shown to be important for certain cognitive functions, including thought, behavior, and emotion (Schmahmann and Sherman, 1997). The cerebellum is composed of the cerebellar cortex, inner white matter, and deep cerebellar nuclei. Neurons of the cerebellar cortex are organized into three layers. The outermost is the molecular layer that contains the basket and stellate interneurons, the distal portion of the excitatory axons from the cerebellar granule neurons (CGNs), and the dendrites of Purkinje neurons. The Purkinje neuron layer is located below the molecular layer and it contains the large cell bodies of these neurons, which constitute the sole output of the cerebellar cortex and are inhibitory GABAergic neurons with axons that project to the deep cerebellar nuclei. The innermost layer of the cerebellar cortex is the granule cell layer, which contains the CGNs and the proximal segments of the ascending portion of their axons. This layer also includes Golgi interneurons and the mossy fibers. The CGNs receive excitatory glutamatergic input from mossy fibers and GABAergic inhibitory input from Golgi cells. This review focuses on the actions of ethanol (EtOH) on GABAergic transmission at CGNs and prior to discussing this topic, a brief overview of EtOH’s effects on cerebellar function is provided.
Numerous studies have shown that short- and long-term EtOH exposure impairs normal development of cerebellar cortical neurons as well as their proper functioning at maturity. Investigators initially measured EtOH’s effect on cerebellar type-A γ-aminobutyric acid receptor (GABAA-R) function using 36Cl- flux assays in microsacs obtained from mature rodents. These studies consistently demonstrated that acute EtOH exposure enhances 36Cl- flux in this preparation (Allan and Harris, 1986; Harris et al., 1995; Proctor et al., 1992a; Proctor et al., 1992b). Most studies on the acute effects of EtOH on the function of specific populations of mature cerebellar neurons have focused on Purkinje neurons (Deitrich et al., 1989). These studies generally found that EtOH acutely depresses Purkinje cell firing (Basile et al., 1983; George and Chu, 1984; Siggins and French, 1979; Sorensen et al., 1980). It was shown that the benzodiazepine ligands, Ro 15-4513 and FG 7142 as well as and the GABAA receptor antagonist, bicuculline, antagonized the EtOH-induced depression of Purkinje neuron firing in anesthetized animals, thereby implicating GABAA-Rs in the mechanism of action of EtOH (Palmer and Hoffer, 1990; Palmer et al., 1988). However, (Lee et al., 1995b) found that Ro15-4513 did not antagonize the acute effects of EtOH on Purkinje neuron firing. It was reported that EtOH did not always potentiate responses evoked by exogenous GABA and that it had a more reliable effect if β-adrenergic, GABAB, or nicotinic receptors were concomitantly activated (Freund and Palmer, 1997; Freund et al., 1993a; Lee et al., 1995a; Lin et al., 1994; Lin et al., 1991; Lin et al., 1993; Palmer and Hoffer, 1990; Yang et al., 1999; Yang et al., 2000). More recently, it was reported that acute EtOH exposure increases quantal GABA release at Purkinje neurons in slices of the cerebellar vermis (Breese et al., 2006; Ming et al., 2006).
The acute effects of EtOH on other neuronal types of the mature cerebellum have only been investigated in a few studies. Single unit recordings showed that intraperitoneal injection of EtOH increases the discharge rate of rat inferior olivary nucleus neurons (Rogers et al., 1986). Acute EtOH exposure was shown to increase firing of inhibitory Golgi interneurons in cerebellar slices (Freund et al., 1993b). The authors of this study stated that this effect would be predicted to increase Golgi neuron input to CGNs and ultimately influence excitatory input to Purkinje neurons.
In the early 1990s, genetic, pharmacological, and functional studies suggested that GABAA-Rs in CGNs are important targets of EtOH. These studies are discussed below in section 2.2.
Effects of EtOH on GABAergic Transmission at CGNs
Overview of GABAergic input to CGNs
CGNs relay excitatory input from mossy fibers to Purkinje neurons via divergent pathways and this process is profoundly modulated by GABAergic transmission. Golgi interneurons provide two types of GABAergic input to CGNs: tonic and phasic. Tonic GABAergic inhibition dominates over phasic inhibition (Hamann et al., 2002). The presence of a tonic GABAergic current in granule cells was first demonstrated by the existence of a bicuculline sensitive “background noise current” that persisted in the presence of glutamate antagonists, tetrodotoxin (TTX), or zero [Ca2+]o (Kaneda et al., 1995). These findings indicated that the current persists in the absence of action potential-dependent GABA release. In rodents, this tonic current appears gradually during development; it is first apparent at P14 and reaches stable levels at ~P30 (Brickley et al., 1996; Tia et al., 1996; Wall and Usowicz, 1997). This current reflects activation of high-affinity extrasynaptic GABAA-Rs by ambient GABA levels as well as spillover of synaptically-released GABA (Rossi et al., 2003). One factor that permits this accumulation of GABA is the unique configuration of the synapse between Golgi and granule cells. This synapse forms part of a glomerulus that contains four elements: 1) the glutamatergic mossy fiber afferent, 2) the axons from Golgi cells, 3) the granule cell dendrites, and 4) a glial sheath. Significantly, this sheath retains released GABA in the glomerulus, increasing the chances of extrasynaptic receptor activation. In addition to the protracted time course of glomerular GABA concentrations, the unique subunit composition of GABAA-Rs present in cerebellar granule cells further contributes to the generation of tonic currents; i.e. extrasynaptic GABAA-Rs contain α6βxδ subunits, which are potently activated by GABA (Hamann et al., 2002). α6βxδ receptors have a relatively low EC50 (~0.2-0.7 μM) for activation by GABA, which is in the range of the lowest level that transporters can maintain extracellular GABA concentrations (~0.16-0.4 μM) (Attwell et al., 1993; Santhakumar et al., 2006a; Saxena and Macdonald, 1996; Wallner et al., 2003). Moreover, GABAA-Rs containing α6 and δ subunits display relatively little desensitization (Saxena and Macdonald, 1994). All of these characteristics make these receptors ideal mediators of tonic currents triggered by low background levels of GABA.
The α6 subunits have been the focus of research interest since they are almost exclusively expressed in the cerebellar granule cells. α6 -/- knockout mice were generated, which also display complete loss of δ subunits due to the preferential receptor partnership of these subunits with α6 subunits in CGNs (Brickley et al., 2001). In cerebellar slices from these mice, the tonic current was absent. Paradoxically, α6 -/- mice exhibit normal motor function which raised questions about the physiological significance of the tonic current. However, deletion of the GABAA receptor subunits in these mice elicited an increase in a leak conductance mediated by the two-pore forming K+ channel TASK-1 (Brickley et al., 2001). The increase in this current compensated for the loss of GABAA receptor subunits and normalized the response of CGNs to excitatory input. The impact of tonic inhibition on CGN excitability was also assessed by (Hamann et al., 2002). Reduction of this current with furosemide increased input resistance, lowered the threshold for action potential generation, and increased the number of action potentials triggered at different levels of current injection. Furosemide increased CGN excitability in response to mossy fiber-to-granule cell synaptic transmission. The studies of (Hamann et al., 2002) estimated that the tonic current mediates over 90% of inhibitory current in granule cells. These authors showed that reduction of the granule cell tonic current with furosemide has a significant impact on Purkinje cell excitability. Specifically, furosemide increases the excitability of Purkinje neurons in response to mossy fiber stimulation of CGNs. However, it does not affect excitability of Purkinje neurons in response to direct stimulation of the parallel fiber. Importantly, an elegant in vivo electrophysiological study recently demonstrated that tonic GABAergic inhibition of CGNs profoundly regulates the transformation of mossy fiber somatosensory input into CGN output (Chadderton et al., 2004).
CGNs also receive phasic GABAergic inhibition in the form of inhibitory postsynaptic potentials (IPSCs) produced by conventional action potential- and Ca2+-dependent vesicular GABA release from Golgi interneurons. This phasic inhibition is mediated in part by receptors containing α1βxγ2 subunits. These IPSCs superimpose on the background tonic current. This form of GABAergic inhibition mediates ~10% of the total inhibition of granule cells and is thought to be important for controlling synchronous rhythmic firing of granule cells (Hamann et al., 2002).
Modulation of GABAergic transmission at CGNs by EtOH
Relatively few studies have characterized the acute effect of EtOH on GABAergic transmission at CGNs. The first indication that EtOH might increase GABAergic input to this neuronal population came from in vivo studies performed with chloralose-anesthetized cats in which EtOH was shown to acutely inhibit responses of CGNs to auditory stimuli (Huang and Huang, 1992; Huang et al., 1991). More recently, a similar finding was reported for CGN responses elicited by visual stimuli (Huang and Huang, 2007). Collectively, these results are consistent with inhibition of mossy fiber-mediated excitation by EtOH. A possible mechanism mediating this effect could be an increase in GABAergic transmission at CGNs. (Engblom et al., 1991) assessed GABAA receptor function in cultured CGNs using the chloride-sensitive fluorescent probe, 6-methoxy-N-(3- sulfopropyl)quinolinium. These investigators found that EtOH at concentrations below 50 mM potentiated Cl- efflux via GABAA-Rs, particularly when low GABA concentrations were used to activate the receptors. In contrast, (Reynolds et al., 1992) found that currents evoked by pressure ejection of GABA onto cultured CGNs were potentiated by concentrations of EtOH between 1-50 mM only in 28% of the neurons examined. Several years later, a study focused on excitatory GABAA-Rs expressed in the axonal rather than the inhibitory GABAA-Rs present in CGN dendrites. (Schmid et al., 1999) found that 50 mM EtOH acutely potentiated the GABA-induced enhancement of [3H]D-aspartate release in synaptosomes enriched for these axonal terminals. Interestingly, EtOH was effective in synaptosomes from Alcohol Non-Tolerant (ANT), but not Alcohol Tolerant (AT) rats.
The first study of EtOH’s action on GABAergic transmission at CGNs using acute cerebellar slices was performed by (Carta et al., 2004), who found that acute EtOH exposure at concentrations as low as 20 mM reversibly increased the frequency of sIPSCs. The amplitude and decay time constants of these events were, however, unaffected. EtOH also increased the magnitude of the tonic GABAergic current mediated by α6βxδ extrasynaptic GABAA-Rs. Importantly, the effect of EtOH on the tonic current was abolished in the presence of the Na+ channel blocker, TTX. Loose-patch cell-attached recordings revealed that EtOH increases spontaneous firing of Golgi cells. Taken together, these findings indicate that EtOH indirectly enhances the tonic current presynaptically via an increase in spillover of spontaneously released GABA from Golgi cells (see Fig 1 for a schematic representation). Our resultss are in general agreement with those of other investigators indicating that low EtOH concentrations do not potentiate currents mediated by extrasynaptic GABAA receptors in cultured CGNs (Casagrande et al., 2007; Yamashita et al., 2006).
Figure 1.
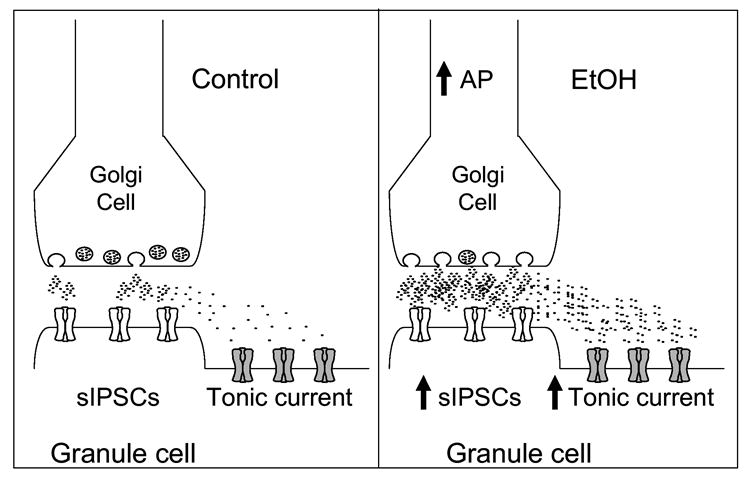
Simplified model of the EtOH-induced enhancement of GABAergic transmission to CGNs. Under control conditions, spontaneous action potential firing of Golgi cells evokes synaptic GABA release and activation of postsynaptic GABAA-Rs. Activation of these receptors generates sIPSCs. Extrasynaptic GABAA-Rs are also activated via GABA spillover, contributing to the tonic current generation together with ambient GABA. Application of EtOH (≥ 20 mM) increases spontaneous action potential (AP) firing in Golgi cells, which elevates GABA levels both at synaptic and extrasynaptic sites. This results in a parallel increase in both sIPSC frequency and the tonic GABAergic current.
Our finding that EtOH increases sIPSC frequency via an increase in Golgi cell firing has been independently reproduced by others (Hanchar et al., 2005; Santhakumar et al., 2006b). However, evidence has been put forward indicating that EtOH directly and very potently enhances the function of EtOH on α6βxδ extrasynaptic GABAA-Rs. (Wallner et al., 2003) reported that EtOH enhances the function of α6β2δ and α6β3δ recombinant receptors expressed in Xenopus oocytes at concentrations ≥30 mM and ≥3 mM, respectively; however, it should be noted that attempts to replicate these findings in a different expression system were unsuccessful (Yamashita et al., 2006) (also see Borghese et al in this issue). Subsequently, (Hanchar et al., 2005) used site-directed mutagenesis and expression in Xenopus oocytes to demonstrate that a substitution from arginine (R) to glutamine (Q) at position 100 in the α6 subunit dramatically enhances the potentiating effect of EtOH on currents mediated by GABAA-Rs containing α6β3δ subunits. These findings generated tremendous interest given that the α6-R100Q polymorphism had long been implicated by studies with genetic rodent models in the mechanism of action of EtOH. Specifically, these initial genetic studies were performed with the AT and ANT rat lines. In the next sections, we discuss genetic, behavioral, and electrophysiological experiments of EtOH actions that have been performed with these rats.
Studies with AT and ANT Rats
α6-R100Q and EtOH-related Behaviors in the AT and ANT Selected Lines
AT and ANT rats were selectively bred for differential ataxic sensitivity to EtOH as measured by performance on the tilting plane test (Eriksson and Rusi, 1981); see Korpi et al., this issue). In this test, each animal was given three trials in which it was placed on the raised end of a mechanical tilting plane that was elevated from a flat horizontal position to an 84-degree angle in 5 s. The average angle at which the animal slid to the base of the plane was recorded (baseline) followed by an intraperitoneal injection of EtOH (2.0 g/kg, 15% w/v in saline). After a 30-minute interval, the average angle obtained on 3 additional trials was recorded. Ataxic sensitivity was measured as the difference between the baseline and post-injection angle of sliding (Sellin and Laakso, 1987). Following more than 35 generations of selection pressure, ataxic sensitivity in the AT and ANT rats differed by over 15 degrees, which is probably the combined effect of at least two possibly genetically-independent sub-traits: a small, but significantly lower baseline angle and a significantly greater angle 30 minutes after 2.0 g/kg EtOH in the AT compared to the ANT rats (Radcliffe et al., 2004; Sarviharju and Korpi, 1993); R. Radcliffe, unpublished results). Other traits, such as anxiety, for which the AT and ANT rats also show a significant difference, may also contribute to the differential ataxic sensitivity (Tuominen et al., 1990).
The AT and ANT rats have been shown to be differentially sensitive to lorazepam and sodium barbital suggesting genetic differences in GABAergic function (Hellevuo et al., 1989) and receptor binding studies with the benzodiazepine ligand [3H]Ro-15-4513 have also implicated GABAergic mechanisms. This ligand binds to two populations of GABAA-Rs: diazepam sensitive and diazepam insensitive. The latter is revealed when the binding assay is conducted in the presence of high concentrations of diazepam. It was found that [3H]Ro-15-4513 binding was normal in the AT, but a large proportion of the ANT rats did not possess the diazepam-insensitive binding sites; i.e., virtually all [3H]Ro-15-4513 binding was blocked in the presence of diazepam (Uusi-Oukari and Korpi, 1990). It was subsequently shown that the binding difference was mediated by the α6-R100Q single nucleotide polymorphism (SNP) (Korpi et al., 1993); also see Korpi et al., this issue). It is likely that this point mutation accounts for at least part of the genetic difference in benzodiazepine sensitivity in the AT and ANT rats (Korpi and Seeburg, 1993).
Given the known interaction between EtOH and GABAA-Rs, it has been speculated by many investigators that the α6-R100Q mutation might be at least partially responsible for the differential EtOH sensitivity of the AT and ANT selected lines, although in the original report it was noted that the mutation did not affect EtOH modulation of α6β2γ2 receptors expressed in a cell culture system (Korpi et al., 1993). Subsequent genotyping of a sample of 116 rats from generations 45-46 showed that the mutant (Q) allele occurred with a frequency of 0.86 in the ANT while the frequency was only 0.09 in the AT (Makela et al., 1995). Among AT and ANT breeders that were transferred to the University of Colorado in 1998 (generation 60), only the mutant allele was found in the ANT, while both alleles were still present in the AT (Radcliffe et al., 2004). These results are consistent with a segregation of the Q allele in the ANT high-sensitivity phenotype. However, if a gene is important in a selected trait, it is expected that after many generations of selection (at least 34 for the AT and ANT; (Sarviharju and Korpi, 1993), alternate alleles of the gene should become fixed (i.e., homozygous) in the selected lines, especially considering the relatively limited number of breeding pairs that are typically maintained in such experiments. However, the alleles were not fixed in either the AT or ANT after at least 45 generations. It is possible that alleles for a gene could be present in a heterozygous state in one of the lines if one of the alleles has a dominant mode of action, although the recessive allele still would be expected to become fixed in the other line. Thus, it has to be concluded that the enrichment of the Q allele in the ANT and of the R allele in the AT, while certainly not proof of the null hypothesis, is not strong evidence in and of itself for an effect of the Q mutation on the tilting plane test, especially considering that a replication of the selection was not conducted (Crabbe et al., 1990).
Six generations of brother-sister matings were carried out with the University of Colorado rats to produce several inbred ANT and AT lines. Although inbreeding typically requires 20 or more generations, genotype testing indicated that the lines were already greater than 90% inbred when they arrived at the University of Colorado (Radcliffe et al., 2004). Care was taken to establish AT lines that carried one or the other of either the Q or R allele; this was not possible for the ANT since all of the University of Colorado ANT rats were homozygous for the Q allele. Studies of the inbred AT lines indicated that the Q mutation did not affect moderate or high dose EtOH sensitivity, including ataxic sensitivity on the tilting plane, consistent with the absence of an EtOH-modulating effect of the Q allele (Radcliffe et al., 2004). It is possible, however, that an interacting ANT allele at a different locus was required for the phenotypic expression of the Q mutation and therefore it would be without effect on a pure AT background.
One way to rule out the possibility of epistasis (“background effect”) is to study the effect of a single gene mutation in a segregating population, or at least on several different strain backgrounds. With this in mind, Korpi and colleagues created an F2 intercross from the AT and ANT phenotypes to more adequately address the question of the involvement of the α6-R100Q mutation in EtOH responses (Korpi and Uusi-Oukari, 1992; Sarviharju and Korpi, 1993). The original tilting plane test and several other behavioral tasks were examined in the F2 rats. [3H]Ro-15-4513 binding in the presence of diazepam was measured as a surrogate marker for the α6-R100Q mutation, which had not yet been discovered. None of the tilting plane measurements correlated to the [3H]Ro-15-4513 binding, nor did measurements of high-dose EtOH sensitivity. Although not definitive, these results suggest that the Gabra6 alleles did not segregate with the tilting plane phenotypes in the F2 generation and therefore were not important in the selected difference between the AT and ANT phenotypes, or in any of the EtOH-related responses that were measured (Korpi and Uusi-Oukari, 1992).
QTL Mapping Studies Related to α6-R100Q in the AT and ANT Selected Lines
Following inbreeding of the AT and ANT lines at the University of Colorado, a large AT X ANT F2 intercross population was generated to conduct quantitative trait locus (QTL) mapping and other genetic studies (Radcliffe et al., 2004). The grandparents of the F2 rats carried both alleles of the α6 mutation – the R allele in the AT and the Q allele in the ANT – to facilitate examination of its role in EtOH-related behaviors. QTL mapping is a type of linkage analysis in which members of a segregating population, such as an F2, are phenotyped for a trait of interest and then genotyped for informative DNA markers, typically throughout the entire genome. It is then determined at which genomic locations phenotype segregates with genotype. If a chromosomal region contains a gene that is polymorphic and that influences the phenotypic expression of the trait, then the markers in that region will correlate to the phenotype (Abiola et al., 2003). Thus, a QTL is a chromosomal region containing a DNA sequence difference(s) that contributes to genetic variance for a trait (Mackay, 2001). We reasoned that if the α6-R100Q mutation was at all responsible for the tilting plane difference in the selected lines, then a QTL should map at approximately 27.5 Mb on chromosome 10, the location of the rat Gabra6 gene.
Over 1200 F2 rats were bred and tested (see (Radcliffe et al., 2004) for complete details of breeding and phenotype testing). In the first week of testing, the animals were assessed for EtOH-induced ataxia in exactly the same manner as that used for the selection of the AT and ANT phenotypes. In fact, the original apparatus was used, courtesy of Dr. Kalervo Kiianmaa. One week later, the animals were again tested on the tilting plane and during this test, the time at which the animal regained its original sliding angle was recorded. Finally, high-dose EtOH sensitivity (3.5 g/kg) was tested in the third week using the loss of righting reflex test (LORR).
QTL mapping is made more efficient by genotyping only the extreme responders in a segregating population, since it is these animals that capture the greatest proportion of the genetic variance (Darvasi and Soller, 1992). Thus, we genotyped 479 F2 animals (approximately equal numbers of males and females) that were in the upper and lower 20% of the distribution for ataxic sensitivity-the selection trait for the AT and ANT phenotypes described above. The average values for ataxic sensitivity for these groups of F2 rats are shown in table 1 along with the average values for the AT and ANT progenitors. A full genome scan was conducted using simple sequence-length polymorphism DNA markers (SSLP) that had been determined to be different between the inbred AT and ANT rats, and spaced at approximately 19 cM on average (see (Radcliffe et al., 2004) for a general description of genotyping procedures). Here we will focus on just chromosome 10, which is where Gabra6 is located. The complete analysis is currently being prepared for publication.
Table 1.
Mean phenotypic scores (± SEM) for the tilting plane test in the AT and ANT progenitors; all ATXANT F2 progeny that were generated for QTL mapping; the ATXANT F2 progeny that were in the upper or lower 20% of the distribution for ataxic sensitivity and that were used for QTL mapping; and additional F2 rats that were genotyped for the Gabra6 mutation
| Subjects | Genotype | n | Baseline angle | Angle at 30’ after 2.0 g/kg EtOH | Ataxic sensitivity (baseline angle minus 30’ angle) |
|---|---|---|---|---|---|
| AT | -- | 87 | 81.2 ± 0.4 * | 82.3 ± 0.3 * | -1.1 ± 0.5 *† |
| ANT | -- | 128 | 83.2 ± 0.1 | 74.3 ± 0.8 | 8.9 ± 0.8 † |
|
| |||||
| All F2 | -- | 1168-1194 | 80.3 ± 0.1 † | 78.7 ± 0.2 | 1.7 ± 0.2 † |
|
| |||||
| F2 (lower 20% for ataxic sensitivity) | -- | 239 | 76.4 ± 0.3 * | 81.4 ± 0.2 * | -5.0 ± 0.2 * |
| F2 (upper 20% for ataxic sensitivity) | -- | 240 | 81.1 ± 0.2 | 71.7 ± 0.3 | 9.5 ± 0.2 |
|
| |||||
| Genotyped F2 | R/R | 19 | 79.7 ± 1.1 | 74.9 ± 1.8 | 4.8 ± 2.3 |
| Genotyped F2 | R/Q | 35 | 79.5 ± 0.8 | 74.8 ± 1.1 | 4.8 ± 1.2 |
| Genotyped F2 | Q/Q | 18 | 80.6 ± 0.8 | 77.6 ± 1.2 | 3.0 ± 1.6 |
AT compared to ANT, or lower F2 compared to upper F2, p<0.001 (one-way ANOVA).
These results were previously published in Radcliffe at al., 2004
The QTL analysis was conducted with the Rqtl package using the Haley-Knott regression method of interval mapping (Broman et al., 2003; Haley and Knott, 1992). Interval mapping is a method with which the location and significance of a QTL can be estimated in the region between markers. The chromosome 10 mapping results for baseline angle, angle at 30 minutes after 2.0 g/kg EtOH, and ataxic sensitivity (baseline angle minus 30 minute angle) are shown in Fig 2. Permutation methods were used to determine empirically-derived suggestive and significant likelihood of the odds ratio (LOD) thresholds; i.e., genome-wide probability of 0.05 and 0.63, respectively (Churchill and Doerge, 1994). These thresholds were very similar for each of the three traits, with LOD scores from 3.2 to 3.3 for significant and from 1.8 to 2.0 for suggestive. For clarity, only the lowest thresholds are shown in the figure. As can be seen, no single locus on chromosome 10 surpassed even the suggestive threshold for any of the three traits that were mapped. Importantly, this included the interval between markers at approximately 18 and 38 Mb within which the Gabra6 gene is located. Interval mapping results for the other EtOH-related behaviors that were tested in weeks 2 and 3 were similar; i.e., all were below the suggestive level (data not shown). These results strongly suggest that the GABAA-R α6-R100Q mutation did not contribute to genetic variation for the behavioral tests that were examined in this study. It can be further concluded that the mutation does not contribute to differential EtOH-mediated ataxic sensitivity in the AT and ANT rats.
Figure 2.
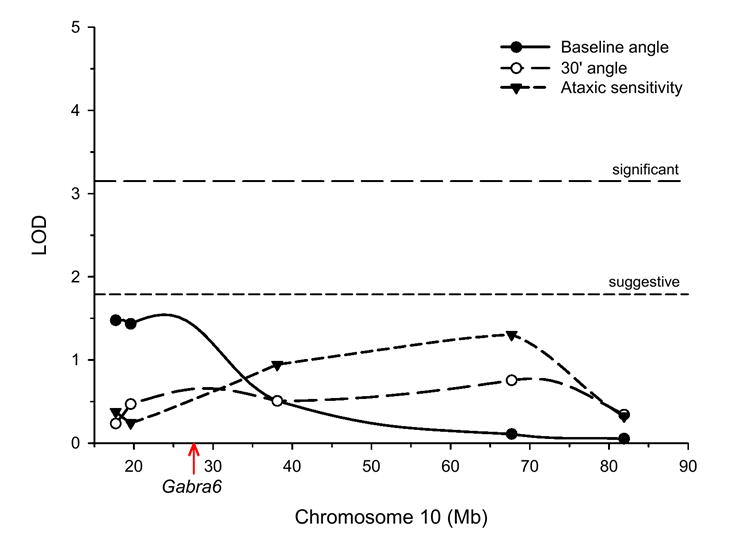
Chromosome 10 QTL map of tilting plane test variables in F2 progeny derived from an AT X ANT intercross. Symbols indicate marker locations and horizontal lines show empirically-derived significance LOD thresholds. Physical location of Gabra6 is indicated by the arrow.
As mentioned above, an epistatic interaction may be necessary for the α6 mutation to be involved in the AT/ANT difference; i.e., the Q allele requires another ANT allele at a different locus for its effect to be expressed. In a segregating population, unlinked alleles are randomly distributed meaning that some proportion of the animals will carry the necessary combination of alleles for the phenotype to be expressed if epistasis is involved. In such cases, QTL mapping procedures are capable of identifying the interacting loci. A pair scan analysis conducted in Rqtl did not reveal any loci interacting with any region of chromosome 10 (results not shown) suggesting that epistasis was not a factor in this population, at least not involving chromosome 10.
It is possible for interval mapping procedures to overestimate a LOD score when the peak is situated somewhere near the middle of two flanking markers, but it is unlikely that it could be underestimated. Nonetheless, a randomly selected cohort of the F2 was genotyped specifically for the Gabra6 SNP to ensure that this was not the case, since Gabra6 is located between markers that were used for the QTL analysis. The Amplification Refractory Mutation System developed for use with the Gabra6 SNP was used to genotype 72 F2 rats (Radcliffe et al., 2004; Saba et al., 2001). The rats were behaviorally tested as described above and the phenotypes were analyzed using one-way ANOVA with the influence of genotype as the factor to be tested. The results of this analysis are shown in Table 1. The distribution of genotypes in the F2 sample was not significantly different from the expected Mendelian ratio of 25%:50%:25% (X2, p>0.9) suggesting that the mutation did not influence fecundity. If the α6-R100Q mutation conferred greater EtOH sensitivity, then it would be predicted that the angle 30 minutes after EtOH would have been smaller while ataxic sensitivity would have been larger in F2 animals with the Q allele compared to those with the R allele. In fact, just the opposite was observed, although there was not a significant effect of genotype for any of the measures (one-way ANOVA, p> 0.3). These results are consistent with the QTL mapping results and do not support the hypothesis that the α6-R100Q mutation mediates any portion of the difference in tilting plane sensitivity in the AT and ANT rats.
Polymorphisms in the rat Gabra6 regulatory region as well as in the Gabra1, Gabrb2, and Gabrg2 genes have been found to be linked to the Q allele of Gabra6 (Congeddu et al., 2003; Saba et al., 2005). It is not known if these polymorphisms have any effect on interactions between GABAA-Rs and EtOH, although it has been postulated that the Gabra6 regulatory region mutations influence α6 expression, especially during the course of chronic EtOH administration (Saba et al., 2005; Sanna et al., 2004). The QTL mapping results further suggest that these or any other linked polymorphisms, if actually present in the ANT rats, do not contribute to the EtOH sensitivity difference between the AT and ANT phenotypes. Otherwise a QTL would have mapped to this region irrespective of which polymorphism was involved.
The α6-100Q mutation has been found in other selected rat lines; specifically, the Sardinian Preferring and Non-Preferring rats (sP, sNP) and the Alko Alcohol and Alko Non-Alcohol lines (AA, ANA). These animals were selected for differential alcohol preference when presented with a choice between an EtOH solution or water (Colombo, 1997; Hilakivi et al., 1984). The non-preferring lines (sNP, ANA) both had a higher frequency of the Q allele compared to that of the preferring lines (sP, AA); the Q allele was not present in other rat lines selected for EtOH drinking behavior (Carr et al., 2003; Saba et al., 2001). There is a degree of consistency in these findings, although there are too few points to allow an actual correlational analysis. It should be noted that in the AT X ANT F2 study of (Sarviharju and Korpi, 1993) EtOH preference did not correlate to tilting plane sensitivity despite a preference difference in the AT and ANT rats. If the Q allele does indeed suppress EtOH preference, which is far from definitive at this time, then this result could be considered additional evidence, albeit indirect, for the absence of an effect of the α6 mutation on the tilting plane test. However, it remains to be seen if any of the GABAA subunit gene polymorphisms that are clustered on chromosome 10, including α6-R100Q, influence genetic variation in EtOH preference.
Electrophysiological Studies
(Valenzuela et al., 2005) investigated the effect of acute EtOH exposure on phasic and tonic GABAergic currents at CGNs in cerebellar slices from male (~30-day-old) ANT and AT rats that were inbred at University of Colorado HSC as discussed above (Radcliffe et al., 2004). Studies were performed on slices from a total of 4 ANT and 7 AT rats. All inbred ANT rats were homozygous for the 100Q genotype and all inbred AT rats were homozygous for the 100R genotype. In slices from these rat lines, we did not detect any differences in the effect of EtOH on tonic GABAergic currents (Valenzuela et al., 2005). Basal tonic current amplitudes were not significantly different in slices from ANT versus AT rats. We also found no significant differences in the acute effect of EtOH on sIPSC frequency in CGNs from ANT and AT rats (Valenzuela et al., 2005). Finally, basal sIPSC frequencies and amplitudes were not significantly different in slices from ANT versus AT rats (Valenzuela et al., 2005). Our results indicate that neither differences in the basal properties of GABAergic transmission at CGNs nor differential modulation of this transmission by acute EtOH exposure are responsible for the phenotypes of ANT and AT rats. These findings are in agreement with the genetic studies discussed above.
Studies with Outbred Sprague-Dawley Rats
A significant effect of the α6-R100Q mutation on EtOH-mediated impairment on the accelerating rotarod was observed in a population of outbred Sprague Dawley rats, which was recently found to carry both the R and Q alleles (Hanchar et al., 2005). This would appear to be at odds with the lack of such an effect on the tilting plane test and other behaviors in the AT and ANT rats, as described above. The authors suggested that the behavioral effect of the mutation is operative only at lower EtOH doses (< 1.5 g/kg), although it was shown that the most robust effect of the Q allele on EtOH enhancement of tonic GABA currents in cerebellar slices occurred at a concentration of 100 mM, which would be equivalent to a brain EtOH concentration produced by a high dose of EtOH (~3.5 g/kg) (Hanchar et al., 2005; Otis et al., 2005). While the effect of α6-R100Q may be population-, dose- and/or task-dependent, the genetic evidence presented in Section 3 and in other reports cited above (especially Korpi et al., 1992) do not support a role for the α6-R100Q mutation in EtOH-induced ataxia either in the AT and ANT selected lines or in segregating crosses derived from them. It is most parsimonious to conclude that the enrichment of the Q allele in the ANT was coincidental and was not because it contributes to genetic variance in the tilting plane test.
(Otis et al., 2005) argued that confounding differences likely arose during selective breeding of the AT and ANT rats and that this could explain the lack of an effect of the α6-R100Q mutation on EtOH sensitivity in these animals as opposed to outbred Sprague- Dawley rats. Differences do arise during selective breeding, but these are minor in comparison to the random differences present in outbred rats. In a group of outbred animals such as the Sprague-Dawley rats used in the study of (Hanchar et al., 2005), it would be unlikely that any two animals would differ only at the α6-100 locus.
In addition to testing behavioral responses to EtOH in the outbred Sprague-Dawley rats, (Hanchar et al., 2005) compared the effect of EtOH on CGN tonic GABAergic currents in slices from rats homozygous for α6-100R versus α6-100Q. These investigators reported that tonic GABAergic currents are enhanced by 30 and 100 mM EtOH to a greater extent in rats with the 100Q genotype. A similar result was found for the effect of these concentrations of EtOH on sIPSC frequency. In the presence of TTX, exogenous GABA, and a GABA transporter inhibitor, these investigators observed enhancement of tonic GABAergic currents in the presence of 10 mM EtOH and this effect was greater in slices from rats with the 100Q genotype. Based on these data, the authors concluded that the α6-R100Q mutation increases both the presynaptic actions of EtOH on Golgi cell firing as well as the modulation of extrasynaptic GABAA-Rs in CGNs. In light of the evidence discussed in Section 3, we attempted to replicate the findings of (Hanchar et al., 2005) using very similar experimental conditions to those used in their study (Botta et al., manuscript in preparation). We genotyped outbred Sprague-Dawley rats from Charles River (Hollister, CA; area H-41) and assessed the effect of EtOH on CGN tonic currents in parasagittal slices of the cerebellar vermis. We found that the effect of 25 mM EtOH on tonic current amplitude and sIPSC frequency was not significantly different in slices from α6-100R versus α6-100Q rats (Botta et al., manuscript in preparation). Importantly, in slices from either α6-100R or α6-100Q rats, we failed to observe effects of 8 or 40 mM EtOH in the presence of TTX, exogenous GABA, and a GABA transporter inhibitor (Botta et al., manuscript in preparation). These findings are in agreement with our previously published data indicating that EtOH indirectly increases the tonic current via a presynaptic mechanism (Carta et al., 2004).
A possible explanation for the lack of consensus between our electrophysiological results and those of (Hanchar et al., 2005) is that there were methodological differences between our studies. One potentially significant difference was the choice of anesthetic. We used ketamine whereas (Hanchar et al., 2005) used the inhalational anesthetic halothane. Ketamine has been shown to enhance the function of GABAA-Rs, and in particular that of CGN extrasynaptic GABAA-Rs containing α6 and δ subunits (Hevers et al., 2006; Lin et al., 1992). Another important difference is that (Hanchar et al., 2005) used slice cutting and storage solutions containing the NMDA receptor antagonist, d,l-APV, whereas we used cutting solution containing ketamine. Although we store our slices in ketamine-free ACSF and pefuse them with ACSF for at least 15 min prior to recording, it is possible that some anesthetic persists in the slices leading to occlusion of EtOH modulation of extrasynaptic GABAA receptors. We therefore compared the effect of EtOH on tonic GABAergic currents in slices prepared from 27-31 day-old male Sprague-Dawley rats using two different anesthetics and cutting/storage solutions. In protocol #1, rats were anesthetized with the inhalation anesthetic isoflurane (halothane is no longer available in our animal research facility) and brains were sliced in solution containing (in mM) 220 sucrose, 26 NaHCO3, 10 glucose, 6 MgSO4, 3 KCl, 1.25 NaH2PO4, 0.2 CaCl2, and 0.001 d,l-APV. Immediately after the slicing procedure, slices were transferred to artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2, 0.001 d,l-APV and 10 glucose equilibrated with 95% O2 plus 5% CO2, and allowed to recover at 36 °C for 45 min followed by storage at room temperature in the same solution. In protocol #2, rats were anesthetized with ketamine (250 mg/kg I.P.). Slices were cut in solution containing (in mM) 220 sucrose, 26 NaHCO3, 10 glucose, 6 MgSO4, 3 KCl, 1.25 NaH2PO4, 0.2 CaCl2 and 0.43 ketamine (Carta et al., 2004; Shuttleworth and Connor, 2001). Immediately after the slicing procedure, slices were transferred to ACSF without d,l-APV and allowed to recover at 36 °C for 45 min, followed by storage at room temperature in the same ACSF. The tonic current was calculated by fitting all-point histograms to a Gaussian distribution (Hanchar et al., 2005) and was defined as the mean steady-state current recorded in the absence minus that recorded in the presence of 10 μM of the GABAA-R antagonist SR95331. The tonic current noise was defined as the standard deviation of the steady-state current recorded in the absence minus that recorded in the presence of 10 μM SR95331. The effect of EtOH was calculated with respect to the average of control and washout responses. To match the conditions of (Hanchar et al., 2005), whole-cell patch-clamp electrophysiological recordings from CGNs were obtained in ACSF at 23 °C with an internal solution containing (in mM): 140 CsCl, 10 HEPES (pH 7.3), 1 EGTA, 4 magnesium ATP, 0.4 GTP, and 4 QX-314 at a holding membrane potential of -70 mV. As shown in Fig 3A-F, 40 mM EtOH increased the tonic current and the tonic current noise to a similar extent in CGNs from slices prepared using protocols #1 and #2. The magnitude of the tonic current was not significantly different between these slices (Fig 3G).
Figure 3.
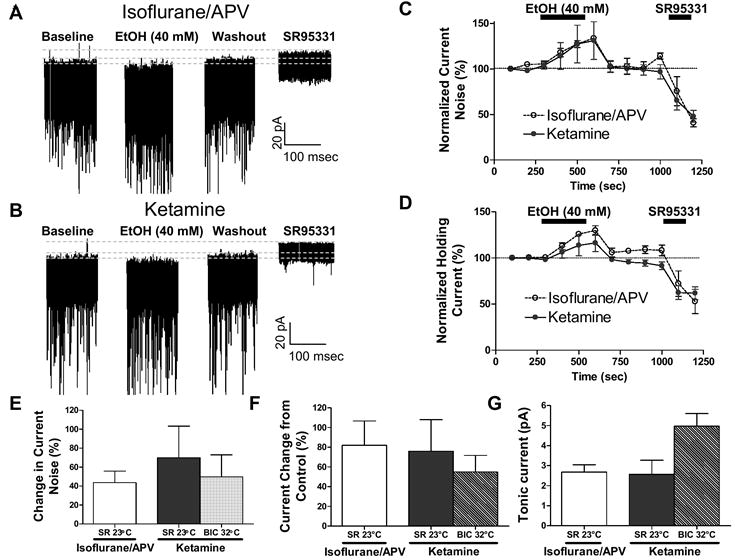
The slice preparation protocol does not influence EtOH modulation of tonic currents in CGNs. (A) Sample trace illustrating the effect of EtOH on tonic currents recorded at 23 °C from slices prepared using isoflurane and d,l-APV (protocol #1-see text). (B) Sample trace illustrating the effect of EtOH on tonic currents recorded at 23 °C from slices prepared using ketamine (protocol #2-see text). (C) Time course of the effect of 40 mM EtOH on tonic current noise recorded at 23 °C from slices prepared as described in A-B (n = 4); data were normalized with respect to the values obtained during the first 100 s of recording. Bin size = 100 s. (D) Same as in C but for holding current amplitude. (E) Summary of the percent change in tonic current noise induced by 40 mM EtOH in slices obtained using isoflurane/ d,l-APV or ketamine; recordings were obtained at 23 °C and SR954331 (SR; 10 μM) was used to block GABAA-Rs at the end of the experiment (n = 4). For comparison, four recordings from the study reported in (Carta et al., 2004) were reanalyzed as described in the text. Slices for those studies were prepared using ketamine (protocol #1); recordings were obtained at 32 °C using 50 mM EtOH and bicuculine (BIC; 20 μM) was used to block GABAA-Rs at the end of the experiments. (F) Same as in E but for EtOH-induced percent change in tonic current. (G) Same as in E but for the basal magnitude of tonic current (steady state current recorded in the absence minus that recorded in the presence of the indicated GABAA-R antagonist); p< 0.05 by unpaired t-test. See text for explanation of all-point histogram analysis used to calculate tonic current amplitude and tonic current noise. Bars represent the mean ± S.E.M.
For comparison, we re-analyzed the data published in (Carta et al., 2004) using all-point histograms. These data were collected using different experimental conditions to those used above. First, the CsCl-based internal solution had a different composition; it contained in (mM) 140 CsCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 HEPES (pH 7.3), 2 Na2-ATP and 4 QX-314. Second, recordings were obtained at 32 °C instead of 23 °C. Finally, 20 μM bicuculline was used instead of 10 μM SR95331 to block GABAA-Rs. Under these conditions, the percent change in tonic current and tonic current noise produced by 50 mM EtOH was similar from that observed in recordings obtained with 40 mM EtOH at 23 °C using slices prepared according to protocols #1 or #2 (Figs 3E-F). However, the magnitude of the tonic current in the experiments of (Carta et al., 2004) was greater (Fig 3G).
Neither basal frequency nor amplitude of the sIPSCs were significantly different in slices prepared using protocols #1 and #2 (Fig 4A-D). Moreover, EtOH equally increased sIPSC frequency (without affecting amplitude) in these slices (Fig 4A, E). The EtOH-induced percent change in sIPSC frequency shown in Fig 4E is similar to that we previously obtained with 50 mM EtOH (Carta et al., 2004).
Figure 4.
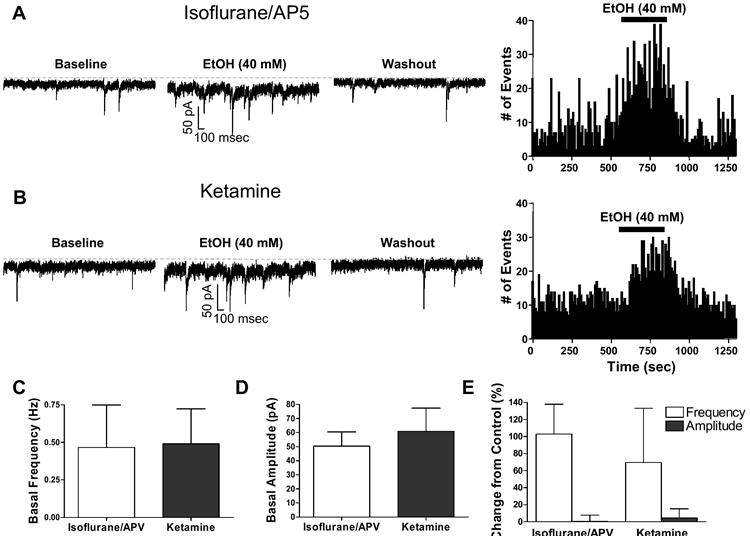
The slice preparation protocol does not influence EtOH modulation of sIPSC frequency in CGNs. (A) Left panels: sample traces illustrating the effect of EtOH on sIPSCs recorded at 23 °C from slices prepared using isoflurane and d,l-APV (protocol #1-see text). Right panel: time course of the effect of EtOH corresponding to the same neuron. (B) Same as in A but for slices prepared using ketamine (protocol #2-see text). (C) Summary of the basal sIPSC frequency recorded from neurons in slices obtained using isoflurane/ d,l-APV (n = 4) or ketamine (n = 3). (D) Same as in C but for basal sIPSC amplitude. (E) Summary of the effect of 40 mM EtOH on sIPSC frequency and amplitude in slices prepared using isoflurane/ d,l-APV (n = 4) or ketamine (n = 3). Recordings were obtained at 23 °C in all cases. Bars represent the mean ± S.E.M.
Based on these findings, we conclude that the choice of anesthetic, neuroprotective agent, recording temperature, internal solutions or GABAA-R antagonist have little influence on the effect of EtOH on sIPSCs and tonic current in CGNs.
Conclusions and Future Directions
EtOH is a difficult drug to study experimentally in part because it has multiple targets and its actions are dependent on a myriad of factors, including intracellular signaling pathways (Lovinger and Crabbe, 2005; Ron and Jurd, 2005). Moreover, EtOH is a solvent that can cause contaminants to be released from tubing, glassware, and other items used in laboratory experiments (Machu et al., 1998; Tully et al., 2000). Therefore, interpretations of EtOH research data are inherently complex and present a great potential for spurious associations. This issue has been particularly evident in investigations of the interaction between EtOH and GABAergic transmission (reviewed in (Criswell and Breese, 2005; Weiner and Valenzuela, 2006). Accordingly, experimental evidence in this area of research is generally received with skepticism, until important findings are replicated by multiple independent investigators using different experimental approaches and analytical methods. In the case of GABAergic transmission, inter-laboratory replication has contributed to the development of a strong case supporting a major presynaptic role in the mechanism of action of EtOH (Breese et al., 2006; Siggins et al., 2005; Weiner and Valenzuela, 2006). This body of evidence includes the demonstration by two independent laboratories that EtOH increases action potential firing of Golgi cells leading to an increase in phasic GABA release onto CGNs (Carta et al., 2004; Hanchar et al., 2005; Santhakumar et al., 2006b; Valenzuela et al., 2005). We have concluded that this effect indirectly increases the tonic current mediated by α6βxδ receptors via enhanced GABA spillover onto extrasynaptic sites (see schematic in Fig 1) (Carta et al., 2004). Interestingly, it has been shown that these presynaptic actions of EtOH on Golgi cell firing are enhanced by the α6-R100Q mutation via a complex circuit-type effect involving changes in CGN excitability (Hanchar et al., 2005; Santhakumar et al., 2006b). However, we have not been able to replicate these findings either in outbred Sprague-Dawley rats (Botta et al., Manuscript in preparation) or AT/ANT rats (Valenzuela et al., 2005). Importantly, the results of our genetic and behavioral studies with AT/ANT rats indicate that the α6-R100Q mutation does not modify the motor-impairing effects of acute EtOH exposure (see Section 3 above).
In contrast to the presynaptic actions of EtOH on GABAergic transmission at Golgi cell- to-CGN synapses, there is a significantly greater degree of uncertainty regarding the possibility that EtOH directly modulates extrasynaptic GABAA-Rs containing α6βxδ subunits (see Sections 3-4). An important issue that has been ignored is that low EtOH concentrations preferentially enhance the function of receptors containing α6β3δ subunits and it is unknown whether such receptors actually exist at extrasynaptic compartments in CGNs (Wisden et al., 1996). Indeed, extrasynaptic GABAA-Rs in CGNs are unlikely to be a homogeneous population, with experimental evidence supporting the possibility that these contain more than one α and/or β subunit subtype (i.e. α1, α6 , β1, β2 or β3) (Jechlinger et al., 1998; Poltl et al., 2003; Sigel and Baur, 2000). Thus, the findings obtained with α6β3δ receptors might not be physiologically relevant, particularly given that the β2 subunit has been demonstrated to associate with δ subunit-containing cerebellar receptors and that it substantially decreases EtOH potency when co-expressed with α6δ subunits (Hanchar et al., 2005; Jechlinger et al., 1998; Poltl et al., 2003; Wallner et al., 2003).
In summary, data independently obtained in two different laboratories indicate that acute EtOH affects GABAergic transmission at CGNs via a presynaptic mechanism. However, there is no consensus as to whether EtOH directly affects CGN extrasynaptic GABAA-Rs, and the reasons behind this discrepancy are unclear at the present time. To address these reasons, a number of studies could be performed. First, given that intracellular signaling cascades are well known to modulate GABAA-R sensitivity to EtOH (Weiner and Valenzuela, 2006), future work should use less invasive experimental approaches (i.e. perforated-patch) to study sensitivity of native CGN extrasynaptic GABAA- Rs to EtOH. Second, prior to pursuing additional studies with recombinant receptors, a more detailed characterization of the subunit composition of native CGN extrasynaptic receptors must be obtained. Finally, it should be independently verified that the α6-R100Q mutation indeed increases behavioral sensitivity to EtOH in outbred rats. Clearly, only by the concerted efforts of several laboratories, will the controversies surrounding the interaction between CGN extrasynaptic GABAA-Rs and EtOH be resolved.
Acknowledgments
Supported by NIH grants R01 AA14973 (to CFV), R01 AA13177 (to RAR), and R01 AA12650 (to RAD). We are grateful to Don Partridge for critically reading the manuscript.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- AA
Alko Alcohol
- ANA
Alko Non-alcohol
- AT
Alcohol Tolerant
- ANT
Alcohol Non-Tolerant
- CGN
cerebellar granule neuron
- dl-AP5
dl-2-amino-5-phosphonovaleric acid
- EtOH
ethanol
- GABA
γ-aminobutyric acid
- type-A GABA receptor
GABAA-R
- IPSC
inhibitory postsynaptic current
- sIPSC
spontaneous IPSC
- LOD
likelihood of the odds ratio
- LORR
loss of righting reflex
- QTL
quantitative trait locus
- sP
Sardinian Preferring
- sNP
Sardinian Non-Preferring
- SNP
single nucleotide polymorphism
- SSLP
simple sequence-length polymorphism
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abiola O, Angel JM, Avner P, Bachmanov AA, Belknap JK, Bennett B, Blankenhorn EP, Blizard DA, Bolivar V, Brockmann GA, et al. The nature and identification of quantitative trait loci: a community’s view. Nat Rev Genet. 2003;4:911–916. doi: 10.1038/nrg1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan AM, Harris RA. Gamma-aminobutyric acid and alcohol actions: neurochemical studies of long sleep and short sleep mice. Life Sci. 1986;39:2005–2015. doi: 10.1016/0024-3205(86)90324-3. [DOI] [PubMed] [Google Scholar]
- Attwell D, Barbour B, Szatkowski M. Nonvesicular release of neurotransmitter. Neuron. 1993;11:401–407. doi: 10.1016/0896-6273(93)90145-h. [DOI] [PubMed] [Google Scholar]
- Basile A, Hoffer B, Dunwiddie T. Differential sensitivity of cerebellar purkinje neurons to ethanol in selectively outbred lines of mice: maintenance in vitro independent of synaptic transmission. Brain Res. 1983;264:69–78. doi: 10.1016/0006-8993(83)91121-6. [DOI] [PubMed] [Google Scholar]
- Breese GR, Criswell HE, Carta M, Dodson PD, Hanchar HJ, Khisti RT, Mameli M, Ming Z, Morrow AL, Olsen RW, et al. Basis of the gabamimetic profile of ethanol. Alcohol Clin Exp Res. 2006;30:731–744. doi: 10.1111/j.0145-6008.2006.00086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;497(Pt 3):753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Carr LG, Spence JP, Peter Eriksson CJ, Lumeng L, Li TK. AA and ANA rats exhibit the R100Q mutation in the GABAA receptor alpha 6 subunit. Alcohol. 2003;31:93–97. doi: 10.1016/j.alcohol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF. Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J Neurosci. 2004;24:3746–3751. doi: 10.1523/JNEUROSCI.0067-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casagrande S, Cupello A, Pellistri F, Robello M. Only high concentrations of ethanol affect GABA(A) receptors of rat cerebellum granule cells in culture. Neurosci Lett. 2007;414:273–276. doi: 10.1016/j.neulet.2006.12.024. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo G. ESBRA-Nordmann 1996 Award Lecture: ethanol drinking behaviour in Sardinian alcohol-preferring rats. Alcohol Alcohol. 1997;32:443–453. doi: 10.1093/oxfordjournals.alcalc.a008279. [DOI] [PubMed] [Google Scholar]
- Congeddu E, Saba L, Porcella A, Sanna A, Marchese G, Lobina C, Gessa GL, Pani L. Molecular characterization of new polymorphisms at the beta2, alpha1, gamma2 GABA(A) receptor subunit genes associated to a rat nonpreferring ethanol phenotype. Brain Res Mol Brain Res. 2003;110:289–297. doi: 10.1016/s0169-328x(02)00660-5. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Kosobud A, Belknap JK. Estimation of genetic correlation: interpretation of experiments using selectively bred and inbred animals. Alcohol Clin Exp Res. 1990;14:141–151. doi: 10.1111/j.1530-0277.1990.tb00461.x. [DOI] [PubMed] [Google Scholar]
- Criswell HE, Breese GR. A conceptualization of integrated actions of ethanol contributing to its GABAmimetic profile: a commentary. Neuropsychopharmacology. 2005;30:1407–1425. doi: 10.1038/sj.npp.1300750. [DOI] [PubMed] [Google Scholar]
- Darvasi A, Soller M. Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor Appl Genet. 1992;85:353–359. doi: 10.1007/BF00222881. [DOI] [PubMed] [Google Scholar]
- Deitrich RA, Dunwiddie TV, Harris RA, Erwin VG. Mechanism of action of ethanol: initial central nervous system actions. Pharmacol Rev. 1989;41:489–537. [PubMed] [Google Scholar]
- Engblom AC, Holopainen I, Akerman KE. Ethanol-induced Cl- flux in rat cerebellar granule cells as measured by a fluorescent probe. Brain Res. 1991;568:55–60. doi: 10.1016/0006-8993(91)91378-e. [DOI] [PubMed] [Google Scholar]
- Eriksson K, Rusi M. Finnish selection studies on alcohol-related behaviors: General outline. Rockville, MD: NIH; 1981. [Google Scholar]
- Freund RK, Palmer MR. Beta adrenergic sensitization of gamma-aminobutyric acid receptors to ethanol involves a cyclic AMP/protein kinase A second-messenger mechanism. J Pharmacol Exp Ther. 1997;280:1192–1200. [PubMed] [Google Scholar]
- Freund RK, van Horne CG, Harlan T, Palmer MR. Electrophysiological interactions of ethanol with GABAergic mechanisms in the rat cerebellum in vivo. Alcohol Clin Exp Res. 1993a;17:321–328. doi: 10.1111/j.1530-0277.1993.tb00770.x. [DOI] [PubMed] [Google Scholar]
- Freund RK, Wang Y, Palmer MR. Differential effects of ethanol on the firing rates of Golgi-like neurons and Purkinje neurons in cerebellar slices in vitro. Neurosci Lett. 1993b;164:9–12. doi: 10.1016/0304-3940(93)90844-b. [DOI] [PubMed] [Google Scholar]
- George F, Chu NS. Effects of ethanol on Purkinje cells recorded from cerebellar slices. Alcohol. 1984;1:353–358. doi: 10.1016/0741-8329(84)90002-8. [DOI] [PubMed] [Google Scholar]
- Haley CS, Knott SA. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity. 1992;69:315–324. doi: 10.1038/hdy.1992.131. [DOI] [PubMed] [Google Scholar]
- Hamann M, Rossi DJ, Attwell D. Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron. 2002;33:625–633. doi: 10.1016/s0896-6273(02)00593-7. [DOI] [PubMed] [Google Scholar]
- Hanchar HJ, Dodson PD, Olsen RW, Otis TS, Wallner M. Alcohol-induced motor impairment caused by increased extrasynaptic GABA(A) receptor activity. Nat Neurosci. 2005;8:339–345. doi: 10.1038/nn1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, McQuilkin SJ, Paylor R, Abeliovich A, Tonegawa S, Wehner JM. Mutant mice lacking the gamma isoform of protein kinase C show decreased behavioral actions of ethanol and altered function of gamma-aminobutyrate type A receptors. Proc Natl Acad Sci U S A. 1995;92:3658–3662. doi: 10.1073/pnas.92.9.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellevuo K, Kiianmaa K, Korpi ER. Effect of GABAergic drugs on motor impairment from ethanol, barbital and lorazepam in rat lines selected for differential sensitivity to ethanol. Pharmacol Biochem Behav. 1989;34:399–404. doi: 10.1016/0091-3057(89)90333-x. [DOI] [PubMed] [Google Scholar]
- Hevers W, Hadley SH, Amin J. Neuroscience Meeting Planner. Atlanta, GA: Society for Neuroscience; 2006. Ketamine selectively potentiates cerebellar GABA-A receptors containing α6 and δ subunits. Online, Program # 405.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilakivi L, Eriksson CJ, Sarviharju M, Sinclair JD. Revitalization of the AA and ANA rat lines: effects on some line characteristics. Alcohol. 1984;1:71–75. doi: 10.1016/0741-8329(84)90040-5. [DOI] [PubMed] [Google Scholar]
- Huang C, Huang R. Intoxication and acute tolerance to ethanol: cerebellar granule neurons. In: Watson RR, editor. Alcohol and neurobiology: brain development and hormone regulation. London: CRC Press; 1992. pp. 45–68. [Google Scholar]
- Huang C, Liu G, Hsiao CF, Huang R. Electrophysiology of ethanol, nitrous oxide, and barbiturate on presumed subtypes of cerebellar granule cells. Ann N Y Acad Sci. 1991;625:264–268. doi: 10.1111/j.1749-6632.1991.tb33845.x. [DOI] [PubMed] [Google Scholar]
- Huang CM, Huang RH. Ethanol inhibits the sensory responses of cerebellar granule cells in anesthetized cats. Alcohol Clin Exp Res. 2007;31:336–344. doi: 10.1111/j.1530-0277.2006.00309.x. [DOI] [PubMed] [Google Scholar]
- Jechlinger M, Pelz R, Tretter V, Klausberger T, Sieghart W. Subunit composition and quantitative importance of hetero-oligomeric receptors: GABAA receptors containing alpha6 subunits. J Neurosci. 1998;18:2449–2457. doi: 10.1523/JNEUROSCI.18-07-02449.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Farrant M, Cull-Candy SG. Whole-cell and single-channel currents activated by GABA and glycine in granule cells of the rat cerebellum. J Physiol. 1995;485(Pt 2):419–435. doi: 10.1113/jphysiol.1995.sp020739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpi ER, Kleingoor C, Kettenmann H, Seeburg PH. Benzodiazepine-induced motor impairment linked to point mutation in cerebellar GABAA receptor. Nature. 1993;361:356–359. doi: 10.1038/361356a0. [DOI] [PubMed] [Google Scholar]
- Korpi ER, Seeburg PH. Natural mutation of GABAA receptor alpha 6 subunit alters benzodiazepine affinity but not allosteric GABA effects. Eur J Pharmacol. 1993;247:23–27. doi: 10.1016/0922-4106(93)90133-t. [DOI] [PubMed] [Google Scholar]
- Korpi ER, Uusi-Oukari M. Cerebellar GABAA receptors and alcohol-related behaviors: focus on diazepam-insensitive [3H]Ro 15-4513 binding. Adv Biochem Psychopharmacol. 1992;47:289–299. [PubMed] [Google Scholar]
- Lee RS, Smith SS, Chapin JK, Shimizu N, Waterhouse BD, Maddus BN, Woodward DJ. Effects of systemic and local ethanol on responses of rat cerebellar Purkinje neurons to iontophoretically applied norepinephrine and gamma-aminobutyric acid. Brain Res. 1995a;687:12–21. doi: 10.1016/0006-8993(95)00286-y. [DOI] [PubMed] [Google Scholar]
- Lee RS, Smith SS, Chapin JK, Waterhouse BD, Shimizu N, Maddux BN, Woodward DJ. Effects of systemic and local ethanol on responses of rat cerebellar Purkinje neurons to iontophoretically applied gamma-aminobutyric acid. Brain Res. 1995b;687:1–11. doi: 10.1016/0006-8993(95)00285-x. [DOI] [PubMed] [Google Scholar]
- Lin AM, Freund RK, Hoffer BJ, Palmer MR. Ethanol-induced depressions of cerebellar Purkinje neurons are potentiated by beta-adrenergic mechanisms in rat brain. J Pharmacol Exp Ther. 1994;271:1175–1180. [PubMed] [Google Scholar]
- Lin AM, Freund RK, Palmer MR. Ethanol potentiation of GABA-induced electrophysiological responses in cerebellum: requirement for catecholamine modulation. Neurosci Lett. 1991;122:154–158. doi: 10.1016/0304-3940(91)90846-l. [DOI] [PubMed] [Google Scholar]
- Lin AM, Freund RK, Palmer MR. Sensitization of gamma-aminobutyric acid-induced depressions of cerebellar Purkinje neurons to the potentiative effects of ethanol by beta adrenergic mechanisms in rat brain. J Pharmacol Exp Ther. 1993;265:426–432. [PubMed] [Google Scholar]
- Lin LH, Chen LL, Zirrolli JA, Harris RA. General anesthetics potentiate gamma-aminobutyric acid actions on gamma-aminobutyric acidA receptors expressed by Xenopus oocytes: lack of involvement of intracellular calcium. J Pharmacol Exp Ther. 1992;263:569–578. [PubMed] [Google Scholar]
- Lovinger DM, Crabbe JC. Laboratory models of alcoholism: treatment target identification and insight into mechanisms. Nat Neurosci. 2005;8:1471–1480. doi: 10.1038/nn1581. [DOI] [PubMed] [Google Scholar]
- Machu TK, Mihic SJ, Dildy-Mayfield JE. Selective actions of a detergent on ligand-gated ion channels expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1998;284:32–36. [PubMed] [Google Scholar]
- Mackay TF. The genetic architecture of quantitative traits. Annu Rev Genet. 2001;35:303–339. doi: 10.1146/annurev.genet.35.102401.090633. [DOI] [PubMed] [Google Scholar]
- Makela R, Wong G, Luddens H, Korpi ER. Phenotypic and genotypic analysis of rats with cerebellar GABAA receptors composed from mutant and wild-type alpha 6 subunits. J Neurochem. 1995;65:2401–2408. doi: 10.1046/j.1471-4159.1995.65062401.x. [DOI] [PubMed] [Google Scholar]
- Ming Z, Criswell HE, Yu G, Breese GR. Competing presynaptic and postsynaptic effects of ethanol on cerebellar purkinje neurons. Alcohol Clin Exp Res. 2006;30:1400–1407. doi: 10.1111/j.1530-0277.2006.00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Hanchar HJ, Dodson PD, Olsen RW, Wallner M. Response to Letter to the Editor. Alcohol Clin Exp Res. 2005;29:1358. [Google Scholar]
- Palmer MR, Hoffer BJ. GABAergic mechanisms in the electrophysiological actions of ethanol on cerebellar neurons. Neurochem Res. 1990;15:145–151. doi: 10.1007/BF00972204. [DOI] [PubMed] [Google Scholar]
- Palmer MR, van Horne CG, Harlan JT, Moore EA. Antagonism of ethanol effects on cerebellar Purkinje neurons by the benzodiazepine inverse agonists Ro 15-4513 and FG 7142: electrophysiological studies. J Pharmacol Exp Ther. 1988;247:1018–1024. [PubMed] [Google Scholar]
- Poltl A, Hauer B, Fuchs K, Tretter V, Sieghart W. Subunit composition and quantitative importance of GABAA receptor subtypes in the cerebellum of mouse and rat. J Neurochem. 2003;87:1444–1455. doi: 10.1046/j.1471-4159.2003.02135.x. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Allan AM, Dunwiddie TV. Brain region-dependent sensitivity of GABAA receptor-mediated responses to modulation by ethanol. Alcohol Clin Exp Res. 1992a;16:480–489. doi: 10.1111/j.1530-0277.1992.tb01405.x. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Soldo BL, Allan AM, Dunwiddie TV. Ethanol enhances synaptically evoked GABAA receptor-mediated responses in cerebral cortical neurons in rat brain slices. Brain Res. 1992b;595:220–227. doi: 10.1016/0006-8993(92)91053-h. [DOI] [PubMed] [Google Scholar]
- Radcliffe RA, Hoffmann SE, Deng XS, Asperi W, Fay T, Bludeau P, Erwin VG, Deitrich RA. Behavioral characterization of alcohol-tolerant and alcohol-nontolerant rat lines and an f(2) generation. Behav Genet. 2004;34:453–463. doi: 10.1023/B:BEGE.0000023650.32243.39. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Prasad A, MacDonald JF. Ethanol modulation of GABA receptor-activated Cl- currents in neurons of the chick, rat and mouse central nervous system. Eur J Pharmacol. 1992;224:173–181. doi: 10.1016/0014-2999(92)90802-b. [DOI] [PubMed] [Google Scholar]
- Rogers J, Madamba SG, Staunton DA, Siggins GR. Ethanol increases single unit activity in the inferior olivary nucleus. Brain Res. 1986;385:253–262. doi: 10.1016/0006-8993(86)91071-1. [DOI] [PubMed] [Google Scholar]
- Ron D, Jurd R. The “ups and downs” of signaling cascades in addiction. Sci STKE. 2005:re14. doi: 10.1126/stke.3092005re14. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Hamann M, Attwell D. Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J Physiol. 2003;548:97–110. doi: 10.1113/jphysiol.2002.036459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba L, Porcella A, Congeddu E, Colombo G, Peis M, Pistis M, Gessa GL, Pani L. The R100Q mutation of the GABA(A) alpha(6) receptor subunit may contribute to voluntary aversion to ethanol in the sNP rat line. Brain Res Mol Brain Res. 2001;87:263–270. doi: 10.1016/s0169-328x(01)00003-1. [DOI] [PubMed] [Google Scholar]
- Saba L, Porcella A, Sanna A, Congeddu E, Marziliano N, Mongeau R, Grayson D, Pani L. Five mutations in the GABA A alpha6 gene 5’ flanking region are associated with a reduced basal and ethanol-induced alpha6 upregulation in mutated Sardinian alcohol non-preferring rats. Brain Res Mol Brain Res. 2005;137:252–257. doi: 10.1016/j.molbrainres.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Sanna A, Congeddu E, Saba L, Porcella A, Marchese G, Ruiu S, Casti P, Saba P, Pani L. The cerebellar GABAA alpha6 subunit is differentially modulated by chronic ethanol exposure in normal (R100R) and mutated (Q100Q) sNP rats. Brain Res. 2004;998:148–154. doi: 10.1016/j.brainres.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Hanchar HJ, Wallner M, Olsen RW, Otis TS. Contributions of the GABAA receptor alpha6 subunit to phasic and tonic inhibition revealed by a naturally occurring polymorphism in the alpha6 gene. J Neurosci. 2006a;26:3357–3364. doi: 10.1523/JNEUROSCI.4799-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Karakossian MH, Otis TS. Neuroscience Meeting Planner. Atlanta, GA: Society for Neuroscience; 2006b. Ethanol enhancement of tonic inhibition in cerebellar granule cells alters circuit activity and underlies ethanol potentiation of Golgi cell GABA release. Online, Program #329.325. [Google Scholar]
- Sarviharju M, Korpi ER. Ethanol sensitivity and consumption in F2 hybrid crosses of ANT and AT rats. Alcohol. 1993;10:415–418. doi: 10.1016/0741-8329(93)90030-r. [DOI] [PubMed] [Google Scholar]
- Saxena NC, Macdonald RL. Assembly of GABAA receptor subunits: role of the delta subunit. J Neurosci. 1994;14:7077–7086. doi: 10.1523/JNEUROSCI.14-11-07077.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NC, Macdonald RL. Properties of putative cerebellar gamma-aminobutyric acid A receptor isoforms. Mol Pharmacol. 1996;49:567–579. [PubMed] [Google Scholar]
- Schmahmann JD, Sherman JC. Cerebellar cognitive affective syndrome. Int Rev Neurobiol. 1997;41:433–440. doi: 10.1016/s0074-7742(08)60363-3. [DOI] [PubMed] [Google Scholar]
- Schmid G, Bonanno G, Raiteri L, Sarviharju M, Korpi ER, Raiteri M. Enhanced benzodiazepine and ethanol actions on cerebellar GABA(A) receptors mediating glutamate release in an alcohol-sensitive rat line. Neuropharmacology. 1999;38:1273–1279. doi: 10.1016/s0028-3908(99)00025-8. [DOI] [PubMed] [Google Scholar]
- Sellin LC, Laakso PS. Effect of ethanol on motor performance and hippocampal population spikes in some standard and selectively outbred rat strains. Alcohol Clin Exp Res. 1987;11:502–505. doi: 10.1111/j.1530-0277.1987.tb01931.x. [DOI] [PubMed] [Google Scholar]
- Shuttleworth CW, Connor JA. Strain-dependent differences in calcium signaling predict excitotoxicity in murine hippocampal neurons. J Neurosci. 2001;21:4225–4236. doi: 10.1523/JNEUROSCI.21-12-04225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E, Baur R. Electrophysiological evidence for the coexistence of alpha1 and alpha6 subunits in a single functional GABA(A) receptor. J Neurochem. 2000;74:2590–2596. doi: 10.1046/j.1471-4159.2000.0742590.x. [DOI] [PubMed] [Google Scholar]
- Siggins GR, French E. Central neurons are depressed by iontophoretic and micropressure application of ethanol and tetrahydropapaveroline. Drug Alcohol Depend. 1979;4:239–243. doi: 10.1016/0376-8716(79)90004-8. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Roberto M, Nie Z. The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther. 2005;107:80–98. doi: 10.1016/j.pharmthera.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Sorensen S, Palmer M, Dunwiddie T, Hoffer B. Electrophysiological correlates of ethanol-induced sedation in differentially sensitive lines of mice. Science. 1980;210:1143–1145. doi: 10.1126/science.7444444. [DOI] [PubMed] [Google Scholar]
- Tia S, Wang JF, Kotchabhakdi N, Vicini S. Developmental changes of inhibitory synaptic currents in cerebellar granule neurons: role of GABA(A) receptor alpha 6 subunit. J Neurosci. 1996;16:3630–3640. doi: 10.1523/JNEUROSCI.16-11-03630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Kupfer D, Dopico AM, Treistman SN. A plasticizer released from IV drip chambers elevates calcium levels in neurosecretory terminals. Toxicol Appl Pharmacol. 2000;168:183–188. doi: 10.1006/taap.2000.9036. [DOI] [PubMed] [Google Scholar]
- Tuominen K, Hellevuo K, Korpi ER. Plus-maze behavior and susceptibility to 3-mercaptopropionate-induced seizures in rat lines selected for high and low alcohol sensitivity. Pharmacol Biochem Behav. 1990;35:721–725. doi: 10.1016/0091-3057(90)90313-7. [DOI] [PubMed] [Google Scholar]
- Uusi-Oukari M, Korpi ER. Diazepam sensitivity of the binding of an imidazobenzodiazepine, [3H]Ro 15-4513, in cerebellar membranes from two rat lines developed for high and low alcohol sensitivity. J Neurochem. 1990;54:1980–1987. doi: 10.1111/j.1471-4159.1990.tb04901.x. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Carta M, Mameli M. Letter to the editor. Alcohol Clin Exp Res. 2005;29:1356–1357. doi: 10.1097/01.alc.0000171926.66397.94. [DOI] [PubMed] [Google Scholar]
- Wall MJ, Usowicz MM. Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur J Neurosci. 1997;9:533–548. doi: 10.1111/j.1460-9568.1997.tb01630.x. [DOI] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW. Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci U S A. 2003;100:15218–15223. doi: 10.1073/pnas.2435171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: The view from the slice. Pharmacol Ther. 2006 doi: 10.1016/j.pharmthera.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Wisden W, Korpi ER, Bahn S. The cerebellum: a model system for studying GABAA receptor diversity. Neuropharmacology. 1996;35:1139–1160. doi: 10.1016/s0028-3908(96)00076-7. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Marszalec W, Yeh JZ, Narahashi T. Effects of ethanol on tonic GABA currents in cerebellar granule cells and mammalian cells recombinantly expressing GABA(A) receptors. J Pharmacol Exp Ther. 2006;319:431–438. doi: 10.1124/jpet.106.106260. [DOI] [PubMed] [Google Scholar]
- Yang X, Criswell HE, Breese GR. Action of ethanol on responses to nicotine from cerebellar Purkinje neurons: relationship to methyllycaconitine (MLA) inhibition of nicotine responses. Neurochem Int. 1999;35:185–194. doi: 10.1016/s0197-0186(99)00060-1. [DOI] [PubMed] [Google Scholar]
- Yang X, Criswell HE, Breese GR. Ethanol modulation of gamma-aminobutyric acid (GABA)-mediated inhibition of cerebellar Purkinje neurons: relationship to GABAb receptor input. Alcohol Clin Exp Res. 2000;24:682–690. [PubMed] [Google Scholar]