

Abstract
The RNA-binding protein HuR regulates the stability of many target mRNAs. Here, we report that HuR associated with the 3'-untranslated region of the mRNA encoding the longevity and stress-response protein SIRT1, stabilized the SIRT1 mRNA, and increased SIRT1 expression levels. Unexpectedly, oxidative stress triggered the dissociation of the [HuR-SIRT1 mRNA] complex, in turn promoting SIRT1 mRNA decay, reducing SIRT1 abundance, and lowering cell survival. The cell cycle checkpoint kinase Chk2 was activated by H2O2, interacted with HuR, and was predicted to phosphorylate HuR at residues Ser-88, Ser-100, and Thr-118. Mutation of these residues revealed a complex pattern of HuR binding, with Ser-100 appearing important for [HuR-SIRT1 mRNA] dissociation after H2O2. Our findings demonstrate that HuR regulates SIRT1 expression, underscore functional links between the two stress-response proteins, and implicate Chk2 in these processes.
Keywords: mRNA turnover, elav, stress response, replicative senescence, ribonucleoprotein complex, posttranscriptional
INTRODUCTION
In cells responding to damaging stimuli, gene expression changes profoundly affect the cellular outcome, directly influencing whether the cell survives or succumbs to the injury. In addition to the stress-induced alterations in gene transcription, changes in mRNA stability (increased or decreased half-lives) also potently influence the steady-state abundance of many transcripts (Garcia-Martinez et al., 2004; Cheadle et al., 2005; Moore, 2005). The posttranscriptional fate of a given mRNA is governed by the interaction of specific mRNA sequences (cis elements) with specific trans factors such as RNA-binding proteins (RBPs) (Wilusz & Wilusz, 2004) and microRNAs (Bhattacharyya et al., 2006). Ribonucleoprotein (RNP) associations have been shown to control the intracellular transport of the mRNA as well as its association with the translation and decay machineries (Keene, 2001; Moore, 2005). Many labile mRNAs have relatively long 3'-untranslated regions (UTRs) featuring characteristic U- or AU-rich stretches that are collectively known as AU-rich elements (AREs) (Chen & Shyu, 1995). Several RBPs have been identified that promote ARE-mRNA decay, including AUF1, BRF1, TTP, and KSRP; their mechanisms of action include the recruitment of the ARE-mRNA to sites of mRNA degradation such as the exosome, proteasome, or P-bodies (Carballo et al., 1998; Laroia et al., 1999; Gherzi et al., 2004; Kedersha et al., 2005). RBPs that stabilize target mRNAs include the elav/Hu proteins, which comprise a family of three primarily neuronal members (HuB, HuC, HuD) and one ubiquitous member, HuR. Although the precise mechanism underlying target mRNA stabilization by HuR, the best-studied Hu protein, remains poorly understood, this process is linked to the cytoplasmic presence of the HuR RNP (Keene, 1999; Brennan & Steitz, 2001). HuR has emerged as a key regulator of genes that are central to the stress response, cell division cycle, immune cell activation, carcinogenesis, and replicative senescence (Brennan & Steitz, 2001; López de Silanes et al., 2004a).
The stress-response and chromatin-silencing factor Sir2 (originally identified as the ‘silencing information regulator 2’ in S. cerevisiae) is an NAD+-dependent histone deacetylase involved in various nuclear events such as transcription, DNA replication, and DNA repair (Blander & Guarente, 2004). In addition, yeast Sir2 and its C. elegans and D. melanogaster homologs have been shown to extend life span (Sinclair & Guarente, 1997; Astrom et al., 2003; Rogina & Helfand, 2004). Since Sir2 activity depends on NAD+ levels, its function links the cell's energy state with the animal life span. Mammalian cells express seven Sir2 homologs, termed sirtuins SIRT1-7 (Frye, 2000), among which SIRT1 shares the highest homology with Sir2 (North & Verdin, 2004). Given its involvement in life span extension in lower eukaryotes, SIRT1 has been the focus of many recent studies. SIRT1 enhances cell survival following exposure to oxidative and genotoxic stresses and these protective effects were attributed to its influence upon several nonhistone deacetylation targets. SIRT1 deacetylates proteins of the FOXO transcription factor family, thereby repressing the transactivation of the proapoptotic factor Bim and increasing the transcription of antiapoptotic protein GADD45 (Brunet et al., 2004; Motta et al., 2004). SIRT1 also deacetylates the tumor suppressor p53, thus reducing its ability to increase the levels of the proapoptotic factor Bax (Luo et al., 2001; Vaziri et al., 2001), and deacetylates Ku70, which further inhibits Bax function by sequestering it in the cytoplasm and preventing its localization in mitochondria to initiate apoptosis (Cohen et al., 2004a).
While the antiapoptotic activity of SIRT1 relies on its altered levels following cell injury, the mechanisms whereby SIRT1 expression is regulated remain largely unknown. SIRT1 transcription is suppressed by p53 in unstimulated rodent cells, although the association of Foxo3a with p53 relieved this suppression (Nemoto et al., 2004). The tumor suppressor HIC1 associates with SIRT1 itself and forms a transcriptional repressor complex which directly binds the SIRT1 promoter (Chen et al., 2005). These reports suggest that SIRT1 expression is subject to a complex control pattern, including negative feedback autoregulation and regulation by p53 and Foxo3a, the very targets of SIRT1 deacetylase function.
An en masse search for HuR target mRNAs (López de Silanes et al., 2004b) identified the SIRT1 mRNA as a putative HuR target and computationally detected several hits of the HuR signature motif in the SIRT1 3'UTR. Here, we set out to investigate whether HuR binds the SIRT1 mRNA, to examine the functional consequences of this association, and to elucidate the underlying molecular pathways controling this process. Given our long-standing interest in studying HuR function in human models of aging and cancer, we examined the effects of HuR upon the SIRT1 mRNA in both transformed and untransformed human cells. HuR was found to associate with the SIRT1 mRNA, to enhance its stability, and to maintain elevated SIRT1 steady-state levels. In untransformed human cells (diploid fibroblasts or HDFs), we documented a concomitant reduction in both HuR and SIRT1 during replicative senescence. Unexpectedly, oxidative stress lowered SIRT1 mRNA and protein expression levels in HDFs and reduced the stability of the SIRT1 mRNA in an HuR-dependent manner. Contrary to the enhanced HuR association with numerous target mRNAs reported in response to other stress agents, HuR dissociated from the SIRT1 mRNA following oxidant treatment. The oxidant-activated, cell cycle checkpoint kinase Chk2 was found to phosphorylate HuR. This posttranslational modification critically influenced the levels of the [HuR-SIRT1 mRNA] RNP complex and underscored a key role for Chk2 as an upstream regulator of HuR binding activity.
RESULTS
SIRT1 mRNA is a direct target of HuR
The 1.8-kb SIRT1 3'UTR contains nine computationally predicted hits of an HuR motif (Lopez de Silanes et al., 2004b) (Fig. 1A), suggesting that SIRT1 mRNA might be a direct target of HuR. We first tested if SIRT1 mRNA associated with HuR by performing immunoprecipitation (IP) assays using anti-HuR antibodies under conditions that preserved RNP integrity. The association of SIRT1 mRNA with HuR was monitored by isolating RNA from the IP material and subjecing it to reverse transcription (RT) and quantitative real-time (q) PCR analysis. As shown in Fig. 1B, the SIRT1 PCR product was dramatically enriched in HuR IP samples compared with control IgG IP samples. The enrichment of a prothymosin α (ProTα) PCR product served as a positive control, since ProTα mRNA is a target of HuR (Lal et al., 2005), while the amplification of UBC and GAPDH PCR products, found in all samples as low-level contaminating housekeeping transcripts (not HuR targets), served to monitor the evenness of sample input. [HuR-SIRT1 mRNA] associations were further tested by using biotinylated transcripts spanning the mRNA regions shown (Fig. 1C, schematic). Following incubation with HeLa cell lysates, the interaction between the biotinylated transcripts and several RBPs was assessed by biotin pulldown followed by Western blot analysis. As shown, HuR formed much more prominent complexes with the SIRT1 3'UTR than with the SIRT1 coding region (CR), while RBPs TIA-1 and NF-90 bound both transcripts at comparably low levels (Fig. 1C). Biotinylated GAPDH 3'UTR, which is not a target of HuR and serves as a negative control transcript, was included to detect background RNP associations. Together, these findings indicate that HuR specifically binds the SIRT1 mRNA, both endogenous and in vitro biotinylated, and that binding occurs in the SIRT1 3'UTR.
Figure 1. HuR binds the SIRT1 mRNA.
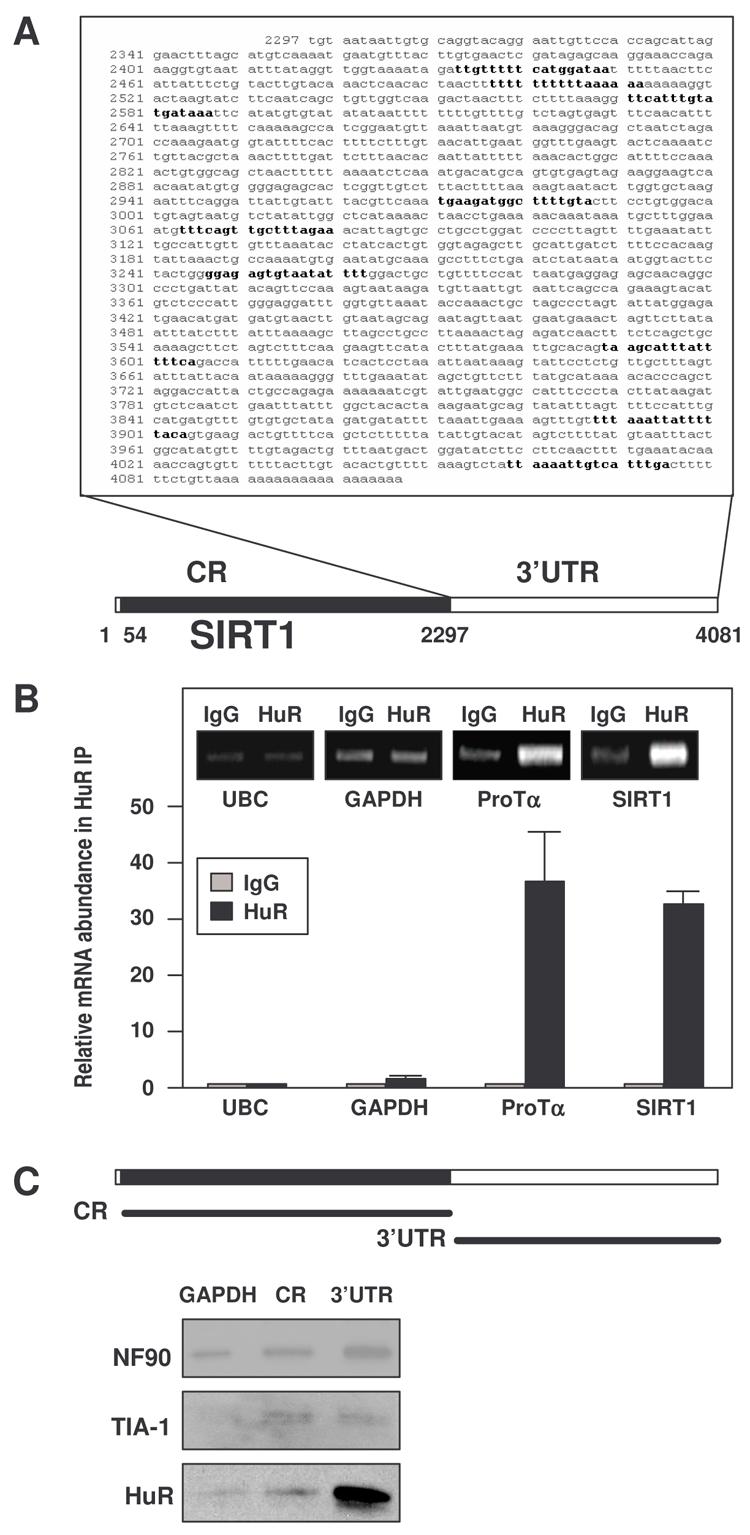
(A) SIRT1 mRNA showing HuR motif hits in the 3'UTR. (B) After IP of RNA-protein complexes from HeLa cell lysates using either anti-HuR antibodies or control IgG1, RNA was isolated and used in RT reactions. Graph, fold differences in transcript abundance in HuR IP compared with IgG IP, as measured by RT-qPCR analysis. Inset, representative qPCR products visualized in ethidium bromide-stained agarose gels; low-level amplification of UBC and GAPDH (housekeeping mRNAs which are not HuR targets) served as negative controls, while ProTα mRNA, a known HuR target, was used as a positive control. The means and standard error of the means (SEM) from 3 independent experiments are represented. (C) Schematic representation of the SIRT1 biotinylated transcripts (CR, 3′UTR) used in biotin pulldown assays; biotinylated GAPDH 3'UTR was included as a negative control. The presence of HuR, NF90, and TIA-1 in the pulldown material was assayed by Western blotting.
HuR stabilizes the SIRT1 mRNA
To assess the functional consequences of [HuR-SIRT1 mRNA] interactions, HuR levels were reduced by RNA interference (RNAi). HeLa cells transfected with small interfering (si)RNA targeting HuR showed <10% of the HuR levels seen in the control transfection group and dramatically reduced SIRT1 abundance, as detected by Western blotting (Fig. 2A). The reduction in SIRT1 was due, at least in part, to the lower SIRT1 mRNA levels in HuR-silenced cells, as measured by Northern blot and RT-qPCR analyses (Fig. 2B). To test the specificity of the decrease in SIRT1 after HuR silencing, a rescue experiment was performed by transfecting cells with HuRU1, an siRNA that silences HuR by targetting its 3'UTR, and simultaneously overexpressing a chimeric HuR protein (HuR-TAP) that lacks the 3'UTR and hence is refractory to HuRU1 siRNA (described in Lal et al., 2005). As shown, silencing of endogenous HuR reduced SIRT1, but HuR-TAP overexpression restored SIRT1 levels, supporting the specificity of HuR's influence upon SIRT1 expression (Fig. 2C). After silencing HuR, the levels of mRNAs encoding other SIRT family members which lacked any predicted HuR motif hits (SIRT2, −3, −6, −7) were unaffected; SIRT4 and −5 mRNAs, each with one putative HuR motif hit in the 3'UTR, were modestly reduced (Fig. 2D).
Figure 2. HuR silencing reduces SIRT1 expression in HeLa cells.

(A) Two days after siRNA transfection, HeLa cells were harvested for Western blot analysis to monitor the expression of HuR, SIRT1, and loading control β-Actin. (B) Cells were transfected as explained in panel (A) and harvested, and RNA was analyzed by either Northern blotting (top), or RT-qPCR (bottom, shown as the means +SEM from 3 experiments). (C) Two days after transfection of an siRNA targetting the HuR 3'UTR (HuRU1) or a control siRNA, along with either TAP- or HuR-TAP-expressing vectors, the levels of endogenous (Endog.) or ectopic (HuR-TAP) HuR, SIRT1, and loading control β-Actin were tested by Western blot analysis. (D) Left, schematic of the mRNAs encoding the Sirt protein family members (SIRT1-7); middle, number of predicted HuR motif hits in each transcript; right, levels of SIRT1-7 mRNAs as determined by RT-qPCR following HuR silencing, compared with control siRNA. (E) SIRT1 mRNA half-life after silencing HuR was measured by incubating cells with actinomycin D, extracting total RNA at the times shown, and measuring SIRT1 and (housekeeping) GAPDH mRNA levels by RT-qPCR analysis. The data were normalized to 18S rRNA levels and represented as a percentage of the mRNA levels measured at time 0, before adding actinomycin D, using a semi-logarithmic scale. The half-lives (indicated) were calculated as the time required for each mRNA decrease to 50% of its initial abundance (discontinuous horizontal line). Inset, representative qPCR reaction products. (F) Northern blot analysis of the mRNAs described in panel D; rRNAs are shown.
To ascertain if the reduction in the SIRT1 mRNA levels was due to changes in mRNA stability, the SIRT1 mRNA half-life (t½) was analyzed following treatment with actinomycin D to inhibit de novo transcription. The levels of SIRT1 mRNA, housekeeping control GAPDH mRNA, and loading control 18S rRNA were monitored by RT-qPCR and Northern blotting (Fig. 2E,F). SIRT1 mRNA was found to be significantly more stable in the control siRNA (Ctrl.) transfection group, with an estimated t½ >8 h, while SIRT1 mRNA stability was markedly reduced in the HuR siRNA group, with t½ ∼1.2 h (Fig. 2E,F). HuR has also been shown to influence the translation of various target mRNAs (Kullmann et al., 2002; Mazan-Mamczarz et al., 2003; Lal et al., 2005), but direct testing of SIRT1 translation did not reveal a significant influence by HuR (Suppl. Fig. S1). In summary, HuR specifically enhanced SIRT1 mRNA stability.
HuR regulates SIRT1 expression in WI-38 HDFs
The functional consequences of the HuR-regulated SIRT1 expression were studied in WI-38 human diploid fibroblasts (HDFs), a widely utilized cell model in which endogenous HuR levels decrease dramatically as cells progress to senescence (Wang et al., 2001). A biomarker of replicative senescence, the activity of a neutral, senescence-associated β-galactosidase [SA-β-gal (Dimri et al., 1995)] was measured in proliferating (early-passage or ‘young’, Y) and senescent (late-passage, S) WI-38 cells [at population doublings (pdls) 18-30 and ∼50, respectively]; as anticipated, SA-β-gal activity was markedly elevated in senescent cells (Fig. 3A). In addition, senescent cultures exhibited lower cyclin-dependent kinase (cdk) activity, diminished rates of 3H-thymidine incorporation, increased G1-phase cells, elevated levels of cyclin D1 and cdk inhibitors p21 and p16, and reduced levels of cyclin A, cyclin B1, and c-fos [data not shown and Wang et al., 2001]. HuR expression levels were reduced in senescent cells [as previously described (Wang et al., 2001)], and the levels p53 protein and p21 mRNA, other hallmarks of senescence, were elevated. Importantly, SIRT1 levels were markedly reduced in senescent HDFs (Fig. 3B).
Figure 3. Reduced SIRT1 levels in senescent or HuR-silenced HDFs.

(A) SA-β-galactosidase activity in proliferating (Young or Y) and Senescent (S) WI-38 HDFs, at 28 and 52 population doublings, respectively. Graph, percentages of SA-β-galactosidase-positive cells. (B) Western blot analysis to monitor the expression of HuR and SIRT1 in Y and S HDFs. p53 and β-Actin levels were tested as positive and loading controls, respectively; p21 mRNA levels were measured by RT-qPCR (graph). HDFs were transfected with the siRNAs indicated and collected for analysis 5 d later. The effect of HuR silencing on HDF protein expression was assessed by Western blotting (C) and its influence on proliferation by measuring 3H-Thymidine incorporation (D). (E) Effect of HuR silencing on the levels of SIRT1-7 mRNAs in HDFs as determined by RT-qPCR in two separate experiments (mean values shown). (F) Half-lives of SIRT1 and GAPDH mRNAs in HDFs. Total RNA was extracted, SIRT1 and GAPDH mRNA levels monitored by RT-qPCR, normalized to 18S rRNA levels, and the half-lives calculated as described in the legend of Fig. 2E. Data represent the means ±SEM from 3 independent experiments.
An siRNA-based intervention to reduce HuR expression levels in early-passage HDFs led to a dramatic decline in SIRT1 levels (Fig. 3C). The silencing of HuR had a growth inhibitory influence on HDFs, as assessed by measuring the incorporation of 3H-thymidine in each transfection group (Fig. 3D) and previously reported (Wang et al., 2001). The specificity of HuR's influence upon SIRT1 expression in HDFs was further tested by monitoring changes in the abundance of the mRNAs encoding other sirtuins. As shown in Fig. 3E, SIRT1 mRNA was specifically reduced following HuR silencing, while the levels of the other mRNAs (SIRT2-7) remained unchanged. That the reduced SIRT1 mRNA levels were associated with a reduction in mRNA stability was evidenced by actinomycin D-based mRNA half-life analysis (Fig. 3F). No specific changes in SIRT1 translation rates were observed when comparing Y and S HDFs (Suppl. Fig. S2).
Oxidative stress dissociates [HuR-SIRT1 mRNA] complexes, lowers SIRT1 expression, and is preferentially toxic to HDFs with low SIRT1 levels
We extended our studies to examine a major process implicating HuR and SIRT1 function, the cellular response to stress. Exposure of HDFs to hydrogen peroxide (H2O2) caused a significant, dose-dependent reduction in SIRT1 mRNA and protein levels, while it increased the H2O2-inducible GADD153 mRNA and had no effect on levels of HuR protein (Fig. 4A,B). Interestingly, following H2O2 treatment of HDFs, the stability of the SIRT1 mRNA was reduced (Fig. 4C), suggesting that the lower SIRT1 mRNA abundance (Fig. 4A) was due, at least in part, to an H2O2-triggered decrease in its half-life.
Figure 4. H2O2 treatment decreased [HuR-SIRT1 mRNA] complexes and SIRT1 expression.

(A) After treating HDFs with the indicated H2O2 doses, RNA was isolated and RT-qPCR performed; as a positive control, GADD153 mRNA levels were monitored. (B) Western blot analysis of SIRT1, HuR, and (loading control) α-Tubulin levels in HDF whole-cell lysates after treatment with H2O2 at the doses and times shown. (C)The half-lives of SIRT1 and GAPDH mRNAs in untreated and H2O2-treated HDFs were quantified by using RT-qPCR and calculated as described in the legend of Fig. 2E; the means ±SEM from 3 independent experiments. (D) IP with anti-HuR or IgG antibodies were performed using lysates that were prepared from either untreated or H2O2-treated HDFs (500 μM, 3 h); HuR abundance in the IP material was unchanged (not shown) and RNA was isolated for RT-qPCR analysis to detect SIRT1 mRNA, GAPDH mRNA (a housekeeping ‘background’ control), and ProTα mRNA (a positive control). Inset, representative qPCR products. (E) Percent SIRT1 mRNA (means and +SEM from 3 independent experiments) remaining in either whole-cell lysates (solid line) or HuR-bound material after RNP IP (dashed line) following H2O2 treatment (500 μM).
To determine if the reduced SIRT1 mRNA stability was linked to changes in its association with HuR, the abundance of these complexes was tested by RNP IP analysis. H2O2 treatment alone did not alter the ability to IP HuR (not shown). However, the amount of SIRT1 mRNA which is constitutively bound to HuR was substantially reduced by the H2O2 treatment, as monitored by RT-qPCR; by contrast, the association of HuR to the control target ProTα mRNA increased (Fig. 4D). The reduction in [HuR-SIRT1 mRNA] complexes was rapid, decreasing to one-half by ∼15 min after H2O2 treatment, whereas total SIRT1 mRNA levels decreased more slowly, with only a 30% decline in transcript levels by 60 min after H2O2 treatment, suggesting that the dissociation of this RNP preceded the decay of SIRT1 mRNA (Fig. 4E). These observations are consistent with a model whereby H2O2 treatment triggers the rapid dissociation of SIRT1 mRNA from HuR, in turn destabilizing the SIRT1 mRNA and reducing SIRT1 protein levels.
To investigate links between HuR levels, SIRT1 expression, and the antiapoptotic influence of these two proteins, we studied the sensitivity of WI-38 cells expressing various levels of HuR and SIRT1. Senescent cells, which expressed low HuR and SIRT1, were more sensitive than young cells with high levels of both HuR and SIRT1 to a range of H2O2 doses. Likewise, exposure to 500 μM H2O2 was progressively more toxic as cells increased in pdl (Fig. 5A) and expressed less HuR and SIRT1 (not shown). Silencing SIRT1 expression by RNAi in young WI-38 cells, did not influence HuR levels (Fig. 5B) but did enhance cell proliferation, as reported (Chua et al., 2005, Suppl. Fig. S3). Overexpression of HuR (Fig. 5C) rescued the H2O2-elicited toxicity, while concomitant silencing of SIRT1 suppressed this protective effect (Fig. 5D). Conversely, SIRT1 overexpression using a vector that contained only the SIRT1 CR [not its 3'UTR (Cohen et al., 2004b), Fig. 5E] conferred protection against H2O2 treatment, while concurrent HuR silencing significantly reduced this survival (Fig. 5F). Taken together, our findings suggest that HuR enhances SIRT1 expression in unstressed cells, that each protein independently promotes survival in response to H2O2, and that the H2O2-elicited toxicity is linked to the dissociation of [HuR-SIRT1 mRNA] complexes and the ensuing reduction in SIRT1 mRNA stability and SIRT1 protein levels.
Figure 5. Protective influence of HuR and SIRT1 in H2O2-treated WI-38 cells.

(A) Y and S cells were treated with the indicated H2O2 doses (left) and cells at the indicated pdls were treated with H2O2 (500 μM, 1 h, right) and survival was monitored 16 h later; data were obtained from two independent experiments. (B) Western blot analysis to monitor protein expression levels by 48 h after siRNA transfection. (C) Cells were transfected with the indicated siRNAs and plasmids [pZeo-HuR (pHuR) or vector control pZeo (V)]; 48 h later, cells were treated with H2O2 (500 μM, 1 h) and collected 16 h later for Western blot analysis. (D) Cell survival in cultures that were transfected and treated as described in panel C. (E) Western blot analysis of cells that were transfected and treated as described in panel C, except that the indicated siRNAs and plasmids [SIRT1 expression vector (pSIRT1) or vector control (V)] were used. (F) Cell survival in cultures that were transfected and treated as described in panel E. Transfection efficiencies were >90%; cell survival (A, D, F) was measured using the MTT assay.
Chk2 phosphorylates HuR
An important lead towards elucidating how H2O2 triggered the dissociation of [HuR-SIRT1 mRNA] complexes came from a yeast two-hybrid screen of a HeLa cell cDNA library that identified HuR as a protein interacting with the checkpoint kinase 2 (Chk2, Suppl. material). We confirmed this interaction in HDFs (and HeLa cells, not shown) by co-IP of HuR followed by Western blotting (Fig. 6A); Chk2 bound HuR in nuclear extracts of control cells, but this interaction was undetectable after silencing either protein (Fig. 6A,B). Chk2 silencing did not affect the total levels or subcellular localization of HuR (Fig. 6B; Suppl. Fig. S4). In an in vitro kinase assay (Fig. 6C), active Chk2 kinase readily phosphorylated purified GST-HuR, as assessed by monitoring [γ-32P]ATP incorporation into the chimeric protein; in parallel reactions, GST alone was not phosphorylated (data not shown). As reported in other cell systems (Leroy et al., 2001; Buscemi et al., 2004), H2O2 treatment triggered the phosphorylation of Chk2 (an activating modification) in WI-38 cells (Fig. 6D). Importantly, inhibition of Chk2 function or Chk2 expression effectively rescued the diminished SIRT1 mRNA (Fig. 6E, top) and protein (Fig. 6F) levels that followed H2O2 treatment of WI-38 cells, supporting the view that Chk2 contributed to lowering SIRT1 expression after H2O2 treatment. The involvement of Chk2 in these effects was further strengthened by the finding that Chk2-deficient cells expressed higher SIRT1 levels before and after H2O2 treatment (Suppl. Fig. S5) and that the H2O2-triggered loss of HuR binding to SIRT1 mRNA was rescued when Chk2 activity was inhibited (Fig. 6E, bottom).
Figure 6. Chk2 interacts with and phosphorylates HuR.

(A) Nuclear and cytoplasmic IPs of HuR were performed after the indicated transfections, followed by HuR or CHK2 Western blot analysis. HC, heavy IgG chain; LC, light IgG chain. (B) Western blot analysis of HuR and Chk2 levels in either control (Ctrl.) or Chk2-silenced cultures; β-Tubulin was used as a cytoplasmic loading control and β-Actin as a loading control for total protein. (C) In vitro kinase assay using active Chk2 kinase and GST-HuR as substrate. The proteins used in the reaction were visualized by SYPRO staining; [γ-32P]ATP incorporation into GST-HuR served to monitor phosphorylation. (D) Western blot analysis of Chk2 phosphorylation at residue Thr-68 (p-Chk2) in WI-38 cells that were treated with 500 μM H2O2 for the times shown; α-Tubulin was tested as loading control. (E) After pretreatment of HDFs with a Chk2 inhibitor (1 μM, 1 h), then with 500 μM H2O2 for 3 h, RNA was isolated for RT-qPCR analysis of total (top) and HuR-bound (bottom) SIRT1 mRNA levels. Data are the means +SEM from 3 independent experiments. (F) Western blot analysis of SIRT1 expression in whole-cell lysates prepared from WI-38 cells that were pretreated with the Chk2 inhibitor (1 μM, 1 h) before treatment with 500 μM H2O2 for 6 h; β-Actin signals served to monitor loading. (G) Top, in vivo HuR phosphorylation, assessed by incubation of WI-38 cells with 32Pi for 16 h, followed by IP with either anti-HuR antibody or IgG (1, 5, and 10 μl of lysate were loaded). Bottom, Western blot analysis of HuR in the IP material. (MW), molecular weight marker. (H) 2-dimensional (2D) Western blot analysis of HuR in WI-38 cells treated with H2O2 (500 μM, 1 h). Before loading for separation in the first-dimension (pI), samples were either left without further treatment (−) or were pretreated with alkaline phosphatase (+CIP) for 1 h at 37°C; Chk2 siRNA, cells were transfected as described in panel B before treatment with H2O2.
Evidence that HuR was phosphorylated in vivo was obtained by incubating WI-38 cells with 32P orthophosphate; subsequent HuR IP analysis yielded a specific, albeit weak ∼36 kDa band (Fig. 6G). Two-dimensional gel electrophoresis followed by Western blotting further supported the notion that HuR was phosphorylated (Fig. 6H). HuR migrated with an apparent pI between 8.5– 9, while it has a predicted unphosphorylated pI value of 9.2. H2O2 treatment shifted HuR signals leftward, indicating a gain in negative charge that was consistent with increased HuR phosphorylation. Treatment with alkaline phosphatase (CIP) caused a rightward shift in HuR signal compared with untreated populations, suggesting that HuR was at least partly dephosphorylated after CIP treatment. In addition, Chk2 silencing by Chk2 siRNA transfection [(Fig. 6H), but not Ctrl. siRNA transfection (not shown)] suppressed this shift in HuR migration, indicating that the gain in HuR negative charge was Chk2-dependent. Pro-Q staining was also consistent with HuR being a phosphoprotein whose phosphorylation increased after H2O2 treatment (Suppl. Fig. S6).
Mutation at Chk2 phosphorylation sites influences HuR RNPs
Examination of the human HuR sequence revealed two serines (S88 and S100) and one threonine (T118) whose surrounding amino acid sequence resembled the consensus for Chk2 kinase phosphorylation (O'Neill et al., 2002). HuR mutants were generated with alanine substitutions at each of the predicted Chk2 phosphorylation sites (Fig. 7A). In vitro kinase assays using recombinant Chk2 showed that all three purified GST-fusion HuR mutants [HuR(S88A), HuR(S100A), HuR(T118A)] were less efficiently phosphorylated than HuR(WT) (Fig. 7B). Similar mutations were created in mammalian vectors expressing HuR-TAP fusion proteins; the corresponding HuR-TAP proteins were produced in transfected HeLa cells, purified, and used for phosphorylation in vitro using recombinant Chk2. Again, HuR(WT)-TAP was phosphorylated more efficiently than were the mutants (Fig. 7C).
Figure 7. Analysis of HuR carrying point mutations at Chk2 phosphorylation sites.

(A) Schematic of point mutations introduced at the three predicted residues of phosphorylation by Chk2. In vitro phosphorylation assays were performed using recombinant purified Chk2 and either GST-HuR fusion proteins made in bacteria (B), or TAP-HuR fusion proteins made in HeLa cells (C). GST-Cdc25C, Chk2 substrate (positive control); (WB), Western blot analysis of HuR-TAP proteins. (D) Chimeric HuR-TAP proteins expressed in transfected WI-38 cells, then untreated or treated with H2O2 (500 μM, 3 h). Binding of chimeric HuR-TAP proteins to the indicated HuR target mRNAs was tested in transfected WI-38 cells (mean +SEM from 3 independent experiments) (E) and HeLa cells (mean of 2 independent experiments yielding similar results) (F) by performing TAP IP followed by RT-qPCR analysis.
Importantly, when the binding of SIRT1 mRNA to each HuR-TAP variant was studied in WI-38 cells (Fig. 7D,E), the association of HuR(T118A) was constitutively lower than that of HuR(WT), while H2O2 treatment could not reduce the binding of HuR(S100A). By contrast, binding to ProTα mRNA was elevated after H2O2 treatment in all groups, particularly in HuR(S100A)-expressing cells. In order to confirm these findings and assay additional targets, the binding of HuR-TAP mutants was also tested in HeLa cells, where larger cell numbers were available for analysis. As shown (Fig. 7F), HuR-TAP complexes displayed two distinct patterns: H2O2 treatment decreased binding to several target mRNAs (those encoding SIRT1, cyclin D1, cytochrome c, cyclin A), but increased binding to other targets (ProTα, p21). Interestingly, binding in untreated and/or H2O2-treated cells was always higher for HuR(S100A), while it was lower and H2O2-inducible for HuR(T118A). In general, HuR(S88A) bound somwhat less than HuR(WT). These results suggest that HuR phosphorylation (constitutive and/or H2O2-inducible) at S100 might reduce, while T118 (and to a lesser extent S88) phosphorylation likely promoted the binding of HuR to target mRNAs. Taken together, these observations support a model whereby Chk2 phosphorylates and thereby regulates HuR binding to target mRNAs.
DISCUSSION
HuR was found to bind the SIRT1 mRNA and to affect its stability, oxidant treatment dissociated [HuR-SIRT1 mRNA] complexes, and Chk2 regulated this dissociation. The molecular characterization of these processes, requiring large amounts of material, was performed in human cervical carcinoma cells. However, given the role of SIRT1 in stress responsiveness and organismal longevity (Lin et al., 2000; Wood et al., 2004; Chua et al., 2005), the functional implications of [HuR-SIRT1 mRNA] RNP complexes were studied in human diploid fibroblasts. In WI-38 HDFs, where both HuR and SIRT1 were highly expressed in young cells but were very low in senescent cells, HuR silencing as well as H2O2-triggered reduction in binding of HuR, each markedly decreased SIRT1 mRNA levels and lowered cell survival after oxidant treatment.
HuR RNPs: association versus dissociation
In light of previous findings that H2O2 treatment of colorectal carcinoma cells promoted the binding of HuR to a p21 3'UTR transcript (Wang et al., 2000b), and the H2O2-enhanced formation of [HuR-ProTα mRNA] complexes (Fig. 4D), the H2O2-triggered dissociation of [HuR-SIRT1 mRNA] complexes was unexpected. However, these results are reminescent of those observed following exposure to another stress, irradiation with UVC (short-wavelength ultraviolet light), which promoted HuR binding to several target transcripts [e.g., p53, p21, ProTα mRNAs (Wang et al., 2000b; Mazan-Mamczarz et al., 2003; Lal et al., 2005)], but decreased binding to another target, the cyclin D1 mRNA, which instead bound more prominently to AUF1 (Lal et al., 2004). These observations support the hypothesis that the changes in HuR association to a given mRNA are determined by the transcript itself, rather than the particular stimulus. This notion is ilustrated by a recent report that endoplasmic reticulum stress also caused a transient dissociation of HuR from the cytochrome c mRNA, while it promoted the binding of TIA-1 to this transcript (Kawai et al., 2006). In another study, the dissociation of RBPs AUF1 and TIAR from the GADD45α mRNA was essential for the increase in GADD45α levels following treatment with methyl methanesulfonate (Lal et al., 2006); in this posttranscriptional derepression paradigm, the genotoxin-elicited dissociation of AUF1 promoted GADD45α mRNA stability, while the dissociation of TIAR enhanced GADD45α translation. In addition, factors other than RBPs may also influence HuR's association with target mRNAs. Recently, Bhattacharyya et al. (2006) showed that the association of microRNA miR-122 with the CAT-1 mRNA repressed its translation. Following exposure to stress agents, HuR associated with the CAT-1 3'UTR, interfered with the binding of miR-122, and relieved the miR-122-imposed translational repression. Emerging from these and other studies is the view that the combinatorial association of trans factors (RBPs and miRNAs) with a target mRNA changes in response to stress agents and influences gene expression posttranscriptionally.
HuR phosphorylation by Chk2
Despite the fact that the cytoplasmic HuR levels actually increase after H2O2 treatment (Suppl. Fig. S7, Wang et al., 2000b), HuR dissociates from the SIRT1 mRNA rapidly (by <15 min after H2O2 treatment), suggesting that posttranslational events triggered this reduction in binding. HuR had not been previously reported to be a phosphoprotein, but our data from 2D Western blotting and from in vivo and in vitro phosphorylation indicate that HuR does appear to be phosphorylated. In fact, some evidence suggests low-level phosphorylation of HuR before H2O2 treatment (Figs. 6F,G, Suppl. Fig. S6), possibly contributing to basal mRNA binding and helping to explain why S88A and T118A mutants bind mRNAs less effectively than WT (Fig. 7E,F). Alternatively, since S88 and T118 lie within RRM1 and RRM2, respectively, local conformational changes caused by the S88A and T118A mutations could reduce the binding. S100 lies between RRM1 and RRM2 and might be implicated in regulating their relative distance; for all mRNAs examined, S100A mutations showed enhanced binding (before and/or after H2O2) and enhanced SIRT1 mRNA stability (Suppl. Fig. S8), suggesting that in WT HuR, S100 phosphorylation was important in reducing HuR complexing with mRNAs. As this analysis moves forward, the study of double and triple point mutants, including mutations that mimic phosphorylation, along with mass spectroscopy and crystallography data will help to fully elucidate the role of HuR phosphorylation at these putative Chk2 target residues.
The checkpoint kinase Chk2/Cds1 was first identified in yeast as a kinase essential for cell cycle arrest following DNA replication blockage (Murakami & Okayama, 1995). In mice, Chk2 deficiency does not affect viability or fertility, unlike the deficiency of the functionally related Chk1, which causes early embryonic lethality (Hirao et al., 2000). Chk2 is activated by ATM-dependent phosphorylation at Thr-68 (Melchionna et al., 2000); after homodimerization and transphosphorylation of the kinase domain, Chk2 is active and can phosphorylate several proteins implicated in growth arrest, gene transcription, and tumorigenesis [BRCA1, Cdc25A, E2F1, Cdc25C, BRCA1, Mdm2, and p53 (reviewed by Bartek & Lukas, 2005)]. Adding to these functions, our results that Chk2 influences HuR function also implicate Chk2 in posttranscriptional gene regulation.
Functional links between Chk2, HuR, and SIRT1
The protective influence of HuR in WI-38 cells treated with H2O2 (Fig. 5) recapitulates earlier observations in UVC-treated HeLa cells, where the HuR-mediated protection was linked to the antiapoptotic properties of ProTα (Lal et al., 2005). Through its ability to regulate several antiapoptotic effector proteins such as p21 (Gorospe et al., 1996; Wang et al., 2000b), ProTα (Lal et al., 2005), and SIRT1 (this report), HuR seems to promote a robust antiapoptotic program. Thus, it is likely that under conditions of elevated HuR expression, such as in actively proliferating fibroblasts and in cancer (Wang et al., 2001; López de Silanes et al., 2003, 2004a), HuR directly supports a prosurvival program via its influence on multiple antiapoptotic target genes. Our findings also indicate that SIRT1 protects against cell death in response to oxidative stress (Fig. 5), in keeping with a previously proposed prosurvival influence for this deacetylase that is likely mediated by several downstream targets of SIRT1 action, including Ku70, p53, and FoxO (Prives & Manley, 2001; Luo et al., 2001; Vaziri et al., 2001; Cohen et al., 2004a; Chen et al., 2005). As Chk2 regulates HuR influence on SIRT1 expression, Chk2 can function as a negative upstream effector of this prosurvival axis.
Finally, Chk2 was shown to be constitutively elevated in senescent cells and to trigger replicative senescence (di Fagagna et al., 2003; Gire et al., 2004). Thus, during senescence, not only is HuR less abundant (resulting in lower SIRT1 mRNA stability), but in addition Chk2 activity is elevated, further impairing HuR binding to SIRT1 mRNA. Together, these two senescence-associated changes in HuR contribute to reducing SIRT1 expression. The influence of HuR function by Chk2 and the effect of HuR upon SIRT1 expression underscore the intricate connections that exist between cellular growth and stress responsiveness, as we seek a better understanding of the molecular underpinnings of organismal longevity.
EXPERIMENTAL PROCEDURES
Cell culture, transfections, and analysis of (SA)β-galactosidase activity, proliferation, and survival
Human cervical carcinoma HeLa cells were cultured in Dulbecco's modified essential medium (Invitrogen) supplemented with 10% fetal bovine serum and antibiotics. WI-38 cells (human diploid fibroblasts or HDFs) were cultured under the same conditions in addition to 0.1 mM (MEM non essential amino acids, Invitrogen). Young (Y) cells were at population doublings (pdl) 18-30, senescent (S) cells were pdl ∼50. H2O2 was added directly to complete medium. Incorporation of [3H]-thymidine was assayed using standard methods (Suppl. material). A senescence-associated (SA) β-galactosidase detection kit was from Cell Signaling. Cell viability was determined using [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT, Sigma), and was represented as a percentage of the cell viability in untreated cultures.
SiRNAs (Qiagen) targeting the HuR coding region (‘HuR’, AAGAGGCAATTACCAGTTTCA), the HuR 3'UTR (‘HuRU1’ AACGACTCAATTGTCCCGATA) or the SIRT1 coding region (GAAGTTGACC TCCTCATTGT), as well as a control siRNA (AATTCTCCGAACGTGTCACGT), were used at 20 nM. To silence Chk2 expression, Chk2-targeting IMG-809-1 and negative control IMG-800-6 siRNA plasmids were used (Imgenex). HeLa cells were transfected either with Oligofectamine (Invitrogen), when only siRNAs were used, or with Lipofectamine 2000 (Invitrogen) when plasmids were included, and were harvested or treated 48 h later. WI-38 cells were transfected using Lipofectamine 2000. pHuR-TAP and pGST-HuR point mutants (S88A; S100A; T118A) were generated by site-directed mutagenesis. The SIRT1 expression vector was kindly provided by D. Sinclair (Cohen et al., 2004b). Chk2 inhibitor [2-(4-(4-Chlorophenoxy)phenyl)-1H-benzimidazole-5-carboxamide] was from Calbiochem.
Northern blotting, Western blotting, and coimmunoprecipitation assay
For Northern blot analysis, RNA was isolated from whole cells or gradient fractions using Trizol (Invitrogen). Oligonucleotides CTATCCGTGGCCTTGGAGTCCAGTCACTAGAGCTTGCATGTGAGG CTCTA and ACGGTATCTGATCGTCTTCGAACC were end-labeled with [α-32P]dATP and terminal transferase and used to detect SIRT1 mRNA and 18S rRNA, respectively.
For Western blot analysis, lysates were size-fractionated by SDS-PAGE and transferred onto PVDF membranes. Monoclonal antibodies recognizing HuR, Chk2 or α-Tubulin (a control cytoplasmic protein), as well as polyclonal antibodies recognizing SIRT1 or hnRNP C1/C2 (a control nuclear protein), and phosphoChk2 (Thr-68) were from Santa Cruz Biotechnology. A monoclonal antibody recognizing β-Actin was from Abcam. After secondary antibody incubations, signals were detected by enhanced chemiluminescence.
For coimmunoprecipitation assays, Protein A-Sepharose beads (Sigma) were precoated with 10 μg IgG1 (BD Pharmingen) or HuR (Santa Cruz Biotech.), washed with NT2 buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM NgCl2, 0.05% Nonidet P-40), and incubated with 0.5 mg protein (16 h, 4°C). After washes with NT2 buffer, samples were denatured, fractionated by SDS-PAGE, and analyzed by Western blotting.
Binding assays: biotin pulldown analysis and immunoprecipitation of RNP complexes
The synthesis of biotinylated transcripts and analysis of RBPs bound to biotinylated RNA were done as previously described (Lal et al., 2004), and is explained in detail in Supplemental material.
Immunoprecipitation (IP) of endogenous RNA-protein complexes was performed as described (Lal et al., 2004; Suppl. material). The RNA isolated from IP material was reverse-transcribed using random hexamers or oligo-dT primer, and SSII Reverse Transcriptase (Invitrogen). Conditions for qPCR and oligomers to amplify GAPDH, and prothymosin α products were described (Lal et al., 2005). Oligomers used for to amplify PCR products are listed (Suppl. material).
HuR phosphorylation assays
In vitro phosphorylation of HuR was performed using recombinant purified GST-HuR proteins [(WT or mutants, as described in Suppl. material)] or using HuR-TAP (WT or mutants) expressed in HeLa cells through transfection of the correponding vectors; active CHK2 (Upstate) and [γ-32P]ATP were used. In vivo HuR phosphorylation was tested by incubating subconfluent HeLa cells (∼107) with 10 mCi for 16 h, followed by IP using either IgG or anti-HuR antibody. After transfering to membranes, radioactive signals were detected using a PhosphorImager and HuR levels by Western blot analysis.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Mazan-Mamczarz, J. L. Martindale, T. D'Souza, C. Sasaki, and F. Bunz for providing reagents and assistance with this work. This research was supported in part by the Intramural Research Program of the NIA, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Astrom SU, Cline TW, Rine J. The Drosophila melanogaster sir2+ gene is nonessential and has only minor effects on position-effect variegation. Genetics. 2003;163:931–937. doi: 10.1093/genetics/163.3.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- Bevilacqua A, Ceriani MC, Capaccioli S, Nicolin A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J. Cell Physiol. 2003;195:356–372. doi: 10.1002/jcp.10272. [DOI] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Brennan CM, Steitz JA. HuR and mRNA stability. Cell. Mol. Life Sci. 2001;58:266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Buscemi B, Perego P, Carenini N, Nakanishi M, Chessa L, Chen J, Khanna K, Delia D. Activation of ATM and Chk2 kinases in relation to the amount of DNA strand breaks. Oncogene. 2004;23:7691–7700. doi: 10.1038/sj.onc.1207986. [DOI] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- Cheadle C, Fan J, Cho-Chung YS, Werner T, Ray J, Do L, Gorospe M, Becker KG. Stability regulation of mRNA and the control of gene expression. Ann. NY Acad. Sci. 2005;1058:196–204. doi: 10.1196/annals.1359.026. [DOI] [PubMed] [Google Scholar]
- Chen CY, Shyu A-B. AU-rich elements: characterization and importance in mRNA degradation. Trends. Biochem. Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Chua KF, et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM, Sinclair DA. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell. 2004a;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004b;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- di Fagagna FdA, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Dimri GP, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez J, Aranda A, Perez-Ortin JE. Genomic run-on evaluates transcription rates for all yeast genes and identifies gene regulatory mechanisms. Mol. Cell. 2004;15:303–313. doi: 10.1016/j.molcel.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Gherzi R, Lee KY, Briata P, Wegmuller D, Moroni C, Karin M, Chen CY. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol. Cell. 2004;14:571–583. doi: 10.1016/j.molcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Gire V, Roux P, Wynford-Thomas D, Brondello JM, Dulic V. DNA damage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 2004;23:2554–2563. doi: 10.1038/sj.emboj.7600259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorospe M, Wang X, Guyton KZ, Holbrook NJ. Protective role of p21(Waf1/Cip1) against prostaglandin A2-mediated apoptosis of human colorectal carcinoma cells. Mol. Cell. Biol. 1996;16:6654–6660. doi: 10.1128/mcb.16.12.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- Kawai T, Lal A, Yang X, Galban S, Mazan-Mamczarz K, Gorospe M. Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol. Cell. Biol. 2006;26:3295–307. doi: 10.1128/MCB.26.8.3295-3307.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fitzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell. Biol. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD. Why is Hu where? Shuttling of early-response-gene messenger RNA subsets. Proc. Natl. Acad. Sci. USA. 1999;96:5–7. doi: 10.1073/pnas.96.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc. Natl. Acad. Sci. USA. 2001;98:7018–7024. doi: 10.1073/pnas.111145598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann M, Gopfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 2002;16:3087–3099. doi: 10.1101/gad.248902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent Versus Individual Binding of HuR and AUF1 to Common Labile Target mRNAs. EMBO J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Kawai T, Yang X, Mazan-Mamczarz K, Gorospe M. Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. EMBO J. 2005;24:1852–1862. doi: 10.1038/sj.emboj.7600661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Abdelmohsen K, Pullmann R, Kawai T, Yang X, Galban S, Brewer G, Gorospe M. Posttranscriptional derepression of GADD45α by genotoxic stress. Mol. Cell. 2006;22:117–128. doi: 10.1016/j.molcel.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Laroia G, Cuesta R, Brewer G, Schneider RJ. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science. 1999;284:499–502. doi: 10.1126/science.284.5413.499. [DOI] [PubMed] [Google Scholar]
- Leroy C, Mann C, Marsolier MC. Silent repair accounts for cell cycle specificity in the signaling of oxidative DNA lesions. EMBO J. 2001;20:2896–2906. doi: 10.1093/emboj/20.11.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- López de Silanes I, Fan J, Yang X, Zonderman AB, Potapova O, Pizer ES, Gorospe M. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene. 2003;22:7146–7154. doi: 10.1038/sj.onc.1206862. [DOI] [PubMed] [Google Scholar]
- López de Silanes I, Lal A, Gorospe M. HuR: Post-transcriptional Paths to Malignancy. RNA Biology. 2004a;1:135–137. doi: 10.4161/rna.2.1.1552. [DOI] [PubMed] [Google Scholar]
- López de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA. 2004b;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Melchionna R, Chen XB, Blasina A, McGowan CH. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat. Cell Biol. 2000;2:762–765. doi: 10.1038/35036406. [DOI] [PubMed] [Google Scholar]
- Mazan-Mamczarz K, Galban S, López de Silanes I, Martindale JL, Atasoy U, Keene JD, Gorospe M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA. 2003;100:8354–8359. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S phase. Nature. 1995;374:817–819. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- North BJ, Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004;5:224. doi: 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill T, et al. Determination of substrate motifs for human Chk1 and hCds1/Chk2 by the oriented peptide library approach. J. Biol. Chem. 2002;277:16102–16115. doi: 10.1074/jbc.M111705200. [DOI] [PubMed] [Google Scholar]
- Prives C, Manley JL. Why is p53 acetylated? Cell. 2001;107:815–818. doi: 10.1016/s0092-8674(01)00619-5. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA. 2004;101:15998–6003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E., Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000a;19:2340–2350. doi: 10.1093/emboj/19.10.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, Holbrook N, Gorospe M. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 2000b;20:760–769. doi: 10.1128/mcb.20.3.760-769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yang X, Cristofalo VJ, Holbrook NJ, Gorospe M. Loss of HuR influences gene expression during replicative senescence. Mol. Cell. Biol. 2001;21:5889–5898. doi: 10.1128/MCB.21.17.5889-5898.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz CJ, Wilusz J. Bringing the role of mRNA decay in the control of gene expression into focus. Trends Genet. 2004;20:491–497. doi: 10.1016/j.tig.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.