

Abstract
Overactivity of glycogen synthase kinase-3 (GSK-3) is associated with insulin resistance of skeletal muscle glucose transport in pre-diabetic and type 2 diabetic rodent models. However, limited information is available concerning the potential molecular mechanisms underlying the role GSK-3 plays in the etiology of insulin resistance in the male Zucker Diabetic Fatty (ZDF) rat, a model of type 2 diabetes. Therefore, we assessed the functionality of proximal and distal insulin signaling elements in isolated type I (slow-twitch oxidative) soleus muscles of ZDF rats following in vitro exposure to a selective GSK-3 inhibitor (1 μmol/L CT98014, Ki<10 nmol/L for GSK-3α and GSK-3β). Moreover, ser307 phosphorylation of IRS-1, which has been implicated in the development of insulin resistance, was also determined in the absence or presence of this GSK-3 inhibitor. Maximally insulin-stimulated (5 mU/mL) GSK-3β serine phosphorylation was significantly less (35%, P<0.05) in soleus muscle of ZDF rats compared to insulin-sensitive lean Zucker rats, indicating GSK-3 overactivity. In the absence of insulin, no effects of GSK-3 inhibition were detected. GSK-3 inhibition led to significant enhancement (28%) of insulin-stimulated glucose transport activity that was associated with significant upregulation of tyrosine phosphorylation of IR (52%) and IRS-1 (50%), and with enhanced Akt ser473 phosphorylation (48%) and GSK-3β ser9 phosphorylation (36%). Moreover, the selective GSK-3 inhibitor induced a significant reduction in the phosphorylation of IRS-1 ser307 (26%) and c-jun N-terminal kinase (JNK1/2) (31%), a mediator of IRS-1 ser307 phosphorylation. These results indicate that selective inhibition of GSK-3 activity in type I skeletal muscle from overtly diabetic ZDF rats enhances IRS-1-dependent insulin signaling, possibly by a decrease in JNK activation and a diminution of the deleterious effects of IRS-1 ser307 phosphorylation.
Introduction
Insulin resistance of the glucose transport process in skeletal muscle is an early and obligatory defect leading initially to the pre-diabetic state and subsequently to the development of overt type 2 diabetes. 1–3 The etiology of skeletal muscle insulin resistance can involve numerous defects in the expression and functionality of several insulin signaling factors that regulate the activation of glucose transport in this tissue (see reviews in refs. 1–3). In particular, defects in the functionality of insulin receptor substrate-1 (IRS-1), arising in many cases as a result of excessive serine phosphorylation, are linked to downstream diminution of insulin action, including the glucose transport process. 1, 4–7
Recent evidence in the literature implicates a potential role of glycogen synthase kinase-3 (GSK-3), a serine/threonine kinase that functions as a distal element of the insulin signaling cascade, in the development of skeletal muscle insulin resistance and as a potential site of intervention for improving insulin action in insulin-resistant states. 8–10 GSK-3 is overactive in conditions of obesity-associated insulin resistance in rodent skeletal muscle 11, 12 and adipose tissue 13 and in human muscle cells. 14 Interestingly, GSK-3 is known to phosphorylate IRS-1 on serine residues and impair its function in insulin signaling. 15, 16 Recent evidence from cultured cells indicates that one site of phosphorylation by GSK-3 is ser332, 16 although other sites are possible.
Importantly, selective inhibition of GSK-3 using substituted aminopyrimidine compounds increases insulin action on glucose transport activity and glycogen synthase activity in skeletal muscle from pre-diabetic obese Zucker rats, 12, 17 overtly type 2 diabetic Zucker Diabetic Fatty (ZDF) rats, 18, 19 and cultured human myocytes. 20 This class of GSK-3 inhibitors enhances post-insulin receptor insulin signaling in muscle from pre-diabetic obese Zucker rats, including IRS-1 tyrosine phosphorylation, IRS-1 associated with the p85 regulatory subunit of PI3-kinase, Akt serine phosphorylation, and GSK-3β serine phosphorylation. 12 However, it is presently unknown whether this regulatory mechanism for this class of GSK-3 inhibitors also functions in conditions of type 2 diabetes.
In the context of the foregoing information, the primary purpose of the present investigation was to determine if selective pharmacological inhibition of GSK-3 in severely insulin-resistant type I (slow-twitch oxidative) skeletal muscle of overtly type 2 diabetic male ZDF rats, which improves insulin action on glucose transport activity, 18, 19 is associated with increases in IRS-1-dependent insulin signaling. An additional purpose of this study was to assess whether this inhibition of GSK-3 in type 2 diabetic skeletal muscle causes a diminution of the phosphorylation of ser307, as increased ser307 phosphorylation has been linked with insulin resistance in a variety of metabolic conditions (see ref. 7).
MATERIALS AND METHODS
Animals
Male Zucker Diabetic Fatty (ZDF/GmiCrl-fa/fa) rats and lean Zucker (Fa/-) rats were purchased from Charles River Laboratories (Wilmington, MA) at the age of 10–11 wk and used in the experiments at 12 wk of age. At the time of their use, the obese Zucker rats weighed 300–350 g, whereas the age-matched lean Zucker rats weighed 180–220 g. We have previously documented that fasting plasma glucose levels in male ZDF rats at this age are typically between 300 and 350 mg/dl.18 All animals were housed in a temperature-controlled room (20–22°C) with a 12:12-h light-dark cycle (lights on from 7AM to 7 PM) at the Central Animal Facility of the University of Arizona. Lean rats had free access to regular lab chow (Teklad, Madison, WI), whereas the ZDF animals were provided a diabetogenic chow (Purina 5008, St. Louis, MO). All procedures were approved by the University of Arizona Animal Use and Care Committee.
GSK-3 inhibitor and in vitro treatments of skeletal muscle
The selective GSK-3 inhibitor CT98014 (kindly provided by Dr. Stephen D. Harrison, Chiron Corporation, Emeryville, CA) has been used previously by our research group 18, 19 and others. 20, 21 CT98014 inhibits both GSK-3α and GSK-3β with Ki values less than 10 nmol/L in an ATP-competitive manner. 9. 10 The compound was >95% pure by HPLC and was in free-base form diluted from a dimethylsulfoxide (DMSO) stock solution. The final DMSO concentration did not exceed 0.5%.
After an overnight food restriction (chow was restricted to 4 g at 5 pm and was consumed immediately), animals were deeply anesthetized at 8 am with an intraperitoneal injection of pentobarbital sodium (50 mg/kg), and strips of soleus muscles (~25–35 mg) were prepared for in vitro incubation in the unmounted state without tension. Each soleus strip was incubated for 60 min at 37°C in 3 mL oxygenated (95% O2-5% CO2) Krebs-Henseleit buffer (KHB) with the NaHCO3 concentration set at 14 mmol/L. This KHB was supplemented with 8 mmol/L glucose, 32 mmol/L mannitol, 0.1% BSA (radioimmunoassay grade, Sigma Chemical), 0.5% DMSO, and the indicated additions of GSK-3 inhibitor or insulin. Thereafter, the muscles were either blotted on filter paper and frozen in liquid nitrogen for the subsequent determination of insulin signaling functionality or were used for assessment of glucose transport activity.
Assessment of glucose transport activity
After the initial 60-min incubation period, the muscles were rinsed for 10 minutes at 37°C in 3 mL of oxygenated KHB containing 40 mmol/L mannitol, 0.1% BSA, GSK-3 inhibitor, and insulin, if present previously. Following the rinse period, the muscles were transferred to 2 mL of KHB containing 1 mmol/L 2-deoxy-[1,2-3H]glucose (2-DG) (300 μCi/mmol; Sigma Chemical), 39 mmol/L [U-14C]mannitol (0.8 μCi/mmol; ICN Radiochemicals, Irvine, CA), 0.1% BSA, GSK-3 inhibitor, and insulin, if previously present. At the end of this final 20-min incubation period at 37°C, the muscles were removed, trimmed of excess fat and connective tissue, quickly frozen between aluminum blocks cooled with liquid nitrogen, weighed, and finally placed in 0.5 mL of 0.5 mmol/L NaOH. After the muscles were completely solubilized, 5 mL of scintillation cocktail were added, and the specific intracellular accumulation of 2-[3H]DG was determined as described previously. 22 This method for assessing glucose transport activity in isolated muscle has been validated. 23
Assessment of insulin signaling factor functionality
Muscles were homogenized in 8 vol of ice-cold lysis buffer (50 mmol/L HEPES, 150 mmol/L NaCl, 20 mmol/L Na pyrophosphate, 20 mmol/L β-glycerophosphate, 10 mmol/L NaF, 2 mmol/L Na3VO4, 2 mmol/L EDTA, 1% Triton X-100, 10% glycerol, 1 mmol/L MgCl2, 1 mmol/L CaCl2, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 0.5 μg/mL pepstatin, and 2 mmol/L PMSF). Homogenates were incubated on ice for 20 min and then centrifuged at 13,000 X g for 20 minutes at 4°C. Total protein concentration was determined using the BCA method (Sigma Chemical). For determination of Akt, GSK-3, AMPK, and p38 MAPK phosphorylation, samples containing equal amounts of total protein were separated by SDS-PAGE on 7.5% or 12% polyacrylamide gels (Bio-Rad Laboratories, Hercules, CA) and transferred to nitrocellulose membranes. Membranes were incubated overnight with antibodies against phospho-Akt ser473, phospho-GSK-3α/β ser21/9, phospho-AMPKα thr172, phospho-p38 MAPK thr180/tyr182, phospho-c-jun N-terminal kinase (JNK1/2), or phospho inhibitor kB kinase (IKKβ) (Cell Signaling, Beverly, MA), or with antibodies against total Akt1/2, GSK-3α/β, AMPKα (all Cell Signaling), p38 MAPK (Santa Cruz Biotechnology, Santa Cruz, CA), JNK and IKKβ (Cell Signaling). In our hands, ser21 phosphorylation of GSK-3α in muscle from ZDF rats is very low (Teachey, M. K., and E.J. Henriksen, unpublished data), and all GSK-3 data in this study are therefore restricted to GSK-3β ser9 phosphorylation. Subsequently, membranes were incubated with secondary goat anti-rabbit antibody conjugated with horseradish peroxidase (Chemicon, Temecula, CA). The proteins were visualized on Kodak X-Omat AR film (Kodak, Rochester, NY) using an enhanced chemiluminescence detection system (GE Healthcare, Piscataway, NJ). The band intensities on the autoradiographs were quantified with a Bio-Rad Model GS-800 imaging densitometer using Bio-Rad Quantity One software. The level of the phosphorylated signaling element was expressed relative to the total amount of that protein from the same sample.
For measurement of tyrosine-phosphorylated IR-β (IR/pY) and IRS-1 (IRS-1/pY), immunoprecipitations and subsequent immunoblotting were performed. Muscles were homogenized in 1 mL of ice-cold lysis buffer and total protein concentration was determined as above. Samples were diluted to 2 mg/ml. For assessment of IR/pY, 0.5 mL of diluted homogenate was immunoprecipitated with 15 μL of recombinant agarose-conjugated anti-phosphotyrosine antibody (4G10, Upstate Biotechnology, Lake Placid, NY). For analysis of IRS-1/pY, 0.5 mL of diluted homogenate was immunoprecipitated with 25 μL of agarose-conjugated anti-IRS-1 antibody (Upstate). After an overnight incubation at 4°C, samples were pulse-centrifuged and the supernatant was removed. The agarose beads were washed three times with ice-cold PBS, mixed with SDS sample buffer, and boiled for 5 min. Equal amounts of the protein of interest were separated by SDS-PAGE on 7.5% polyacrylamide gels and transferred to nitrocellulose membranes. For detection of IR/pY, membranes were incubated with the appropriate dilution of an antibody against insulin receptor β-subunit (Upstate). For analysis of IRS-1/pY, the nitrocellulose membrane was incubated in anti-phosphotyrosine antibody (PY99, Santa Cruz). For assessment of ser307 phosphorylation of IRS-1, the nitrocellulose membrane was incubated with antibody against IRS-1 phosphorylated on the ser307 residue. Thereafter, the membranes were incubated with secondary goat anti-mouse antibody conjugated with horseradish peroxidase (Santa Cruz). Protein bands of interest were exposed, visualized, and quantified as described above.
Statistical analysis
All values are expressed as means ± SE. All experiments were done with paired muscles from the same animal incubated with or without the GSK-3 inhibitor. Therefore, differences between the two groups due to the GSK-3 inhibitor alone (in the absence or presence of insulin) were analyzed using a paired Student’s t-test. A level of P<0.05 was set for statistical significance.
RESULTS
GSK-3 serine phosphorylation in soleus muscle from lean Zucker and ZDF rats
In order to assess the activity state of GSK-3 in skeletal muscle of type 2 diabetic ZDF rats, GSK-3β ser9 phosphorylation, which is inversely related to GSK-3 activity, was measured (Fig. 1). In the absence of insulin, GSK-3β phosphorylation was 36% less (P<0.05) in the ZDF soleus compared to the lean Zucker soleus. Likewise, insulin-stimulated (5 mU/mL) GSK-3β phosphorylation was 35% less in the ZDF soleus compared to the soleus from lean Zucker muscle. Protein expression of GSK-3β in soleus was not different between lean and ZDF groups (data not shown). These results indicate that GSK-3β activity is greater in skeletal muscle from the type 2 diabetic ZDF. In addition, insulin-stimulated ser473 phosphorylation of Akt, the immediate upstream kinase from GSK-3, was also significantly less (45%, P<0.05) in ZDF soleus compared to lean soleus (data not shown), consistent with the insulin-resistant state of the ZDF muscle.
Figure 1.
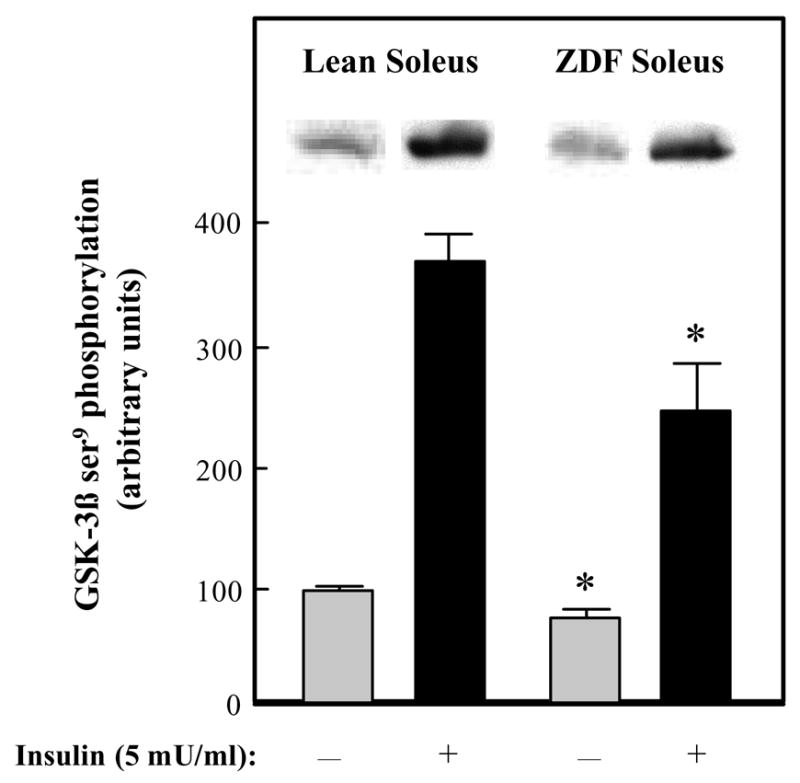
Basal and insulin-stimulated GSK-3β ser9 phosphorylation in skeletal muscle of lean Zucker and Zucker Diabetic Fatty rats. Soleus strips were incubated without (
 ) or with 5 mU/ml insulin (■) for 60 min. Assessment of the phosphorylation of ser9 on GSK-3β was then performed as described in Materials and Methods. The mean value in the lean basal grouo was arbitrarily set at 100%. Representative autoradiographic bands are shown. Values are means ± SE for 4–6 muscles per group. *P<0.05 ZDF vs. lean group of the same incubation condition.
) or with 5 mU/ml insulin (■) for 60 min. Assessment of the phosphorylation of ser9 on GSK-3β was then performed as described in Materials and Methods. The mean value in the lean basal grouo was arbitrarily set at 100%. Representative autoradiographic bands are shown. Values are means ± SE for 4–6 muscles per group. *P<0.05 ZDF vs. lean group of the same incubation condition.
Effects of GSK-3 inhibition on glucose transport activity
In the absence of insulin, incubation of soleus muscle with the selective GSK-3 inhibitor CT98014 (1 μmol/L) had no effect on glucose transport activity, as assessed by 2-deoxyglucose uptake (Fig. 2). In contrast, GSK-3 inhibition enhanced the effect of 5 mU/mL insulin on soleus glucose transport activity by 28% (P<0.05). This is in agreement with our previous in vitro findings using this selective GSK-3 inhibitor with type I skeletal muscle from male ZDF rats 18, 19 and with type I skeletal muscle from pre-diabetic obese Zucker rats. 12 The rate of insulin-stimulated glucose transport activity in the soleus from age-matched male lean Zucker rats is typically ~800 pmol/mg/min, 18, 19 so the GSK-3 inhibitor overcomes about 40% of the in vitro insulin resistance observed in type 2 diabetic soleus muscle.
Figure 2.
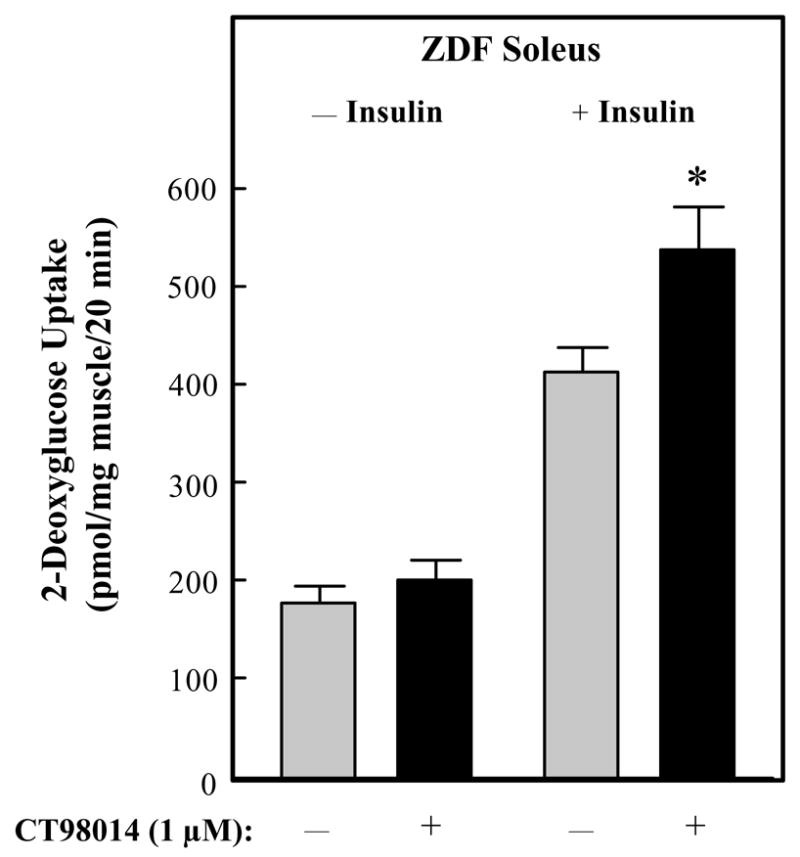
Effects of GSK-3 inhibitor CT98014 on basal or insulin-stimulated glucose transport activity in skeletal muscle of Zucker Diabetic Fatty rats. Soleus muscle strips were incubated without (
) or with CT9804 (1 μM) (■) in the absence or presence of insulin (5 mU/ml) for 60 min. 2-Deoxyglucose uptake was then performed as described in Materials and Methods. Values are means ± SE for 4–6 muscles per group. *P<0.05 vs. paired muscle group in the absence of GSK-3 inhibitor.
Effects of GSK-3 inhibition on insulin signaling functionality
The effects of selective GSK-3 inhibition on insulin signaling was initially assessed in upstream aspects of this signaling cascade in soleus muscle of male ZDF rats. In the absence of insulin, CT98014 did not alter tyrosine phosphorylation of the IR ßsubunit (Fig. 3). However, insulin-stimulated IR β-subunit tyrosine phosphorylation was increased by 52% (P<0.05). Likewise, basal tyrosine phosphorylation of IRS-1 was not affected by the GSK-3 inhibitor, whereas insulin-stimulated IRS-1 tyrosine phosphorylation was enhanced by 50% (P<0.05) in the presence of CT98014 (Fig. 4). The phosphorylation of IRS-1 specifically on ser307 was assessed under these same conditions (Fig. 5). Basal IRS-1 ser307 phosphorylation was not significantly changed by selective GSK-3 inhibition. In contrast, this parameter was decreased by 26% (P<0.05) by CT98014 in the presence of insulin.
Figure 3.
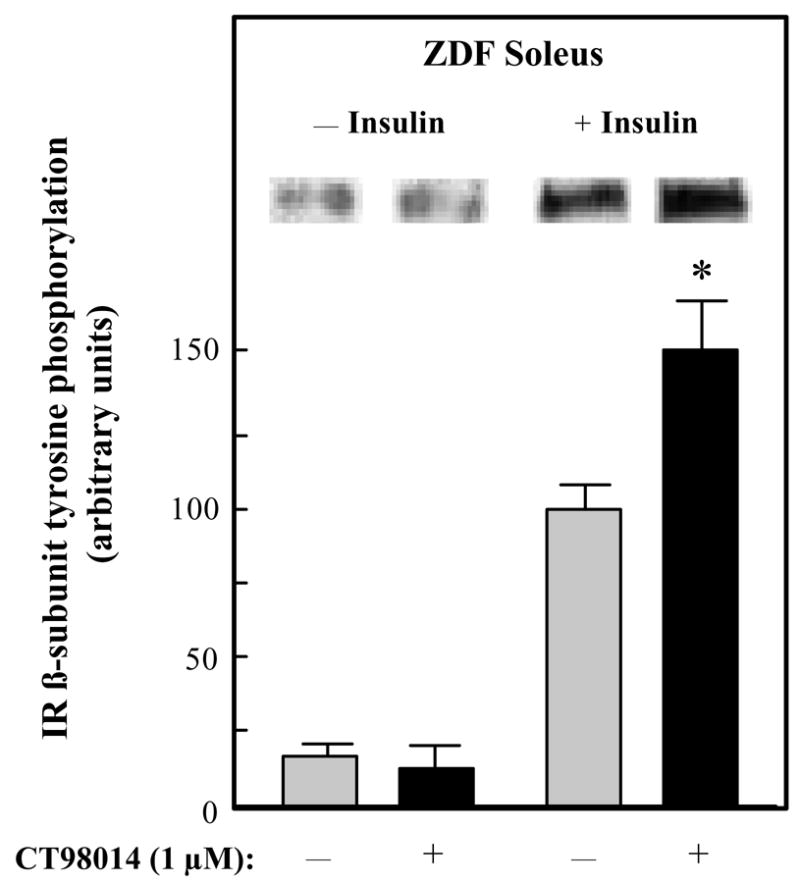
Effect of GSK-3 inhibitor CT98014 on basal or insulin-stimulated IR-β tyrosine phosphorylation in skeletal muscle of Zucker Diabetic Fatty rats. Soleus muscle strips were incubated without (
) or with CT98014 (1 μM) (■) in the absence or presence of insulin (5 mU/ml) for 60 min. Tyrosine phosphorylation of the β-subunit of IR was then performed as described in Materials and Methods. The insulin-stimulated value in the absence of CT98014 was arbitrarily set at 100%. Representative autoradiographic bands are shown. Values are means ± SE for 4–6 muscles per group. *P<0.05 vs. paired muscle group in the absence of GSK-3 inhibitor.
Figure 4.
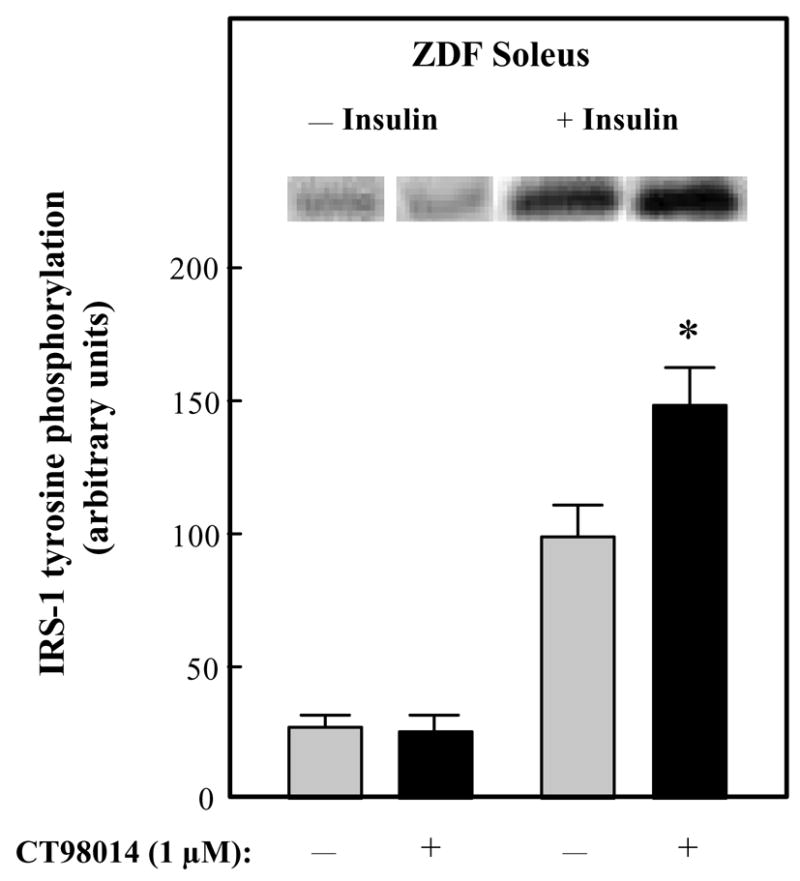
Effect of GSK-3 inhibitor CT98014 on basal or insulin-stimulated IRS-1 tyrosine phosphorylation in skeletal muscle of Zucker Diabetic Fatty rats. Soleus muscle strips were incubated without (■) or with CT98014 (1 μM) (
) in the absence or presence of insulin (5 mU/ml) for 60 min. Tyrosine phosphorylation of IRS-1 was then performed as described in Materials and Methods. The insulin-stimulated value in the absence of CT98014 was arbitrarily set at 100%. Representative autoradiographic bands are shown. Values are means ± SE for 4–6 muscles per group. *P<0.05 vs. paired muscle group in the absence of GSK-3 inhibitor.
Figure 5.
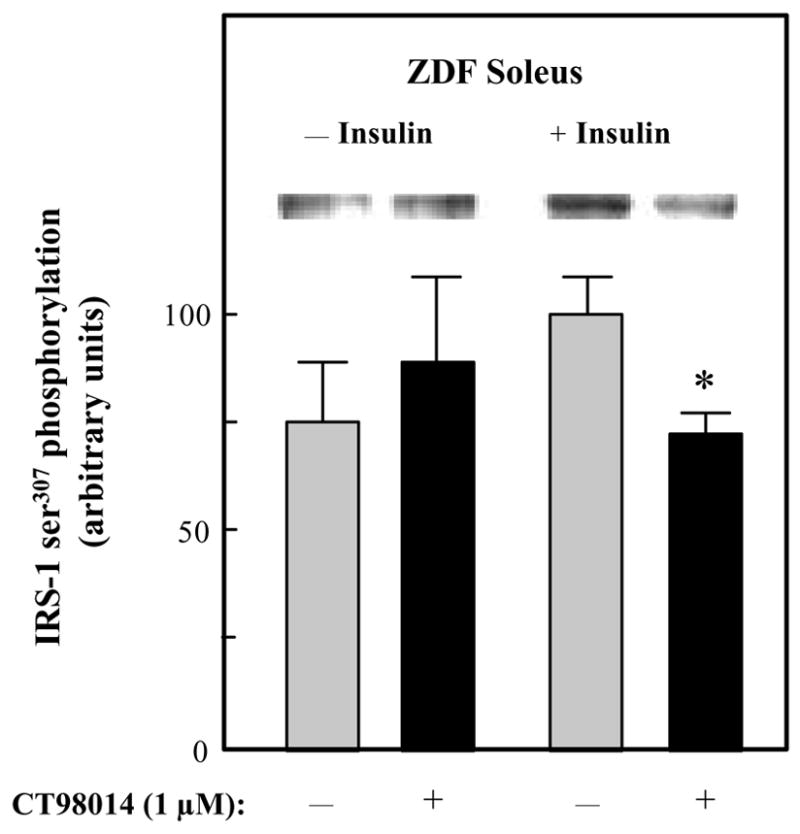
Effect of GSK-3 inhibitor CT98014 on basal or insulin-stimulated IRS-1 ser307 phosphorylation in skeletal muscle of Zucker Diabetic Fatty rats. Soleus muscle strips were incubated without (■) or with CT98014 (1 μM) (
) in the absence or presence of insulin (5 mU/ml) for 60 min. Phosphorylation of ser307 on IRS-1 was then performed as described in Materials and Methods. The insulin-stimulated value in the absence of CT98014 was arbitrarily set at 100%. Representative autoradiographic bands are shown. Values are means ± SE for 4–6 muscles per group. *P<0.05 vs. paired muscle group in the absence of GSK-3 inhibitor.
The impact of selective GSK-3 inhibition on distal elements of the insulin signaling in ZDF soleus muscle was determined next. The phosphorylation of ser473 on Akt was minimally detectable and was not altered by the GSK-3 inhibitor (Fig. 6). In contrast, insulin-stimulated Akt ser473 phosphorylation was increased by 48% (P<0.05) by CT98014. Finally, GSK-3β ser9 phosphorylation was assessed (Fig. 7). As with the other insulin signaling elements, CT98014 had no effect on this variable in the ZDF soleus. However, in the presence of insulin, this selective GSK-3 inhibitor caused a 36% (P<0.05) enhancement of GSK-3β ser9 phosphorylation.
Figure 6.
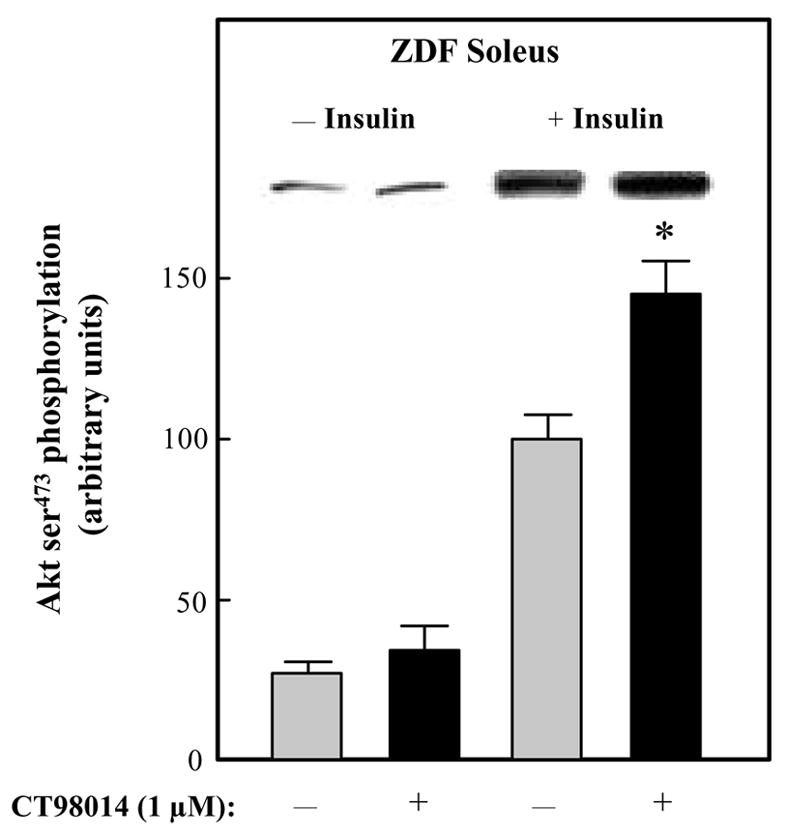
Effect of GSK-3 inhibitor CT98014 on basal or insulin-stimulated Akt ser473 phosphorylation in skeletal muscle of Zucker Diabetic Fatty rats. Soleus muscle strips were incubated without (■) or with CT98014 (1 μM) (
) in the absence or presence of insulin (5 mU/ml) for 60 min. Assessment of the Akt ser473 phosphorylation was then performed as described in Materials and Methods. The insulin-stimulated value in the absence of CT98014 was arbitrarily set at 100%. Representative autoradiographic bands are shown. Values are means ± SE for 4–6 muscles per group. *P<0.05 vs. paired muscle group in the absence of GSK-3 inhibitor.
Figure 7.
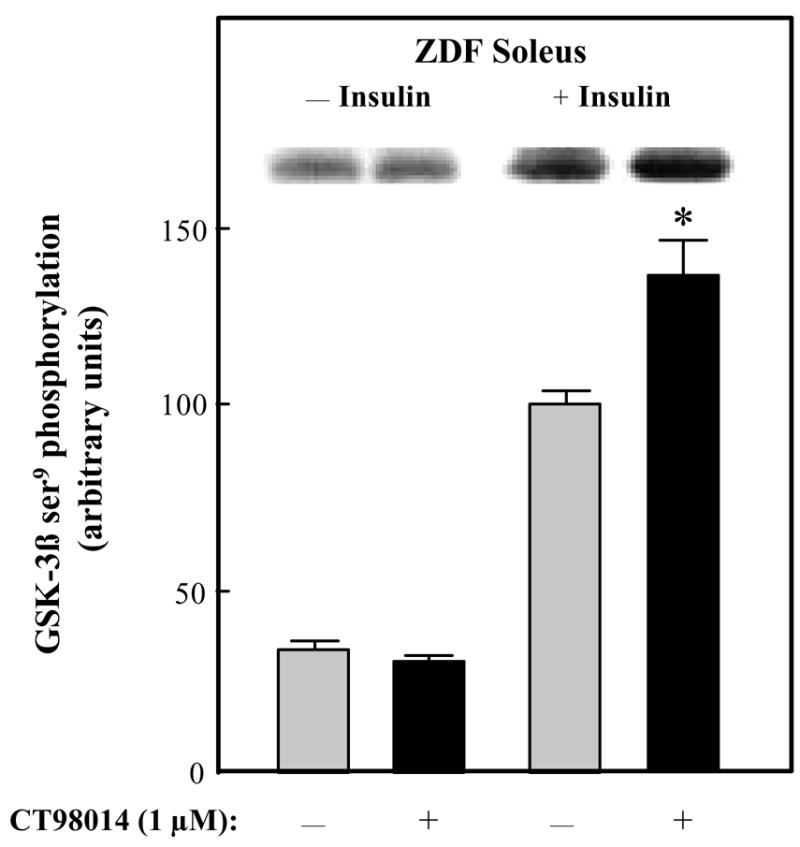
Effect of GSK-3 inhibitor CT98014 on basal or insulin-stimulated GSK-3β ser9 phosphorylation in skeletal muscle of Zucker Diabetic Fatty rats. Soleus muscle strips were incubated without (
) or with CT98014 (1 μM) (■) in the absence or presence of insulin (5 mU/ml) for 60 min. Assessment of the GSK-3β ser9 phosphorylation of was performed as described in Materials and Methods. The insulin-stimulated value in the absence of CT98014 was arbitrarily set at 100%. Representative autoradiographic bands are shown. Values are means ± SE for 4–6 muscles per group. *P<0.05 vs. paired muscle groups in the absence of GSK-3 inhibitor.
We also assessed the effect of GSK-3 inhibition on the phosphorylation of JNK and IKKβ, as these kinases are known to phosphorylate IRS-1 at ser307.5, 6, 24, 25 Whereas GSK-3 inhibition in the presence of insulin had no effect on phosphorylation of IKKβ (data not shown), it did induce a 31% decrease (P<0.05) in the phosphorylation of JNK1/2 (100 ± 14 vs. 69 ± 12 units, n=4).
No effect of the selective GSK-3 inhibitor CT98014 was observed for other signaling events that are known to impact the glucose transport system in skeletal muscle, either in the absence or in the presence of insulin, including thr172 phosphorylation of AMPKα or thr180/tyr182 phosphorylation of the p38 MAPK (data not shown).
DISCUSSION
In the present study, we report the novel finding that the acute in vitro treatment of type I soleus muscle from overtly type 2 diabetic ZDF rats with a selective inhibitor of GSK-3 (the substituted aminopyrimidine compound CT98014) induces a potentiation of insulin action on insulin signaling, including enhanced insulin-stimulated tyrosine phosphorylation of the IR β-subunit (Fig. 1) and IRS-1 (Fig. 2) and serine phosphorylation of Akt (Fig. 6) and the GSK-3β isoform (Fig. 7). This enhanced insulin signaling was associated with increased insulin action on glucose transport activity in type 2 diabetic skeletal muscle (Fig. 2), confirming findings in the ZDF rat reported previously by our research group. 18, 19 There is likely a mechanistic connection between the upregulation of IRS-1-dependent insulin signaling elicited by CT98014 and the previously reported effect of this selective GSK-3 inhibitor to increase insulin-mediated GLUT-4 translocation in skeletal muscle of ZDF rats. 19
The enhanced IRS-1 tyrosine phosphorylation due to the GSK-3 inhibition was associated with a significant decrease in ser307 phosphorylation of IRS-1 (Fig. 5). Moreover, selected interventions that induce insulin resistance in insulin-sensitive cells, such as low-grade oxidative stress in L6 myocytes and 3T3 L1 adipocytes, also enhance ser307 phosphorylation of IRS-1. 26, 27 Numerous investigations have documented the ability of various kinases to induce phosphorylation of IRS-1 on ser307, including JNK 5, 6, 24 and IKKβ. 25 In the present investigation, we have demonstrated that the decrease in IRS-1 ser307 phosphorylation was associated with a similar diminution of JNK phosphorylation, but not IKKβ phosphorylation. Based on this new evidence, GSK-3 can be added to the list of intracellular factors and enzymes that, directly or indirectly, can induce this deleterious alteration in IRS-1 phosphorylation state, possibly by an effect on the activation of JNK.
We provide in the present investigation further evidence that GSK-3 is overactive in type I skeletal muscle of the type 2 diabetic ZDF rats, as insulin-stimulated ser9 phosphorylation of GSK-3β is significantly less in soleus muscle of ZDF rats compared to the lean Zucker group (Fig. 1). These data are in agreement with previous studies demonstrating a greater GSK-3 activity in insulin-resistant skeletal muscle from obese pre-diabetic rats, 12 obese type 2 diabetic rats 11 and obese type 2 diabetic humans, 14 although no differences in GSK-3 activity in human skeletal muscle of non-diabetic controls and type 2 diabetic subjects have also been reported. 28 It appears that GSK-3 overactivity is a prerequisite for the effectiveness of these GSK-3 inhibitors, as treatment of insulin-sensitive muscle and non-muscle tissues with GSK-3 inhibitors is without effect (Ring et al., 2003; Henriksen 2003, Coughlin 2000, Macaulay 2003). 18, 19, 29 Interestingly, overexpression of GSK-3β in mouse skeletal muscle is associated with marked derangements of glucose tolerance and significant hyperinsulinemia and dyslipidemia associated with an increase in central adiposity. 30 These negative metabolic characteristics are very similar to those displayed by the ZDF rat, supporting the concept that GSK-3 overexpression and overactivity in skeletal muscle is one factor that can be associated with the development of glucose dysregulation.
For the most part, these effects of the GSK-3 inhibitor to mediate upregulation of insulin signaling functionality in skeletal muscle of this type 2 diabetic rodent model are similar to those we have previously reported for skeletal muscle from a pre-diabetic rodent model, the obese Zucker rat. 12 Increases in insulin-stimulated IRS-1 tyrosine phosphorylation and serine phosphorylation of Akt and GSK-3β were induced by the GSK-3 inhibitor CT118637, which has essentially the same chemical structure and possesses the same mode of action and pharmacokinetics as CT98014. 12, 17 However, in the present investigation, CT98014 did induce an increase in IR tyrosine phosphorylation (Fig. 3). There are important distinctions between these two investigations that may account for the differences in the GSK-3 inhibitor-induced response of IR tyrosine. In the present study, the skeletal muscle from obtained from overtly type 2 diabetic male animals, whereas in the previous investigation 12 the muscle was from pre-diabetic female animals. Aside from the obvious gender difference, there may be a potential role of the continuous and marked hyperglycemia of the ZDF rat that allows for the GSK-3 inhibitor to enhance the action of insulin on IR tyrosine phosphorylation. Data from the present investigation indicate that GSK-3 may also act on IR as well as on IRS-1 in the regulation of insulin signaling, at least in skeletal muscle from male type 2 diabetic rats.
The effects of the substituted aminopyrimidine class of GSK-3 inhibitors used in the present investigation to upregulate insulin action are similar to the actions of other, structurally distinct GSK-3 inhibitors. For example, the research group of Eldar-Finkelman and colleagues has developed synthetic phosphorylated peptides that are substrate-competitive inhibitors of GSK-3. 8, 32 These compounds are selective and sensitive, inhibiting GSK-3β with an IC50 of 40 μmol/L. 32 Moreover, they enhance in vitro glucose transport activity in isolated mouse adipocytes, 32 and improve glucose tolerance when given acutely to obese, insulin-resistant mice on a high-fat diet 32 or when administered chronically to ob/ob mice. 33 Interestingly, the chronic GSK-3 inhibition in ob/ob mice given these synthetic phosphorylated peptides leads to upregulation of GLUT-4 protein expression in skeletal muscle, 33 a finding we did not make in obese Zucker rats treated chronically with the substituted aminopyrimidine class of GSK-3 inhibitors. 17 Taken collectively, these findings support the concept that selective GSK-3 inhibitors can be used to beneficially modulate insulin action on the glucose transport system in conditions of insulin resistance.
We have shown previously that there is a beneficial effect of selective, aminopyrimidine-based GSK-3 inhibitors on the glucose transport system of type IIb (fast-twitch glycolytic) skeletal muscle, but these effects, in absolute terms, are only ~25% of those observed in type I skeletal muscle. 12, 19 We have also shown that CT98014 increases insulin action in type IIb epitrochlearis muscle of ZDF rats (Teachey and Henriksen, unpublished data), but because these effects were marginal (only at the threshold of detectability) and distinctly less than in the soleus muscle, we did not pursue a complete analysis of the insulin signaling factors reported for the soleus muscle in the present investigation. It is clear that the beneficial effects of GSK-3 inhibitors are much more pronounced in oxidative muscle than in glycolytic muscle types.
In summary, we have demonstrated that the acute in vitro inhibition of GSK-3 in insulin-resistant type 1 skeletal muscle of type 2 diabetic ZDF rats is characterized by upregulation of the functionality of both upstream (IR and IRS-1) and downstream (Akt and GSK-3β) elements of the insulin signaling pathway. Moreover, the enhanced IRS-1 functionality following acute GSK-3 inhibition in diabetic skeletal muscle is associated with decreased ser307 phosphorylation, possibly mediated by a diminution of JNK activation. These responses of the insulin signaling pathway to acute GSK-3 inhibition likely account for the increased action of insulin to stimulate glucose transport activity in skeletal muscle of type 2 diabetic ZDF rats.
Acknowledgments
Supported by Grant R01 DK063967 from the National Institute of Diabetes Digestive and Kidney Diseases to E.J.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zierath JR, Krook A, Wallberg-Henriksson H. Insulin action and insulin resistance in human skeletal muscle. Diabetologia. 2000;43:821–835. doi: 10.1007/s001250051457. [DOI] [PubMed] [Google Scholar]
- 3.Henriksen EJ. Invited Review: Effects of acute exercise and exercise training on insulin resistance. J Appl Physiol. 2002;93:788–796. doi: 10.1152/japplphysiol.01219.2001. [DOI] [PubMed] [Google Scholar]
- 4.Mothe I, Van Obberghen E. Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J Biol Chem. 1996;271:11222–11227. doi: 10.1074/jbc.271.19.11222. [DOI] [PubMed] [Google Scholar]
- 5.Aguirre V, Uchida T, Yenush L, Davis RJ, White MF. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 6.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 7.Draznin B. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85α. The two sides of a coin Diabetes. 2006;55:2392–2397. doi: 10.2337/db06-0391. [DOI] [PubMed] [Google Scholar]
- 8.Eldar-Finkelman H, Ilouz R. Challenges and opportunities with glycogen synthase kinase-3 inhibitors for insulin resistance and type 2 diabetes treatment. Expert Opin Invest Drugs. 2003;12:1511–1519. doi: 10.1517/13543784.12.9.1511. [DOI] [PubMed] [Google Scholar]
- 9.Wagman AS, Johnson KW, Bussiere DE. Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes. Curr Pharmaceut Design. 2004;10:1105–1137. doi: 10.2174/1381612043452668. [DOI] [PubMed] [Google Scholar]
- 10.Henriksen EJ, Dokken BB. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr Drug Targ. 2006;7:1435–1442. doi: 10.2174/1389450110607011435. [DOI] [PubMed] [Google Scholar]
- 11.Brozinick JT, Misener EA, Ni B, Ryder JW, Dohm GL. Impaired insulin signaling through GSK3 in insulin resistant skeletal muscle (abstract) Diabetes. 2000;49 (Suppl 1):A326. [Google Scholar]
- 12.Dokken BB, Sloniger JA, Henriksen EJ. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in pre-diabetic insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E1188–E1194. doi: 10.1152/ajpendo.00547.2004. [DOI] [PubMed] [Google Scholar]
- 13.Eldar-Finkelman H, Schreyer SA, Shinohara MM, LeBoeuf RC, Krebs EG. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes. 1999;48:1662–1666. doi: 10.2337/diabetes.48.8.1662. [DOI] [PubMed] [Google Scholar]
- 14.Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49:263–271. doi: 10.2337/diabetes.49.2.263. [DOI] [PubMed] [Google Scholar]
- 15.Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci. 1997;94:960–964. doi: 10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liberman Z, Eldar-Finkelman H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J Biol Chem. 2005;280:4422–4428. doi: 10.1074/jbc.M410610200. [DOI] [PubMed] [Google Scholar]
- 17.Dokken BB, Henriksen EJ. Chronic selective glycogen synthase kinase-3 inhibition enhances glucose disposal and muscle insulin action in pre-diabetic obese Zucker rats. Am J Physiol Endocrinol Metab. 2006;291:E207–E213. doi: 10.1152/ajpendo.00628.2005. [DOI] [PubMed] [Google Scholar]
- 18.Ring D, Johnson KW, Henriksen EJ, et al. Selective GSK3 inhibitors potentiate insulin action on glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–595. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- 19.Henriksen EJ, Kinnick TR, Teachey MK, et al. Modulation of muscle insulin resistance by selective glycogen synthase kinase-3 inhibition in Zucker Diabetic Fatty rats. Am J Physiol Endocrinol Metab. 2003;284:E892–E900. doi: 10.1152/ajpendo.00346.2002. [DOI] [PubMed] [Google Scholar]
- 20.Nikoulina SE, Cirialdi TP, Mudaliar S, Carter L, Johnson K, Henry RR. Inhibition of glycogen synthase kinase-3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes. 2002;51:2190–2198. doi: 10.2337/diabetes.51.7.2190. [DOI] [PubMed] [Google Scholar]
- 21.Cline GW, Johnson K, Regittnig W, et al. Effects of a novel glycogen synthase kinase-3 inhibitor on insulin-stimulated glucose metabolism in Zucker Diabetic Fatty (fa/fa) rats. Diabetes. 2002;51:2903–2910. doi: 10.2337/diabetes.51.10.2903. [DOI] [PubMed] [Google Scholar]
- 22.Henriksen EJ, Halseth AE. Early alteration in soleus GLUT-4, glucose transport, and glycogen in voluntary running rats. J Appl Physiol. 1994;76:1862–1867. doi: 10.1152/jappl.1994.76.5.1862. [DOI] [PubMed] [Google Scholar]
- 23.Hansen PA, Gulve EA, Holloszy JO. Suitability of 2-deoxyglucose for in vitro measurement of glucose transport activity in skeletal muscle. J Appl Physiol. 1994;76:1862–1867. doi: 10.1152/jappl.1994.76.2.979. [DOI] [PubMed] [Google Scholar]
- 24.Rui L, Aguirre V, Kim J, et al. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest. 2001;107:181–189. doi: 10.1172/JCI10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao Z, Hwang D, Bataille F, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277:48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- 26.Bloch-Damti A, Potashnik R, Tanti JF, Le Marchand-Brustel Y, Rudich A, Bashan N. IRS-1 protein degradation induced by oxidative stress is associated with distinct time-course of phosphorylation on serine 307 and serine 632. Diabetologia. 2003;46 (Suppl 2):A209. [Google Scholar]
- 27.Bloch-Damti A, Bashan N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxidants Redox Signaling. 2005;7:1553–1567. doi: 10.1089/ars.2005.7.1553. [DOI] [PubMed] [Google Scholar]
- 28.Højlund K, Staehr P, Hansen BF, et al. Increased phosphorylation of skeletal muscle glycogen synthase at NH2-terminal sites during physiological hyperinsulinemia in type 2 diabetes. Diabetes. 2003;52:1393–1402. doi: 10.2337/diabetes.52.6.1393. [DOI] [PubMed] [Google Scholar]
- 29.MacAulay K, Hajduch E, Blair A, Coghlan MP, Smith SA, Hundal HS. Use of lithium and SB-415286 to explore the role of glycogen synthase kinase-3 in the regulation of glucose transport and glycogen synthase. Eur J Biochem. 2003;270:3829–3838. doi: 10.1046/j.1432-1033.2003.03777.x. [DOI] [PubMed] [Google Scholar]
- 30.Pearce NJ, Arch JRS, Clapham JC, et al. Development of glucose intolerance in male transgenic mice overexpressing human glycogen synthase kinase-3 on a muscle-specific promoter. Metabolism. 2004;53:1322–1330. doi: 10.1016/j.metabol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Coghlan MP, Culbert AA, Cross DAE, et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;24:1–11. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- 32.Plotkin B, Kaidanovich O, Talior I, Eldar-Finkelman H. Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J Pharmacol Exper Therapeut. 2003;305:974–980. doi: 10.1124/jpet.102.047381. [DOI] [PubMed] [Google Scholar]
- 33.Kaidanovich-Beilin O, Eldar-Finkelman H. Long-term treatment with novel glycogen synthase kinase-3 inhibitor improves glucose homeostasis in ob/ob mice: molecular characterization in liver and muscle. J Pharmacol Exper Therapeut. 2006;316:17–24. doi: 10.1124/jpet.105.090266. [DOI] [PubMed] [Google Scholar]