

Abstract
While there are many reviews which examine the group of proteins known as protein kinase C (PKC), the focus of this article is to examine the cellular roles of two PKCs that are important for stress responses in neurological tissues (PKCγ and ε) and in cardiac tissues (PKCε). These two kinases, in particular, seem to have overlapping functions and interact with an identical target, connexin 43 (Cx43), a gap junction protein which is central to proper control of signals in both tissues. While PKCγ and PKCε both help protect neural tissue from ischemia, PKCε is the primary PKC isoform responsible for responding to decreased oxygen, or ischemia, in the heart. Both do this through Cx43.
It is clear that both PKCγ and PKCε are necessary for protection from ischemia. However, the importance of these kinases has been inferred from preconditioning experiments which demonstrate that brief periods of hypoxia protect neurological and cardiac tissues from future insults, and that this depends on the activation, translocation, or ability for PKCγ and/or PKCε to interact with distinct cellular targets, especially Cx43.
This review summarizes the recent findings which define the roles of PKCγ and PKCε in cardiac and neurological functions and their relationships to ischemia/reperfusion injury. In addition, a biochemical comparison of PKC γ and PKC ε and a proposed argument for why both forms are present in neurological tissue while only PKC ε is present in heart, are discussed. Finally, the biochemistry of PKCs and future directions for the field are discussed, in light of this new information.
Keywords: protein kinase C epsilon, protein kinase C gamma, ischemia, heart, neural tissues
Introduction
Protein Kinase C –Part of the ABC Kinase Superfamily
The Protein Kinase C (PKC) family of proteins is part of the larger ABC protein kinases which includes Protein Kinase A (PKA), Protein Kinase B (PKB, synonymous to Akt), and Protein Kinase C (PKC) [1-3]. All members of this superfamily have an N-terminal regulatory region and a conserved C-terminal kinase core that contains two conserved phosphorylation sites. These sites are known as the turn motif and hydrophobic motif. The regulatory regions of ABC kinases have two functions. One is to bind to the plasma membrane or other cellular targets, and the other is to inhibit the active site of the enzyme. The C-terminal region functions as the substrate binding site and phosphor-acceptor/donor site.
All ABC kinases contain an activation loop threonine that must be phosphorylated by upstream Phosphoinositide Dependent Kinase-1 (PDK-1), except protein kinase A (PKA) which is recognized by PDK-1, in vitro, but does not require PDK-1 activity in vivo. PDK-1 is constantly active, but, it's downstream substrates must be in an open conformation in order to become targets. For example, when the pleckstrin homology (PH) domain of protein kinase B (PKB) encounters Phosphatidylinositol (3,4,5)-trisphosphate (PIP3), this opens the conformation, by disassociation of the PH domain, with the kinase core, revealing the activation loop. Once activated, PKB remains in this state. In contrast, PKCs are phosphorylated at the activation loop, but this only primes them. They still must interact with secondary messengers or other signals before becoming activated.
The Protein Kinase C Family of Proteins
Biochemical Features
There are at least 10 isoforms of PKC that are divided into 3 groups related through their primary structure [3]. In general, PKCs contain a regulatory domain, with a pseudo substrate region, as well as the elements necessary to respond to second messengers (Fig. 1). The N-terminal regulatory domain is followed by a hinge region and the kinase core which contains the substrate and ATP binding sites in the C-terminus of PKC. PKCs, once processed in the cell by priming phosphorylation events by PDKI, remain in an auto-inhibited state until second messengers bind their C1 and/or C2 domains [4]. The PKC family of proteins is divided into three groups based on variations in the arrangement or existence of C1 and C2 domains. Conventional (α,βI,βII,γ) PKCs have repeat cysteine rich C1 domains which act as diacylglycerol (DAG) sensors and C2 domains which bind to Ca+2. Within the C1B domain of PKCs, cysteine residues combine with key histidines to form two zinc fingers. In the case of PKCγ, and discussed in this review, this type of C1 domain affords PKCγ an alternative mode of activation. In this case, oxidation of cysteine residues, in the C1B domain, by oxidative stress, leads to the formation of disulfide bonds and it is thought that this provides a direct link between PKCγ activation and increased oxidative or reactive oxygen species (ROS) [66]. A similar example of this occurs in Hsp33, a bacterial chaperone protein which becomes activated by oxidation of its zinc finger domain, forms dimers, and, then, has chaperone activity[5].
Figure 1.
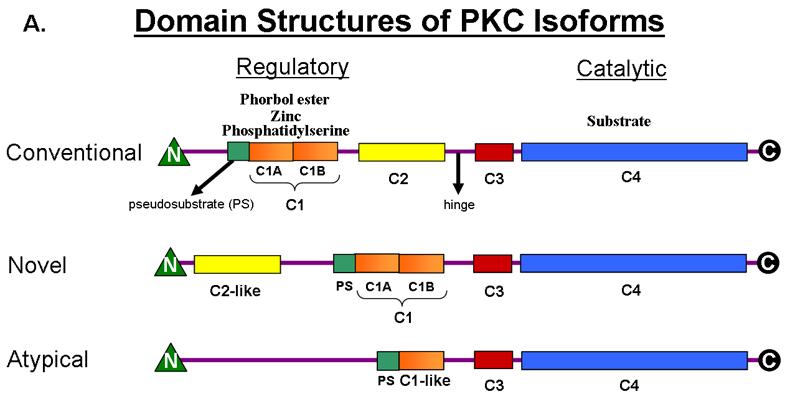

A) Domain Structures of PKC Isoforms. PKCγ is a conventional PKC. PKCε is a novel PKC.
B) Sequences of C1B Domains of PKCγ and PKCε.
Novel PKCs (δ,ε,η,θ) contain C1 and C2 domains but they are inverted in the primary structure (Fig. 1). In addition, novel PKCs have C2 domains which do not bind to known ligands, and,thus, are not activated by calcium signals. It is thought that without the calcium sensor, novel PKCs translocate and activate differently than conventional PKCs [6]. PKCε is a member of this group.
Atypical PKCs (ζ,ι/λ) do not have C2 domains or typical C1 domains, and, they lack a conserved Ser/Thr residue in the C terminus which is an important phosphor-acceptor site in conventional and novel PKCs (Fig. 1).
Once fully processed and phosphorylated, PKCs can respond to second messengers, depending on their C1 or C2 domains, and engage and phosphorylate downstream targets. Activated PKCs are subject to phosphatase activity and down-regulation by ubiquitination and proteasomal degradation, after prolonged activation with tumor promoting phorbol esters [7]. The domain architectures of the PKC isoform groups are presented in fig. 1 and show the striking similarities in their structure, yet subtle differences which allow members of this diverse family of proteins to display different sensitivities towards activators. As a result, PKCs translocate and interact with varying targets depending on the isoform, tissue, and their location in the cell. Due to the available structural information for the C1, C2, and the kinase domains of PKCs, and the extensive biochemical information, a general mechanism of activation for PKCs can be described in detail [3].
A General Mechanism for Processing and Activation of PKCs
The newly synthesized PKCs associate with membranes through interactions with C1 and/or C2 domains. Together, these weak interactions contribute enough binding energy to hold PKC in a membrane bound, open conformation. PDK-1 is able to access its binding site at the hydrophobic motif in the kinase domain and, once docked there, it phosphorylates PKCs on the PKC activation loop. The release of PDK-1 from PKC is thought to be rate-limiting [8] but the mechanism of release is unknown. Once phosphorylated on the activation loop (Thr514 in PKCγ) PKCs auto-phosphorylate at the turn motif and the hydrophobic motif. Evidence suggests that phosphorylation at the turn motif precedes the hydrophobic motif since mutations at the turn motif abolish phosphorylation at the hydrophobic motif [9]. It is likely that these events are linked to undocking of the PDK-1 from the hydrophobic motif of PKC, and, it is clear that phosphorylation in the C-terminal kinase core of PKC stabilizes the enzyme in a closed conformation. This allows the N-terminal pseudosubstrate region of the PKC to contact the substrate binding site in an inhibitory loop, thus, inhibiting the enzyme until activation by second messengers. In this conformation the PKC is released from the membrane and it localizes to the cytoplasm where it encounters scaffolding proteins, docking proteins such as 14-3-3, remaining in this cellular compartment until activated.
Once calcium levels rise in the cell (in the case of PKC α, βI, and βII), Ca2+ binds to the C2 domain and the C1 region now interacts at the membrane with diacylglycerol (DAG). The phorbol esters, used in research labs, are analogs of DAG and are used, in this manner, to activate PKCs for experimental purposes.
In contrast, PKCγ, as well as the novel and atypical PKC isoforms, do not respond to calcium as their primary signal, but, rather, display different sensitivities to DAG and membrane lipids such as phosphatidylserine, based upon the specific structure of their C1 domains. This provides a biochemical explanation for why a specific PKC can translocate to different cellular components upon activation.
Because of their ability to interact with partner proteins there is yet another way to regulate PKC activity. Since PKCs often interact with distinct docking/scaffolding proteins (14-3-3 for PKCγ and RACK5 [receptor for activated C-kinase] for PKCε) peptides derived from the sequences of different PKC isoforms make potent inhibitors and activators [10]. Once activated or released from their docking partner PKCs can interact with numerous cellular components. PKCs can interact with the plasma membrane [11], golgi [12], and cytoskeleton [13]. More recently, it has become evident that some PKCs interact with mitochondria [14] and cell nuclei [15]. As a consequence, PKCs are involved in such diverse functions as differentiation, cell growth, apoptosis, gene expression, muscle contraction, metabolism, and endocytosis [16]. Thus, the PKCs are involved in memory and learning, development, alcohol dependence, adaptation to pain, cancer metastasis, tumor growth, hypothermia, hyperthermia, starvation and other physiological stresses.
Lipid Activation Signals
Since both PKCγ and PKCε have functional C1 domains they are both activated by lipid signals. The specificity of these interactions allows each to translocate to different cellular compartments. PKCs with functional C1 domains are activated primarily by diacylglycerol (DAG), a product of phospholipase C. Phorbol esters, which mimic DAG, have been used extensively to study the broad ranging effects of activation of conventional and novel PKCs. Though reactive oxygen species (ROS) are known to activate some PKCs at their C1 domains, it is not clear if all isoforms are oxidative stress sensitive. PKCε, which is activated during ischemia, is thought to be directly activated by ROS but the mechanism is unclear. Similarly, PKCγ is activated during neural ischemia, but whether PKCγ activation depends directly on changes in the levels of oxidative species or rather on upstream events is unknown. Because these isoforms contain C1 domains, they each have the potential to undergo major conformational rearrangements under conditions which favor the formation of disulfide bonds within their C1 domains (Fig. 1B for sequences).
Because DAG is a product of phospholipase C (PLC) activity, and is generated by a diverse set of ligand/receptor interactions, PKCs are connected to many signal transduction pathways. For example, phospholipase activation within the low density lipoproteins (LDL) favors the formation of arterial plaques. Phospholipase can modify LDL and this confers a survival response towards oxidative stress which was found to be dependent upon the activation of several PKC isoforms [17]. There are other membrane lipids which interact with the C1 domain and activate either PKCγ or PKCε. A linoleic acid derivative with a cyclopropane ring, instead of a cis double bond was found to activate PKC ε, and to a lesser extent PKC γ, possibly through interaction with the phosphatidylserine binding site of the C1 domain [18]. Another lipid that activates PKCε was found to protect against endoplasmic reticulum (ER) dependent cell death. The ability of dilinoleoyl-phosphatidylethanolamine (DLPE) in a neuronal cell line to protect against ER-mediated cell death was verified, but the mechanism of activation was not clear [19]. Pyrroloquinoline quinone (PQQ), which acts as a growth factor and promotes DNA synthesis, was shown to activate both extracellular signal regulated kinase (ERK) as well as PKCε. Because nitric oxide (NO) was found to reduce cell viability in PQQ treated cells, this pathway is considered to be NO sensitive. Again, the role of PKCε in PQQ mediated cell growth is unknown [20]. A pro-inflammatory agent lipopolysaccharide (LPS) that induces astrocyte stellation has enhanced activity in the presence of PKCε. This effect is blocked by expression of constitutively active Rho A, demonstrating that PKC activity is important for inactivating Rho A dependent pathways [21]. LPS signaling mechanisms are unclear but recently it was shown that TRAM (Toll-like receptor 4 adaptor molecule) is directly phosphorylated by PKCε on Ser16. This places PKCε firmly upstream of TRAM [22, and see 23-29].
Targets of PKCs
PKCs are expressed in a tissue specific manner. For example, PKCγ, a conventional isoform, is expressed mainly in brain cells, neuronal tissues, retina, and lens. Neuronal tissues also contain the ubiquitous α isoform as well as all others except λ. In epidermis, α,δ,ε,η,ζ are co-expressed and play roles in the complex differentiation and cell death program which exists in skin tissue [30]. In heart, PKCα, β, δ, and ε are expressed and ε in particular has been implicated in a protective role for cardiac tissue while γ is absent in heart cells. PKCε is found in many tissues but primarily in neuronal, immune, hormonal, and heart cells. Aside from neuronal tissue, in which γ and ε are co-expressed, they are also co-expressed in the lens, retina, and the vascular endothelium that constitutes the blood brain barrier [31]. PKCα, γ, λ, ε, and μ have been detected in lens and the blood/brain barrier is thought to express ten isoforms.
Differences and similarities in the structures of PKCγ and PKCε
It was shown using a reporter assay that the magnitude and duration of PKC activation is largely controlled by the location of the PKC, persistence of DAG, and the levels of phosphatase activity at a particular location in the cell [32]. But , additional studies clearly show that PKCγ and PKCε respond differently to the same level of DAG. It was recently discovered that a single residue in the C1B domain of PKC's determines why some PKCs become activated and translocate with only a DAG signal. Dries et al. ([33] found that PKCs with Tyr at this key position bind DAG with much less affinity than PKCs containing Trp. PKCγ (Tyr122) required Ca2+ binding at C2 for DAG activation and translocation to the plasma membrane, while PKCε (Trp191) more readily responded to DAG. Furthermore, it was found that PKCs with Tyr have a 2-fold selectivity for phosphatidylserine (PS), a plasma membrane lipid, but localized to the cytoplasm until activated [33]. PKCε, with a lower lipid headgroup specificity requirement and higher affinity for DAG, is able to translocate to various membrane targets under conditions when PKCs with Tyr, such as PKCγ, cannot. Consistent with these findings, when comparing the headgroup selectivity of PKCε and PKCδ, it was found that while PKCδ required PS to untether the C1 domain, PKCε displayed higher conformational flexibility and a lack of need for a specific lipid headgroup. Because of the increased flexibility, PKCε translocated much faster to various cellular compartments while the need for PS directed PKCδ to the plasma membrane [34]. Tethering of C1 domains has been attributed to two electrostatic and one aromatic interaction in PKCα. Mutants lacking these interdomain interactions had higher basal membrane affinity and translocated faster to subcellular compartments than wildtype PKCα [34].
Studies comparing PKCα to PKCγ, demonstrated that both C1 domains of PKCγ had comparably high affinities for DAG, while the C1B domain of PKCα had high affinity for phorbol ester. In vitro activity assays and monolayer penetration analysis showed that PKCα is auto-inhibited and does not translocate to the plasma membrane at basal calcium concentrations. Although PKCγ is also a conventional isoform, the conformational flexibility of the C1A and C1B domains in PKCγ were shown to be quite flexible compared to PKCα. In fact, PKCγ translocated to the plasma membrane in the presence of Ca2+ concentrations which were insufficient to activate PKCα [35].
PKCε and PKCγ display highly flexible C1 domains which have high affinity for DAG and phorbol ester. However, they have different subcellular localization patterns and modes of activation. It is evident that neither kinase is tethered like PKCα or PKCδ, which results in easier access to DAG and faster activation. PKCγ, however, contains Tyr122 while PKCε has Trp191 in their C1B domains [36]. Hence, PKCγ requires PS because of increased lipid headgroup specificity, in contrast to PKCε. This provides a structural explanation for how these two kinases are localized to sometimes overlapping, but often different subcellular locations. Protein/protein interactions are also reported to play a major role in determining whether the binding sites for DAG are accessible in C1 domains [36]. A proteomics approach has been used to identify some of the proteins which bind to PKCε [37], but a similar study has not been done for PKCγ.
Substrate Specificity of PKCε and PKCγ
The structural differences in these two PKC isoforms also translate into differences in substrate specificity. DNA microarray analyses on the PKCγ knockout mice show changes in activity towards transerythrin, a thyroxin and retinol transport protein [98]. In brain, PKCγ directly interacts with and phosphorylates the GluR4 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor subunit [100]. PKCε, on the other hand, has specificity for GAP-43 (growth-associated protein of 43 kDa), a neuronal calmodulin binding protein [99].
PKCs are normally inhibited by the binding of the N-terminal pseudosubstrate inhibitory sequence to the catalytic domain. Synthetic peptides which correspond to these regions can be used to alter functions of each PKC and this gives some indication of the substrate sequence specificity of each isoform. The pseudosubstrate region of PKCε is: ERMRPRKRQGSVRRRV. The pseudosubstrate region of PKCγ is: FCRKGALRQ. The peptides which is commercially used as a PKC substrate is called Kemptide and has the sequence: LRRASLG [101]. These synthetic peptides have been used to determine the preferred substrate consensus sequences of the different PKC isoforms. In general, PKCs phosphorylate S or T residues with basic amino acids spaced on either side of the target residue.
Their have been several studies detailing the unique substrate consensus sequences for PKCε and PKCγ. Synthetic peptides based upon the Ser 7 of glycogen synthase 1 (GS1), the EGF receptor region (VRKRILRRL), pp60c-src, and myelin basic protein-1 were used to demonstrate differences in substrate consensus region recognition [102]. PKCγ had a Km for GS1 of 53 μM while the PKCα Km was 550 μM. Using peptides with varied distance between the positively charged amino acids (either lysine or arginine) and the S phosphorylation site, it was determined that PKCε required R at either two or three or six residues from the S. In contrast, the best synthetic peptide substrates for PKCγ required R's (Arg) on either side of the S phosphorylation site [102]. Thus, PKCγ appears to be more specific for substrate choice when compared to PKCε. Examination of the residues surrounding the S368 of Cx43 would, therefore, predict that this would be a PKCγ phosphorylation site while S262 would be a PKCε site. This has been found to be the case [56-59].
Two PKCs which respond to stress
PKCε in the heart
PKCε has been implicated as a major factor in the phenomena of cardiac preconditioning [38]. Under hypoxic conditions PKCε translocation to Cx43 and to mitochondria and subsequent protection from ischemia are dependent upon the presence of increased mitochondrial reactive oxygen species (ROS). The requirement for PKCε was confirmed using the PKCε null mouse hearts which do not develop tolerance to antimycinA-induced ischemia [39].
Cardiac preconditioning, which has been demonstrated in many species, can be induced through a variety of methods including hypoxia/ischemia [40], anesthetics [41], glucose [42], phorbol esters [43], cannabinoids [44], and heat-stress [45]. It is assumed that these act in a unified manner and activation of PKCε is, most likely, key to this control [46].
If this proves to be the case, what proteins is PKCε binding to and phosphorylating? One target for hypoxia activated PKCε is the mitochondrial permeability transition pore (MPTP). PKCε is thought to interact, first, with the mitochondrial KATP channel which leads to an influx of K+. This triggers a rise in pH and the subsequent formation of reactive oxygen species (ROS), which further activates PKCε, now localized in the mitochondria, resulting in inhibition of the MPTP [47]. The functional association of the mitochondrial KATP channels with PKCε were recently described using an in vitro proteoliposome system [48]. PKCε also interacts with at least one component of the electron transport chain. Cytochrome C oxidase (CytCox), which serves as the last component in the electron transport chain, is key in regulating ATP production via oxidative phosphorylation. It also serves as a direct sensor of the oxygen levels available in the cell and nitric oxide (NO) competes with binding of oxygen to CytCox [49]. It has been shown in studies using cardiac myocytes that PKCε, when activated by hypoxia, interacts with subunit IV (COIV), increasing the activity of CytCox [50]. This presumably increases respiration and energetics and protects the cell from ROS caused by electron leakage at complex I and III.
Another cardiac signaling complex which PKCε interacts with is Akt-eNOS. Upon activation of either PKCε or Akt (Protein Kinase B), the association of the three proteins is enhanced and eNOS (endogenous Nitric Oxide Synthase) and Akt activity are reported to be activated directly by PKCε [51]. However, it was also reported that, in cardiovascular endothelial cells, PKCε inhibits eNOS activity [52]. In addition, PKC isoforms α,δ, and ε were overexpressed in keratinocytes and both PKCδ and ε were found to be potent inhibitors of Akt [53]. While it is not clear whether activation or inhibition occurs, it is clear that this provides a direct link between Akt, PKCε, and eNOS. Because NO is a direct activator of PKCε, its association with eNOS demonstrates a feedback loop in which PKCε activity can be modulated. Aside from this, it seems there is also a control point at CytCox where O2, NO, and PKCε respond to the cellular levels of oxygen and regulate respiration accordingly. In transgenic mice that express constitutively active PKCε (aPKCε), aPKCε induces augmentation of mitochondrial respiration. Specifically, an increase in adenosine nucleotide translocase activity, a decrease in cytochrome c release, and stabilization of the inner mitochondrial membrane potential were attributed to increased activity of PKCε and correlated with cell survival after UV induced apoptosis [54]. Another mitochondrial target for PKCε is Bcl-2 associated death domain protein (BAD). PKCε plays an anti-apoptotic role under some conditions by inhibition of BAD [55].
Besides it's role in mitochondrial protection, PKCε interacts with gap junctions in heart tissue. In heart, functional Cx43 gap junctions are essential for conductance of electrical impulses and stabilization of synchrony. In attempts to identify which kinases phosphorylate Cx43, it was shown that both PKCα and PKCε phosphorylate Cx43 at multiple serines (S365, S368, S369, S372, and S373) in the C-terminus of Cx43 [56,57]. The antiarrhythmic peptide, Rotigaptide [97, see Fig.4], postponed dephosphorylation of Ser297 and Ser368 [58]. Dephosphorylation of these residues was correlated with an extended time to asystole following 30 minutes of ischemia due to preservation of conductance currents through gap junctions. (see Fig. 2 for phosphorylation sites) In contrast, phosphorylation of Ser368, in cell culture, is associated with decreased cell coupling. This inconsistency is likely due to the nature of cell lines. Regardless, it is clear that Ser368 represents an important regulatory site for cardiac PKCε. Another PKCε phosphorylation site on Cx43 is Ser262. Phosphorylation of Ser262 leads to reversal of the marked decrease in DNA synthesis in cardiac myocytes associated with over-expression of Cx43 [59]. Recently, it became evident that Cx43 is also present in heart mitochondria and that preconditioning against ischemia, by treatment of heart tissue with diazoxide, depends on Cx 43 localization to the inner mitochondrial membrane [60]. Signals propagated through gap junctions are implicated in the induction of apoptosis, differentiation, metabolism, preconditioning against ischemia, formation of cataracts and other physiological processes. Like Cytcox, gap junctions may also be control points regulated by PKCε.
Figure 4.

Structure of Rotigaptide from [97].
Figure 2.

Sequence of Cx43-C-Terminus.
PKCγ and PKCε in neural tissues
PKCγ, is not expressed in the heart, but is widely expressed in the brain and eye tissues such as retina and lens. These tissues contain both PKCγ and PKCε which have been implicated in protective roles against stroke and neural ischemia [61]. During and after middle cerebral artery occlusion, PKCγ migrates to synaptic membranes where it possibly modulates neurotransmission and cell survival [62]. Studies on primary neurons in rats show that insulin could mediate inhibition of necrotic cell death following ischemia/reperfusion injury. Furthermore, by decreasing PKCγ expression with antisense oligonucleotides, the insulin induced protection from insult was abolished [63]. For a comprehensive review of PKCs in cerebral ischemia, see [61,64].
Mutations in the PKCγ C1B domain have been implicated in the movement and cognitive disorder spinocellebellar ataxia type 14 (SCA14) [65]. Because it has been demonstrated that these mutations occur in the C1B region of PKCγ, it is assumed that this would prevent activation by oxidative signals [66]. When expressed in cells in culture, these mutations cause ER stress, induce apoptosis, and have a dominant effect upon endogenous PKCγ [96]. The mutant enzymes were not activated by oxidative stress and this resulted in a failure to control gap junctions. Caspase-3 linked apoptosis in these cells was prevented by a gap junction inhibitor [96]. This clearly demonstrates that there is a direct link between the ability to oxidatively activate PKCγ and control gap junctions.
Another mutation in PKCγ has been associated with retinitis pigmentosa and occurs at the very C-terminus of the protein [67]. In studies using chinese hampster ovary (CHO) cell culture, the R659S mutant induced cell death more readily than did the wildtype kinase [68]. PKCγ is also suspected of mediating ER stress dependent neuronal cell death. In mouse neuroblastoma cell viability assays, in the presence of tunicamycin, it was discovered that dilinoleoyl-phophatidylethanolamine (DLPE) has a neuroprotective effect. Based on structural analysis of DLPE, it was hypothesized and then demonstrated that DLPE activates both PKCγ as well as PKCε [19].
PKCγ is also expressed at relatively high levels in the cerebellum. In primary cerebellar neurons, PKCγ directly phosphorylates Ser890 of the N-methyl-D-aspartate receptor (NMDAR). Activation of type I metabotropic glutamate receptors (mGluR) which leads to phosphorylation of NMDAR with 3,5-dihyidroxy-phenylglycine (DHGP) activates PKCγ [69]. PKCγ therefore lies downstream of mGluR and upstream of NMDAR. It has been reported recently that PKCγ activation via iGluR (i for inducible) and mGluR requires both channels in order to get NMDAR activation, implying that both a lipid and calcium signal are necessary in this case [70].
Other cases in which PKCγ is important for neural function are related to sensitivity to alcohol, opiates, and pain. Pain sensitivity after alcohol administration is associated with altered levels of PKCγ in the cytoplasm of rat motor neurons [71]. In related work, withdrawal-induced hyperalgesia was reversibly inhibited with PKCε oligodeoxynucleotide antisense [72]. Morphine tolerance studies in rats show that PKCγ expression increases substantially after eight days of morphine treatment [73]. In more recent work, it was demonstrated that PKCγ contributes to the induction of tolerance to morphine. PKCγ knock out mice had similar nocioceptive processing regardless of morphine treatment [74]. In other work, PKCγ knock out mice treated with morphine did not exhibit spinal cord astroglial hypertrophy or proliferation which is normally associated with development of morphine tolerance [75].
Roles for PKCε in protecting neurons from stroke and ischemia are also well documented. PKCε is broadly expressed in neural tissues, retina, and lens and has been implicated in many signaling pathways in these tissues. Under conditions of glucose/oxygen starvation PKCε activator peptides reduced cortical neuron and astrocyte injury after ischemia/reperfusion while specific inhibition of PKCε abolished this effect [76]. When ischemia is chemically induced, transductional levels of PKCε increase dramatically after ischemia and these levels are maintained or even potentiated during one hour of ischemic reperfusion [77]. In l-buthionine-S,R-sulfoximine (BSO) treated neurons, a burst of ROS leads to neuronal cell death. In this case, inhibition of PKCε correlated with neuron survival rather than cell death [78]. In a rodent stroke model, moderate hypothermia was reported to preserve PKCε activity [79]. An observed loss of PKCε during ischemia was not dependent on caspase-3 activation but was prevented by hypothermia by an unknown mechanism. In another hypothermia study in rat hearts, N(2-Mercaptopropionyl) glycine or PKCε inhibitors abolished the protective effects of temperature preconditioning, demonstrating a primary role for ROS [80]. Consistent with this, ROS illicits cardioprotection in wildtype mouse hearts but is lost in PKCε −/−mice[39].
Activation of PKCε mediated pathways during ischemic injury involves extracellular signal-regulated kinases (ERKs). Both activation of N-methyl-D-Aspartate receptors (NMDAR) and glucose/oxygen deprivation induces neuronal protection in mice. This protective effect is dependent on activation of upstream NMDA receptors (NMDAR) and downstream ERKs. Specific inhibition of PKCε or ERKs results in loss of neuroprotection [84]. In neocortical explants, activation of PKCε mimics estrogen induced ERK activation. As reported, inhibition of PKCε diminished estrogen mediated ERK phosphorylation [85]. These results directly link PKCε mediated signal transduction to ERKs, which are important for neural protection, stress sensing, cell growth, and differentiation. PKCε activity has also been shown to be critical for the process of neural growth cone collapse [86], as well as neurite outgrowth [13].
In mouse hippocampal neurons, PKCε reduces Na+ channel current by modulating phosphorylation sites on Na+ channel subunits after stimulation of muscarinic acetycholine receptors, possibly by a direct interaction. Furthermore, PKCε knock out mice do not display a reduction in sodium channel currents in response to PKCε activation, indicating that, in mouse hippocampal neurons, PKCε is the PKC isozyme responsible for voltage gated sodium channel modulation [87]. Altered regulation of these channels is associated with many neurological disorders such as epilepsy. Interestingly, PKCε modulates the activity of the anticonvulsant, topiramate in rat neurons treated with tetrodotoxin. Topiramate activity is dependent on the phosphorylation state of the sodium channel, indicating that PKCε phosphorylation of the channel blocks the hyperpolarizing effects of topiramate [88]. Aside from Na+ channels, it is also reported that amyloid beta peptide directly inhibits PKCε phosphorylation and translocation to membranes [89].
Both PKCγ and PKCε, interact with the same gap junction protein,Cx43, in neuronal tissues. In astrocytes, Cx43 is widely expressed, and mutations in the Cx43 primary structure are associated with the development of oculodentodigital dysplasia, a rare but devastating abnormality of craniofacial and limb growth [90]. In lens tissues, PKCγ has been implicated in the disassembly of connexin 46/50 gap junctions and this communication pathway is critical for lens homeostasis, especially the communication between mature fiber cells and differentiating fiber cells [91]. PKCγ directly phosphorylates Cx46 and phosphorylates other connexins in the lens, such as Cx50 and Cx43 [92]. There is one report of PKCε in the lens [93], and this work demonstrates that PKCε is not activated by the well known PKCγ activator, 12-O-tetradecanoylphorbol-13-acetate (TPA). Since both PKCγ and PKCε act on Cx43, a future area of research will be to determine the effects of having both PKC's in the same cell.
Why two stress sensor kinases in neural tissues?
Unlike the oxygen rich environment of the heart where only PKCε is found, both PKCγ and PKCε are expressed in brain. Brain tissue is highly susceptible to decreased levels of oxygen. Unlike cardiac myocytes which may survive 20-40 minutes of ischemia, neurons begin dying during cerebral ischemia in minutes. Damaged areas are characterized by necrosis as a result of depolarization and the resulting influx of ions leads to rapid, immediate increases in infarct size. The spread of damage is through gap junctions [94, 95]. Neural tissues most likely contain both stress-sensing kinases in order to protect against this damage. Under conditions of hypoxia which activates PKCε, the PKCε could translocate to the mitochondria and this may help prevent mitochondrial-induced apoptosis from occurring. The PKCγ may then become activated by rising ROS and translocate to the plasma membrane to phosphorylate and inhibit gap junctions. This could help to prevent the spread of apoptotic signals to adjacent neurons by the “Bystander Effect.” Prolonged ischemia would overwhelm this protection mechanism but it could provide extra protection under normal oxidative stress. (Fig.3)
Figure 3.
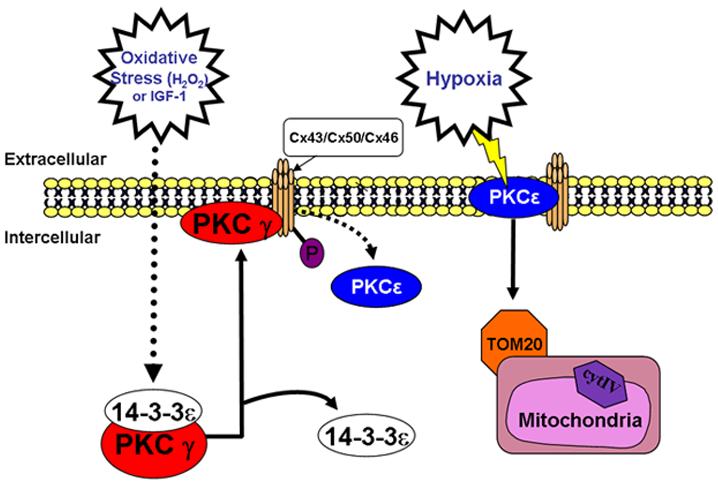
Model of Functions of PKCγ and PKCε During Oxidative and Hypoxic Stress.
In heart where PKCε helps to maintain electrical conductance via modulation of Cx43, PKCγ does not exist. In neural tissues which express both isoforms, PKCγ appears to be the primary regulator of gap junctions after oxidative stress. It could be that in these tissues, PKCγ displaces PKCε from gap junctions as a means of protection that is not required in more resilient tissues such as heart. While metabolic dependence on glucose is an important factor in neural ischemia, it is thought that the brain's intercellular communication pathways play a major role in causing ischemic injury [94,95]. Although knockout mouse models exist for both PKCγ and PKCε a double knockout does not exist and this would be an ideal model to determine the requirements for these two stress sensing kinases.
Conclusions
Both PKCγ and PKCε have open and easily activated C1 domains which allow them to become activated by oxidative signals and ROS, without a DAG or calcium signal. These PKC isoforms play a key role in the control of both mitochondria and gap junctions during ischemic stress. This stress control appears to involve, mainly, PKCε in heart, but the additional PKCγ in neural tissues. It is not certain if both PKCs control the Cx43 gap junction protein. However, the use of the available knockout models together with structural information and studies of mutants should provide the necessary tools to determine how each of these PKC isoforms function, when alone or together, to provide protection from ischemia. This information will be critical to the development of drugs to treat stroke and cardiac arrhythmias.
Acknowledgements
This review was made possible by NIH grant number P20-RR016475 (to D. Madgwick) from the INBRE Program of the National Center for Research Resources, and NIH grant number RO1-EY13421 (to D. Takemoto).
List of Abbreviations
- PKC
protein kinase C
- Cx43
connexin 43
- C1B
region of diacylglycerol-binding domain of PKC
- ROS
reactive oxygen species
- Cx46
connexin 46
- Cx50
connexin 50
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanks SK, Hunter T. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 2.Balendran A, Hare GR, Kieloch A, Williams MR, Alessi DR. FEBS Lett. 2000;484:217–223. doi: 10.1016/s0014-5793(00)02162-1. [DOI] [PubMed] [Google Scholar]
- 3.Newton AC. Biochem. J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parekh DB, Ziegler W, Parker PJ. EMBO J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graumann J, Lilie H, Tang X, Tucker KA, Hoffmann JH, Vijayalakshmi J, Saper M, Bardwell JC, Jakob U. Structure. 2001;9:377–387. doi: 10.1016/s0969-2126(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 6.Giorgione JR, Lin JH, McCammon JA, Newton AC. J. Biol. Chem. 2006;281:1660–1669. doi: 10.1074/jbc.M510251200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leithe E, Cruciani V, Sanner T, Mikalsen SO, Rivedal E. Carcinogenesis. 2003;24:1239–1245. doi: 10.1093/carcin/bgg066. [DOI] [PubMed] [Google Scholar]
- 8.Sonnenburg ED, Gao T, Newton AC. J. Biol. Chem. 2001;276:45289–45297. doi: 10.1074/jbc.M107416200. [DOI] [PubMed] [Google Scholar]
- 9.Edwards AS, Faux MC, Scott JD, Newton AC. J. Biol. Chem. 1999;274:6461–6468. doi: 10.1074/jbc.274.10.6461. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen TA, Takemoto LJ, Takemoto DJ. J. Biol. Chem. 2004;279:52714–52725. doi: 10.1074/jbc.M403040200. [DOI] [PubMed] [Google Scholar]
- 11.Spitaler M, Cantrell DA. Nat. Immunol. 2004;5:785–790. doi: 10.1038/ni1097. [DOI] [PubMed] [Google Scholar]
- 12.Schultz A, Ling M, Larsson C. J. Biol. Chem. 2004;279:31750–31760. doi: 10.1074/jbc.M313017200. [DOI] [PubMed] [Google Scholar]
- 13.Zeidman R, Troller U, Raghunath A, Pahlman S, Larsson C. Mol. Biol. Cell. 2002;13:12–24. doi: 10.1091/mbc.01-04-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH. Mol. Cell Biol. 1999;19:8547–8558. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eitel K, Staiger H, Rieger J, Mischak H, Brandhorst H, Brendel MD, Bretzel RG, Haring HU, Kellerer M. Diabetes. 2003;52:991–997. doi: 10.2337/diabetes.52.4.991. [DOI] [PubMed] [Google Scholar]
- 16.Jaken S, Parker PJ. Bioessays. 2000;22:245–254. doi: 10.1002/(SICI)1521-1878(200003)22:3<245::AID-BIES6>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 17.Preiss S, Namgaladze D, Brune B. Cardiovasc. Res. 2007;73:833–840. doi: 10.1016/j.cardiores.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 18.Kanno T, Yamamoto H, Yaguchi T, Hi R, Mukasa T, Fujikawa H, Nagata T, Yamamoto S, Tanaka A, Nishizaki T. J. Lipid Res. 2006;47:1146–1156. doi: 10.1194/jlr.M500329-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Nagai K, Chiba A, Nishino T, Kubota T, Kawagishi H. J. Nutr. Biochem. 2006;17:525–530. doi: 10.1016/j.jnutbio.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Kumazawa T, Hiwasa T, Takiguchi M, Suzuki O, Sato K. Int. J. Mol. Med. 2007;19:765–770. [PubMed] [Google Scholar]
- 21.Burgos M, Calvo S, Molina F, Vaquero CF, Samarel A, Llopis J, Tranque P. Eur. J. Neurosci. 2007;25:1069–1078. doi: 10.1111/j.1460-9568.2007.05364.x. [DOI] [PubMed] [Google Scholar]
- 22.McGettrick AF, Brint EK, Palsson-McDermott EM, Rowe DC, Golenbock DT, Gay NJ, Fitzgerald KA, O'Neill LA. Proc. Natl. Acad. Sci. U. S. A. 2006;103:9196–9201. doi: 10.1073/pnas.0600462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meier M, Menne J, Park JK, Holtz M, Gueler F, Kirsch T, Schiffer M, Mengel M, Lindschau C, Leitges M, Haller H. J. Am. Soc. Nephrol. 2007;18:1190–1198. doi: 10.1681/ASN.2005070694. [DOI] [PubMed] [Google Scholar]
- 24.Lu D, Huang J, Basu A. J. Biol. Chem. 2006;281:22799–22807. doi: 10.1074/jbc.M603390200. [DOI] [PubMed] [Google Scholar]
- 25.Pardo OE, Wellbrock C, Khanzada UK, Aubert M, Arozarena I, Davidson S, Bowen F, Parker PJ, Filonenko VV, Gout IT, Sebire N, Marais R, Downward J, Seckl MJ. EMBO J. 2006;25:3078–3088. doi: 10.1038/sj.emboj.7601198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaughnessy LM, Lipp P, Lee KD, Swanson JA. Cell Microbiol. 2007 doi: 10.1111/j.1462-5822.2007.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steinhart R, Kazimirsky G, Okhrimenko H, Ben-Hur T, Brodie C. Glia. 2007;55:224–232. doi: 10.1002/glia.20454. [DOI] [PubMed] [Google Scholar]
- 28.Rathore R, Zheng YM, Li XQ, Wang QS, Liu QH, Ginnan R, Singer HA, Ho YS, Wang YX. Biochem. Biophys. Res. Commun. 2006;351:784–790. doi: 10.1016/j.bbrc.2006.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MY, Kim MJ, Yoon IS, Ahn JH, Lee SH, Baik EJ, Moon CH, Jung YS. Br. J. Pharmacol. 2006;149:1059–1070. doi: 10.1038/sj.bjp.0706922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds NJ, Baldassare JJ, Henderson PA, Shuler JL, Ballas LM, Burns DJ, Moomaw CR, Fisher GJ. J. Invest Dermatol. 1994;103:364–369. doi: 10.1111/1523-1747.ep12394957. [DOI] [PubMed] [Google Scholar]
- 31.Fleegal MA, Hom S, Borg LK, Davis TP. Am. J. Physiol Heart Circ. Physiol. 2005;289:H2012–H2019. doi: 10.1152/ajpheart.00495.2005. [DOI] [PubMed] [Google Scholar]
- 32.Gallegos LL, Kunkel MT, Newton AC. J. Biol. Chem. 2006;281:30947–30956. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- 33.Dries DR, Gallegos LL, Newton AC. J. Biol. Chem. 2007;282:826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 34.Stahelin RV, Digman MA, Medkova M, Ananthanarayanan B, Melowic HR, Rafter JD, Cho W. J. Biol. Chem. 2005;280:19784–19793. doi: 10.1074/jbc.M411285200. [DOI] [PubMed] [Google Scholar]
- 35.Ananthanarayanan B, Stahelin RV, Digman MA, Cho W. J. Biol. Chem. 2003;278:46886–46894. doi: 10.1074/jbc.M307853200. [DOI] [PubMed] [Google Scholar]
- 36.Colon-Gonzalez F, Kazanietz MG. Biochim. Biophys. Acta. 2006;1761:827–837. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Edmondson RD, Vondriska TM, Biederman KJ, Zhang J, Jones RC, Zheng Y, Allen DL, Xiu JX, Cardwell EM, Pisano MR, Ping P. Mol. Cell Proteomics. 2002;1:421–433. doi: 10.1074/mcp.m100036-mcp200. [DOI] [PubMed] [Google Scholar]
- 38.Ytrehus K, Liu Y, Downey JM. Am. J. Physiol. 1994;266:H1145–H1152. doi: 10.1152/ajpheart.1994.266.3.H1145. [DOI] [PubMed] [Google Scholar]
- 39.Kabir AM, Clark JE, Tanno M, Cao X, Hothersall JS, Dashnyam S, Gorog DA, Bellahcene M, Shattock MJ, Marber MS. Am. J. Physiol Heart Circ. Physiol. 2006;291:H1893–H1899. doi: 10.1152/ajpheart.00798.2005. [DOI] [PubMed] [Google Scholar]
- 40.Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL. Circulation. 2003;108:79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- 41.Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Br. J. Anaesth. 2003;91:551–565. doi: 10.1093/bja/aeg205. [DOI] [PubMed] [Google Scholar]
- 42.Awan MM, Makaula S, Forresti S, Sack MN, Opie LH. Mol. Cell Biochem. 2000;211:111–121. doi: 10.1023/a:1007143531328. [DOI] [PubMed] [Google Scholar]
- 43.Gray MO, Karliner JS, Mochly-Rosen D. J. Biol. Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- 44.Lepicier P, Bibeau-Poirier A, Lagneux C, Servant MJ, Lamontagne D. J. Pharmacol. Sci. 2006;102:155–166. doi: 10.1254/jphs.crj06011x. [DOI] [PubMed] [Google Scholar]
- 45.Joyeux M, Baxter GF, Thomas DL, Ribuot C, Yellon DM. J. Mol. Cell Cardiol. 1997;29:3311–3319. doi: 10.1006/jmcc.1997.0556. [DOI] [PubMed] [Google Scholar]
- 46.Liu H, Zhang HY, Zhu X, Shao Z, Yao Z. Am. J. Physiol Heart Circ. Physiol. 2002;282:H1380–H1386. doi: 10.1152/ajpheart.00348.2001. [DOI] [PubMed] [Google Scholar]
- 47.Costa AD, Jakob R, Costa CL, Andrukhiv K, West IC, Garlid KD. J. Biol. Chem. 2006;281:20801–20808. doi: 10.1074/jbc.M600959200. [DOI] [PubMed] [Google Scholar]
- 48.Jaburek M, Costa AD, Burton JR, Costa CL, Garlid KD. Circ. Res. 2006;99:878–883. doi: 10.1161/01.RES.0000245106.80628.d3. [DOI] [PubMed] [Google Scholar]
- 49.Mason MG, Nicholls P, Wilson MT, Cooper CE. Proc. Natl. Acad. Sci. U. S. A. 2006;103:708–713. doi: 10.1073/pnas.0506562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ogbi M, Johnson JA. Biochem. J. 2006;393:191–199. doi: 10.1042/BJ20050757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Baines CP, Zong C, Cardwell EM, Wang G, Vondriska TM, Ping P. Am. J. Physiol Heart Circ. Physiol. 2005;288:H954–H961. doi: 10.1152/ajpheart.00756.2004. [DOI] [PubMed] [Google Scholar]
- 52.Michell BJ, Chen Z, Tiganis T, Stapleton D, Katsis F, Power DA, Sim AT, Kemp BE. J. Biol. Chem. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- 53.Li L, Sampat K, Hu N, Zakari J, Yuspa SH. J. Biol. Chem. 2006;281:3237–3243. doi: 10.1074/jbc.M512167200. [DOI] [PubMed] [Google Scholar]
- 54.McCarthy J, McLeod CJ, Minners J, Essop MF, Ping P, Sack MN. J. Mol. Cell Cardiol. 2005;38:697–700. doi: 10.1016/j.yjmcc.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 55.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. Circ. Res. 2002;90:390–397. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 56.Doble BW, Ping P, Fandrich RR, Cattini PA, Kardami E. Cell Commun. Adhes. 2001;8:253–256. doi: 10.3109/15419060109080733. [DOI] [PubMed] [Google Scholar]
- 57.Bowling N, Huang X, Sandusky GE, Fouts RL, Mintze K, Esterman M, Allen PD, Maddi R, McCall E, Vlahos CJ. J. Mol. Cell Cardiol. 2001;33:789–798. doi: 10.1006/jmcc.2000.1349. [DOI] [PubMed] [Google Scholar]
- 58.Axelsen LN, Stahlhut M, Mohammed S, Larsen BD, Nielsen MS, Holstein-Rathlou NH, Andersen S, Jensen ON, Hennan JK, Kjolbye AL. J. Mol. Cell Cardiol. 2006;40:790–798. doi: 10.1016/j.yjmcc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 59.Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E. J. Cell Sci. 2004;117:507–514. doi: 10.1242/jcs.00889. [DOI] [PubMed] [Google Scholar]
- 60.Rodriguez-Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, RuizMeana M, Konietzka I, Miro E, Totzeck A, Heusch G, Schulz R, Garcia-Dorado D. Circ. Res. 2006;99:93–101. doi: 10.1161/01.RES.0000230315.56904.de. [DOI] [PubMed] [Google Scholar]
- 61.Chou WH, Messing RO. Trends Cardiovasc. Med. 2005;15:47–51. doi: 10.1016/j.tcm.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 62.Matsumoto S, Shamloo M, Matsumoto E, Isshiki A, Wieloch T. J. Cereb. Blood Flow Metab. 2004;24:54–61. doi: 10.1097/01.WCB.0000095920.70924.F5. [DOI] [PubMed] [Google Scholar]
- 63.Hamabe W, Fujita R, Ueda H. J. Pharmacol. Exp. Ther. 2005;313:1027–1034. doi: 10.1124/jpet.104.082735. [DOI] [PubMed] [Google Scholar]
- 64.Bright R, Mochly-Rosen D. Stroke. 2005;36:2781–2790. doi: 10.1161/01.STR.0000189996.71237.f7. [DOI] [PubMed] [Google Scholar]
- 65.Chen DH, Brkanac Z, Verlinde CL, Tan XJ, Bylenok L, Nochlin D, Matsushita M, Lipe H, Wolff J, Fernandez M, Cimino PJ, Bird TD, Raskind WH. Am. J. Hum. Genet. 2003;72:839–849. doi: 10.1086/373883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin D, Takemoto DJ. J. Biol. Chem. 2005;280:13682–13693. doi: 10.1074/jbc.M407762200. [DOI] [PubMed] [Google Scholar]
- 67.Al-Maghtheh M, Vithana EN, Inglehearn CF, Moore T, Bird AC, Bhattacharya SS. Am. J. Hum. Genet. 1998;62:1248–1252. doi: 10.1086/301819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mochizuki H, Seki T, Adachi N, Saito N, Mishima HK, Sakai N. Neurochem. Int. 2006;49:669–675. doi: 10.1016/j.neuint.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 69.Sanchez-Perez AM, Felipo V. Neurochem. Int. 2005;47:84–91. doi: 10.1016/j.neuint.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 70.Codazzi F, Di CA, Chiulli N, Albanese A, Meyer T, Zacchetti D, Grohovaz F. J. Neurosci. 2006;26:3404–3411. doi: 10.1523/JNEUROSCI.0478-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li HF, Mochly-Rosen D, Kendig JJ. Br. J. Pharmacol. 2005;144:301–307. doi: 10.1038/sj.bjp.0706033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dina OA, Messing RO, Levine JD. Eur. J. Neurosci. 2006;24:197–204. doi: 10.1111/j.1460-9568.2006.04886.x. [DOI] [PubMed] [Google Scholar]
- 73.Mao J, Price DD, Phillips LL, Lu J, Mayer DJ. Brain Res. 1995;677:257–267. doi: 10.1016/0006-8993(95)00161-i. [DOI] [PubMed] [Google Scholar]
- 74.Zeitz KP, Malmberg AB, Gilbert H, Basbaum AI. Pain. 2001;94:245–253. doi: 10.1016/S0304-3959(01)00353-0. [DOI] [PubMed] [Google Scholar]
- 75.Narita M, Suzuki M, Narita M, Yajima Y, Suzuki R, Shioda S, Suzuki T. Eur. J. Neurosci. 2004;19:479–484. doi: 10.1111/j.0953-816x.2003.03119.x. [DOI] [PubMed] [Google Scholar]
- 76.Wang J, Bright R, Mochly-Rosen D, Giffard RG. Neuropharmacology. 2004;47:136–145. doi: 10.1016/j.neuropharm.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 77.Selvatici R, Falzarano S, Franceschetti L, Cavallini S, Marino S, Siniscalchi A. Neurochem. Int. 2006;49:729–736. doi: 10.1016/j.neuint.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 78.Jung YS, Ryu BR, Lee BK, Mook-Jung I, Kim SU, Lee SH, Baik EJ, Moon CH. Biochem. Biophys. Res. Commun. 2004;320:789–794. doi: 10.1016/j.bbrc.2004.05.217. [DOI] [PubMed] [Google Scholar]
- 79.Shimohata T, Zhao H, Steinberg GK. Stroke. 2007;38:375–380. doi: 10.1161/01.STR.0000254616.78387.ee. [DOI] [PubMed] [Google Scholar]
- 80.Khaliulin I, Clarke SJ, Lin H, Parker JE, Suleiman MS, Halestrap AP. J. Physiol. 2007 doi: 10.1113/jphysiol.2007.130369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao T, Newton AC. J. Biol. Chem. 2002;277:31585–31592. doi: 10.1074/jbc.M204335200. [DOI] [PubMed] [Google Scholar]
- 82.Coaxum SD, Griffin TM, Martin JL, Mestril R. Am. J. Physiol Heart Circ. Physiol. 2007 doi: 10.1152/ajpheart.01080.2006. [DOI] [PubMed] [Google Scholar]
- 83.Aziz MH, Manoharan HT, Verma AK. Cancer Res. 2007;67:1385–1394. doi: 10.1158/0008-5472.CAN-06-3350. [DOI] [PubMed] [Google Scholar]
- 84.Jia J, Wang X, Li H, Han S, Zu P, Li J. J. Neurosurg. Anesthesiol. 2007;19:18–24. doi: 10.1097/01.ana.0000211020.88431.e2. [DOI] [PubMed] [Google Scholar]
- 85.Setalo G, Jr., Singh M, Nethrapalli IS, Toran-Allerand CD. Neurochem. Res. 2005;30:779–790. doi: 10.1007/s11064-005-6871-y. [DOI] [PubMed] [Google Scholar]
- 86.Mikule K, Sunpaweravong S, Gatlin JC, Pfenninger KH. J. Biol. Chem. 2003;278:21168–21177. doi: 10.1074/jbc.M211828200. [DOI] [PubMed] [Google Scholar]
- 87.Chen Y, Cantrell AR, Messing RO, Scheuer T, Catterall WA. J. Neurosci. 2005;25:507–513. doi: 10.1523/JNEUROSCI.4089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Curia G, Aracri P, Colombo E, Scalmani P, Mantegazza M, Avanzini G, Franceschetti S. Br. J. Pharmacol. 2007 doi: 10.1038/sj.bjp.0707144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee W, Boo JH, Jung MW, Park SD, Kim YH, Kim SU, Mook-Jung I. Mol. Cell Neurosci. 2004;26:222–231. doi: 10.1016/j.mcn.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 90.Lai A, Le DN, Paznekas WA, Gifford WD, Jabs EW, Charles AC. J. Cell Sci. 2006;119:532–541. doi: 10.1242/jcs.02770. [DOI] [PubMed] [Google Scholar]
- 91.Zampighi GA, Planells AM, Lin D, Takemoto D. Invest Ophthalmol. Vis. Sci. 2005;46:3247–3255. doi: 10.1167/iovs.04-1504. [DOI] [PubMed] [Google Scholar]
- 92.Saleh SM, Takemoto LJ, Zoukhri D, Takemoto DJ. Mol. Vis. 2001;7:240–246. [PubMed] [Google Scholar]
- 93.Berthoud VM, Westphale EM, Grigoryeva A, Beyer EC. Invest Ophthalmol. Vis. Sci. 2000;41:850–858. [PubMed] [Google Scholar]
- 94.Saito N, Shirai Y. J. Biochem. (Tokyo) 2002;132:683–687. doi: 10.1093/oxfordjournals.jbchem.a003274. [DOI] [PubMed] [Google Scholar]
- 95.Lee JM, Grabb MC, Zipfel GJ, Choi DW. J. Clin. Invest. 2000;106:723–731. doi: 10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin D, Shanks D, Prakash O, Takemoto D. Exp.Eye. Res. 2007 doi: 10.1016/j.exer.2007.03.007. DOI:10.1016. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nattel S, Carlsson L. Nat. Rev. Drug Disc. 2006;5:1034–1049. doi: 10.1038/nrd2112. [DOI] [PubMed] [Google Scholar]
- 98.Smith A, Bowers B, Radcliffe R, Wehner J. Behavioral Genetics. 2006;36:869–881. doi: 10.1007/s10519-006-9083-6. [DOI] [PubMed] [Google Scholar]
- 99.Oehrlein SA, Parker PJ, Herget T. Biochem. J. 1996;317:219–224. doi: 10.1042/bj3170219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liyanage M, Frith D, Livneh E, Stabel S. Biochem. J. 1992;283:781–787. doi: 10.1042/bj2830781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schaap D, Parker PJ. J. Biol. Chem. 1990;265:7301–7307. [PubMed] [Google Scholar]
- 102.Marais RM, Nguyen O, Woodgett JR, Parker PJ. FEBS J. 1990;277:151–155. doi: 10.1016/0014-5793(90)80831-3. [DOI] [PubMed] [Google Scholar]