

Abstract
The immunoglobulin degrading enzyme of Streptococcus pyogenes, IdeS, is an unusual cysteine protease produced by group A streptococci for which the only known substrate is immunoglobulin G (IgG). To date, IdeS has not been found to cleave any of the known synthetic substrates that other cysteine proteases hydrolyse, thus making the development of an IdeS detection assay difficult. Furthermore, at high doses of substrate, product generation is inhibited potentially due to the need for a dimeric enzyme complex with IgG. In this study we have developed a mass spectral assay for IdeS activity based on the detection of a Mr~25,300 Fc fragment that retains the ability to bind streptococcal protein G. Using this assay procedure, evidence for a multimeric enzyme-substrate complex was obtained as well as identifying isolated heavy chains as a non-substrate inhibitor of IdeS activity. Under appropriate experimental conditions the assay could be used to detect IdeS activity in bacterial culture media or in human plasma without a requirement for purified reactants. The availability of a rapid and sensitive assay for IdeS should facilitate the detailed biochemical characterization of this unusual bacterial cysteine protease.
1. Introduction
Bacterial cysteine proteases are virulence factors that have been studied extensively for their role in promoting the pathogenesis and pathophysiology of infections (Otto and Schirmeister, 1997; Lasakosvitsch et al., 2003). In general, these enzymes, such as the streptococcal cysteine protease SpeB, have broad substrate specificity and, as a consequence, these gene products are usually translated as zymogen forms that become activated following secretion (Elliott, 1945; Saouda et al., 2001; Kagawa, et al., 2000). Recently, a number of investigators have described a novel cysteine protease expressed by certain group A streptococcal isolates (immunoglobulin degrading enzyme of Streptococcus pyogenes, IdeS) that demonstrates high substrate selectivity for human and certain other species of mammalian IgG (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen et al., 2003; Vincents et al., 2004; Wenig et al., 2004). Studies by Kawabata and colleagues also identified a secreted form of an anchorless immunoglobulin-binding protein from a group A streptococcus that could bind IgG, IgA and IgM (Kawabata et al., 2002). This protein, encoded by the sib38 gene, has an identical nucleotide sequence to that reported for the ides gene. These studies did not, however, study the ability of the protein to mediate immunoglobulin cleavage.
IdeS was also independently and concurrently identified as Mac-1 by Lei and colleagues in a comprehensive proteomic analysis of culture supernatants from a SpeB negative variant of a group A isolate (Lei et al., 2000). Mac-1 demonstrates homology to eukaryotic CD11b (Lei et al., 2001a). CD11b interacts with the FcγIIb receptor (CD16) and therefore, it is not surprising that IdeS (Mac-1) and the products it generates by cleavage of IgG can inhibit opsonophagocytosis (Agniswamy et al., 2004). Analysis of the ides gene in 67 S. pyogenes isolates of multiple M serotypes revealed the presence of a conserved RGD integrin binding domain as well as evidence for two sequence variants (Lei et al., 2003). Based on these various properties of IdeS, a variety of roles for this enzyme in S. pyogenes pathogenesis have been proposed (Lei et al., 2001b; von Pawel-Rammingen et al., 2001; Frank, 2001; von Pawel-Rammingen and Bjorck, 2003).
X-ray crystallographic studies coupled with molecular genetic approaches and functional analysis all support the conclusion that IdeS is a cysteine protease (Wenig et al., 2004; Akesson et al., 2006; Agniswamy et al., 2006; Lei et al., 2003). Unlike other cysteine proteases, e.g. papain or SpeB (Otto and Schirmeister, 1997), IdeS is highly specific in the substrates it will cleave (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen and Bjorck, 2003; Vincents et al., 2004). To date, the only known substrate is human and related mammalian sources of IgG (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen and Bjorck, 2003; Vincents et al., 2004). Attempts to create a synthetic peptide substrate that encompassed the identified cleavage site in IgG failed to yield a suitable candidate (Vincents et al., 2004). This finding has lead to the suggestion that the binding between IgG and IdeS may involve a conformational change that results in the appropriate orientation of the enzyme with the susceptible peptide bond in the IgG substrate (Vincents et al., 2004; Wenig et al., 2004). Alternatively, as suggested from a recent crystallographic study, a dimer of the IdeS protein may be required for catalytic activity (Agniswamy et al., 2006). This may in turn explain the observation that at high concentrations of substrate IgG no detectable Fc product is generated in the presence of the active enzyme (Vincents et al., 2004).
Further analysis of IdeS is complicated by the absence of a synthetic substrate assay and the need for a protein separation technique to monitor enzymatic activity (Vincents et al., 2004). IdeS is also unusual among bacterial cysteine proteases in its failure to be inhibited by E-64 or a number of other well characterized cysteine protease inhibitors (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen and Bjorck, 2003; Vincents et al., 2004). Current methods to monitor IdeS activity involve either SDS-PAGE, FPLC or Biacore and require high concentrations of homogeneous reactants and are slow and technically challenging (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen and Bjorck, 2003; Vincents et al., 2004; Wenig et al., 2004; Lei et al., 2004; Lei et al., 2001a; Agniswamy et al., 2004). In previous studies of post translational modification of bacterial secreted and surface proteins by SpeB, our laboratory has demonstrated SELDI-TOF protein chip mass spectrometry can be used efficiently (Saouda et al., 2002; Romer and Boyle, 2003; Rezcallah et al., 2004; Hess et al., 2005; Hess and Boyle, 2006). In this study we have evaluated the use of a modified SELDI-TOF analysis as a rapid, sensitive method to analyze the activity of IdeS based on the ability to monitor the generation of a Mr~25,300 Fc product. Key to the method is a selective capture enhancement step which uses bacterial bound protein G to capture the Fc product of IdeS activity prior to transfer to a gold protein chip for mass spectral analysis.
2. Materials and Methods
2.1. Bacterial Strains
Streptococcus pyogenes isolate AP1 used in this study was originally obtained from the World Health Organization Collaborating Centre for References and Research on Streptococci, Institute of Hygiene and Epidemiology, Prague, Czech Republic.
2.2. Recombinant IdeS preparation
Based on the published gene sequence for IdeS (von Pawel-Rammingen et al., 2002) primers were designed to amplify the gene using chromosomal DNA from S. pyogenes isolate AP1 as template [These studies were approved by the Juniata College recombinant DNA committee]. The selected forward primer was 5′-CACCGATAGTTTTTCTGCTAAT-3′ and the reverse primer was 5′-CTATCAACCTTAGTCTGGTTA-3′. Based on the reported sequence for the ides gene in this isolate (von Pawel-Rammingen et al., 2002), these primers would amplify the entire open reading frame coding for IdeS minus the leader peptide. PCR was performed using Accupol Taq polymerase (PGC Scientifics, Frederick, MD) and 30 cycles of 95 °C for 60 seconds, 45.3 °C for 60 seconds, and 72 °C for 60 seconds. The resulting 930 bp blunt-ended PCR product (see Figure 1A) was directionally cloned into the pBAD expression vector (Invitrogen, Carlsbad, CA), which adds a thioredoxin tag on the amino-terminus of the IdeS polypeptide and a polyhistidine tag on the carboxy-terminus. After cloning, the construct DNA was sequenced to confirm its identity. The sequence of the recombinant DNA is shown in Figure 1B and was >99% identical to the previously published ides gene from this isolate (von Pawel-Rammingen et al., 2002).
Fig. 1.
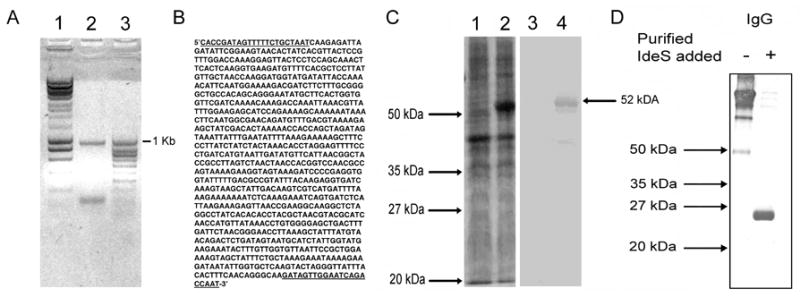
Generation and characterization of recombinant IdeS. Panel A: PCR product generated using ides primers described in the Methods. Panel B: sequence of ~1 Kb PCR fragment. The nucleotides in bold and underlined correspond to the PCR primers used. Panel C: Lanes 1 and 2: Coomassie blue stain. Lanes 3 and 4: immunoblot of crude E. coli lysates probed with horseradish peroxidase-conjugated anti-polyhistidine tag antibody. Lanes 1 and 3 before induction with arabinose. Lane 2 and 4 after 6 hours of induction with 0.05% arabinose. Panel D: Analysis of enzyme activity of recombinant IdeS was carried out as described in the Methods.
The construct was transformed into chemically competent E. coli. Following induction with 0.05% arabinose, IdeS was expressed and the recombinant protein was detected by immunoblotting using a horseradish peroxidase conjugated mouse antipolyhistidine monoclonal (IgG2b) antibody reporter system (Invitrogen). The recombinant protein was purified by metal chelate affinity chromatography using the Probond® purification system (Invitrogen). The resulting product was a single band on SDS- PAGE (see Figure 1C). The thioredoxin (15 kDa) and the polyhistidine (3 kDa) tags, cause the molecular weight of IdeS to increase to 52 kDa from its expected native 34 kDa (see Figure 1C). The concentration of the recombinant protein was determined from its theoretical extinction coefficient of 62280 M−1 cm−1 which was calculated using the ProtParam tool (http://www.expasy.org/tools/protparam.html). The enzymatic activity of purified IdeS was determined using an IgG cleavage assay and immunoblotting (see below).
2.3. Immunoblot assay to measure IdeS activity
For the IgG cleavage reaction, 1 μg of purified recombinant IdeS or 9 μl of crude recombinant lysate was added to 12.5 μg of human immunoglobulin G (IgG) (Athens Research & Technology, Athens, GA) in a final volume of 70 μl in phosphate buffered saline (PBS), pH 7.4, and was incubated at 37°C for 30 minutes. Seven microliters of this mixture was mixed with sample buffer and boiled before loading onto 12.5% SDS-PAGE gels. Samples were analyzed by SDS-PAGE under non-reducing conditions. Immunoblots were probed with horseradish peroxidase conjugated protein G (Sigma, St. Louis, MO). Immunoblots were developed using a TMB substrate system (KPL, Gaithersburg, MD). The enzymatic activity of the cloned protein was confirmed using this method by the ability to cleave human IgG and generate a Mr~25,000 Fc fragment that retained the ability to react with streptococcal protein G using a western immunoblot technique (see Figure 1 D). Similar studies with each human IgG subclass (data not shown) confirmed that the recombinant protein had the expected enzymatic function (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen and Bjorck, 2003; Vincents et al., 2004).
2.4. Protein G capture and mass spectrometry assay for IdeS activity
For the IgG cleavage reaction, 1 μg of purified recombinant IdeS was added to 12.5 μg of human immunoglobulin G (IgG) (Athens Research & Technology) in a final volume of 25 μl in 100mM Tris buffered saline (TBS), pH 7.4, and was incubated at 37°C for 30 minutes. The procedure used to measure the generation of protein G-binding Fc fragments was a modification of the immunoproteomic protocol previously described by our laboratory (Saouda et al., 2002; Romer and Boyle, 2003; Rezcallah et al., 2004; Hess et al., 2005; Hess and Boyle, 2006). Essentially, 10 μl of the reaction mixture containing the Fc IgG fragment product of IdeS cleavage of human IgG was mixed with 50 μl of a 10% (w/v) heat-killed suspension of protein G-expressing bacteria in a final volume of 1 ml of TBS. The mixture was rocked at ambient temperature for 30 minutes. After incubation, the bacterial suspension was centrifuged at 10,000 x g for 2 minutes. The resulting pellet was washed once with 500 μl of TBS and then resuspended in 50 μl of TBS. One microliter of the suspension was spotted directly onto a gold Ciphergen™ protein chip. After drying, each spot was coated with an energy-absorbing matrix consisting of saturated 4- hydroxy-3,5-dimethoxycinnamic acid in 50% acetonitrile and 0.05% trifluoroacetic acid and then transferred to the Ciphergen PBSII protein chip mass spectrometer for analysis. The high voltage detector sensitivity and laser intensity were determined empirically to obtain the optimal signal in the targeted mass range. Ciphergen Protein Chip Software, Version 3.0.2, was used to identify protein peaks, assign masses, and calculate intensities. All data were generated by averaging 65 laser shots on different positions of each protein chip spot.
2.5. Detection of IdeS activity in bacterial culture media
Most studies of IdeS activity require purified reagents (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen and Bjorck, 2003; Vincents et al., 2004; Wenig et al., 2004; Lei et al., 2004; Lei et al., 2001a; Agniswamy et al., 2004). To assess the ability of the protein G capture method to detect IdeS activity in crude bacterial culture media, isolate AP1 was grown as a stationary culture in Todd-Hewitt broth at 37°C for varying times. A 10 ml aliquot of media was harvested from the culture at different times and bacteria removed by centrifugation at 2,000xg for 15 minutes. The bacteria free culture media was mixed with 40 μg of human IgG in a final reaction volume of 1 ml and the reaction mixture was incubated at 37°C for 30 minutes and the reaction stopped by addition of iodoacetate (20 mM final concentration). Control samples harvested at the time of inoculation were included in each assay as well as samples to which either 20 mM iodoacetate or 0.01 mM E64 were also included. Iodoacetate inhibits the majority of cysteine protease including IdeS while E64 is also a broad cysteine protease inhibitor that fails to inhibit IdeS (Vincents et al., 2004). Following the incubation period, an aliquot of each sample (10 μl) was added to 50 μl of a 10% (w/v) heat-killed suspension of protein G-expressing bacteria in 1 ml of PBS, and was incubated at ambient temperature with gentle agitation for 30 minutes. Samples were then processed and assayed as described earlier.
2.6. Detection of IdeS activity in plasma samples
To assess the ability of IdeS to cleave IgG in plasma, 2 μg of IdeS was incubated with 70 μl of a 10% v/v dilution of either 10% or 1% human plasma. The reaction mixture was incubated at 37°C for 30 minutes and the reaction stopped by addition of iodoacetate (20 mM final concentration). An aliquot of the sample (10 μl) was added to 50 μl of a 10% (w/v) heat-killed suspension of protein G-expressing bacteria in 1 ml of PBS, and was incubated at ambient temperature with gentle agitation for 30 minutes. Samples were then processed and assayed as described earlier.
3. Results
3.1. Activity of recombinant IdeS
In preliminary studies, using western immunoblotting techniques, we confirmed that the recombinant form of IdeS cloned and expressed as described in the Methods could cleave human IgG generating an Mr~25,300 fragment that represented the Fc portion of the antibody based on its ability to bind streptococcal protein G, see Figure 1. This observation suggested that the product could be captured using an immobilized form of this bacterial IgG-binding protein. Previous studies from our laboratory have demonstrated that protein G-expressing bacteria can be used to capture antibodies via their Fc region and facilitates transfer to a protein chip for analysis of bound antigens by mass spectrometry (Saouda et al., 2002). This immunoproteomic assay has proved to be an efficient, sensitive and reproducible method for monitoring antigen-bound to their cognate immobilized antibody (Romer and Boyle, 2003; Rezcallah et al., 2004; Hess et al., 2005; Hess and Boyle, 2006). Consequently, we hypothesized that a modification of this technique could be used to follow the generation of the unique IdeS-dependent Fc product.
3.2. Detection of IdeS activity using protein G capture and mass spectrometry
In the initial experiments to test this prediction, human IgG (12.5 μg) was incubated with recombinant IdeS (1 μg) in a final volume of 70 μl 100 mM Tris buffered saline, pH 7.4 (TBS) for 30 minutes at ambient temperature. At the end of the incubation period, aliquots were removed and mixed with protein G-expressing bacteria as described in the Methods. Following a 30-minute incubation period, the bacteria were pelleted and washed twice before resuspending in 50 μl of TBS. One μl of the suspension was transferred to a spot on a gold chip and was air-dried. The spot was then coated with one μl of an energy absorbing matrix (EAM), dried, and then analyzed by mass spectrometry. The results presented in Figure 2 A and B demonstrated the generation of a unique Mr~ 25,300 Fc fragment that was not detected if 20 mM iodoacetate was included in the reaction mixture. The appearance of the protein G binding, Fc fragment (Mr~25,300) was accompanied by the disappearance of the Mr~147,000 IgG substrate peak and the Mr~74,000 double charged form of IgG, compare Figure 2 A and B. The initial conditions of laser energy chosen (13.5μJoules) for these experiments were designed to monitor both intact IgG and the Fc fragment generated by IdeS action. In subsequent studies the assay was optimized to detect only fragments in the Mr~25,300 range. This enabled the laser energy to be reduced and the signal to noise ratio of samples in this molecular mass range to be maximized (see Figure 2 C and D).
Fig. 2. Mass spectral analysis of IgG treated with IdeS following capture by bacterial bound protein G.
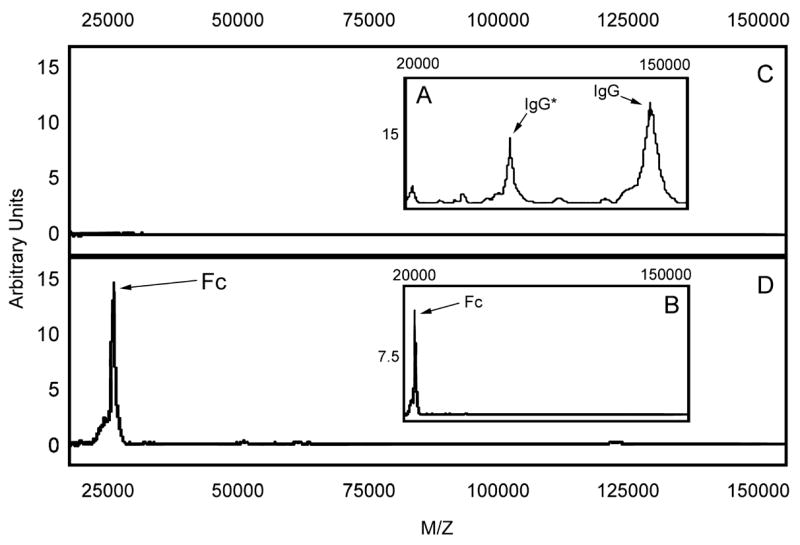
The mass spectral analysis was carried out at two laser settings. The high setting (13.5 μJoules) was used for the untreated (panel A) and IdeS treated sample (panel B) to allow the substrate IgG and the Fc product to be monitored. A lower energy laser setting (6.1 μJoules) was used to maximize the signal to noise ratio for the Fc fragment which was the product of IdeS activity (compare panel C and D). Panel C represents the IgG sample incubated with buffer and panel D represents the IgG sample incubated with IdeS. IgG* represents the double charge peak for IgG.
Studies were conducted to establish the relationship between Fc fragment concentration, peak height, and area under the peak using the lower laser energy setting (6.1 μJoules). In these studies, we observed a proportional change in peak height and area varied for intensity values of less than 12.5 arbitrary units. Using the optimized assay conditions to detect the Fc fragment, a series of kinetic studies were performed. The results presented in Figure 3 demonstrate the appearance of the Mr~25,300 Fc fragment occurs rapidly and that under the experimental conditions used an endpoint was reached within 15 minutes. The reaction was totally inhibited by addition of 20 mM iodoacetate (data not shown). If the enzyme digestion was continued for more than 30 minutes the height of the Mr~25,300 peaks declined indicating that additional degradation of the Fc fragment could occur. This could be prevented by addition of iodoacetate (data not shown).
Fig. 3.
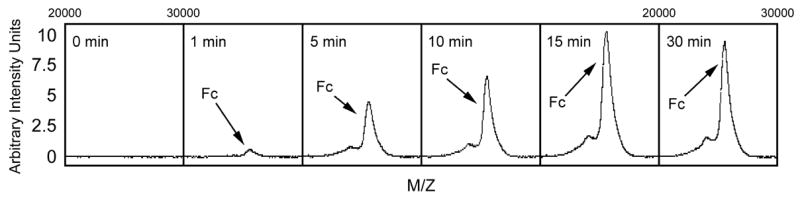
Kinetic anaylsis of Fc fragment generation by IdeS. Human IgG (12.5 μg) was incubated with recombinant IdeS (1 μg) for 0, 1, 5, 10, 15 or 30 minutes at 37°C before adding iodoacetate to a final concentration of 20 mM. An aliquot of the reaction mixture from each time point was assayed for Fc product generation using protein G-capture and mass spectral analysis as described in the Methods.
3.3. Effects of IgG concentration on IdeS activity
Previous studies of IdeS activity had reported that at high substrate concentrations the generation of Fc fragments by IdeS could be inhibited (von Pawel-Rammingen et al. 2002, Vincents et al. 2004). Consequently the experiments described in Figure 3 were repeated in which the concentration of IdeS was held constant and the concentration of IgG varied. For these experiments, the analysis was conducted without protein G capture. Although this increased the background noise in the spectra, the modification of the experimental protocol was used to ensure that the generated Fc fragment would not be out competed from binding to protein G expressing bacteria when measuring the area under the peak corresponding to the Fc fragment (Mr~ 25,300). This was achieved using the Ciphergen PBS II computer software. The results of a representative experiment are presented in Figure 4 and suggest that there was a critical ratio of IgG to IdeS. Similar results were obtained using a western immunoblotting assay (data not shown). The results of these experiments are consistent with two or more IdeS molecules being required to bind to IgG in order to generate the observed Fc fragment and suggested some form of multimeric binding of IdeS to IgG was required for enzymatic activity to be observed. This would be consistent with a dimer of IdeS being required (Agniswamy et al., 2006) and potentially suggest that that structure could only be achieved in the native S-S linked dimer of heavy chains present in the Fc region of native IgG.
Fig. 4.
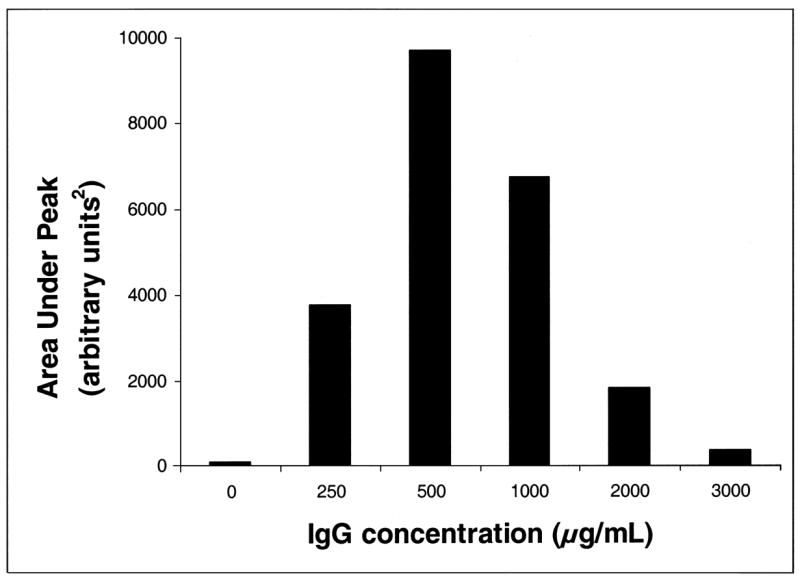
Effect of substrate IgG concentration on IdeS activity. Varying concentrations of human IgG were incubated with recombinant IdeS for 30 minutes at 37°C. After the incubation, iodoacetate was added to a final concentration of 20 mM to stop the reaction and the generation of Fc fragment determined, without protein G capture, as described in the Methods. The area under the Mr~ 25,300 peak was determined by Ciphergen Protein Chip Software, Version 3.0.2, and was used as a semi-quantitative measure of the amount of IdeS-generated Fc product.
3.4. Inhibitory effects of isolated γ heavy chain on IdeS activity
To address this prediction, we tested the ability of IdeS to directly cleave isolated human heavy chain. In a series of preliminary experiments, conducted over a wide range of γ heavy chain concentrations, no detectable cleavage was observed (data not shown). If the IdeS binding region on the heavy chain could bind a single, catalytically inactive molecule of the enzyme, we predicted that addition of the heavy chain to a reaction between IgG and IdeS could result in inhibition of enzyme activity. The results of these studies are presented in Figure 5 and suggest that the heavy chain can act as an inhibitor of the IgG cleaving potential of IdeS. [The protein G capture step was not included in this assay because of the differences in affinity of protein G for γ chain and intact IgG]. The unique Fc fragment generated by IdeS was detectable and it was also possible to demonstrate that IdeS failed to modify the γ heavy chain over a wide range of heavy chain to IdeS concentrations (data not shown).
Fig. 5.
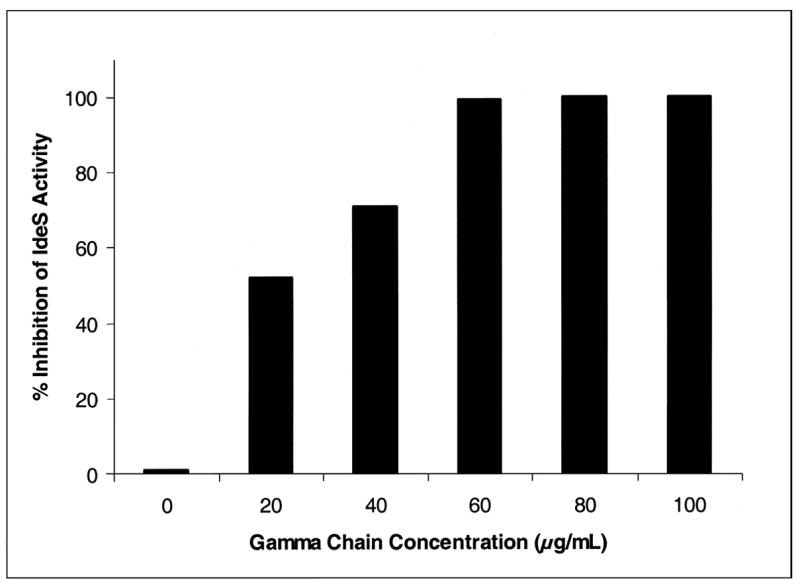
Effect of γ heavy chain on IdeS activity. Human IgG was incubated with varying concentrations of human IgG γ chain and recombinant IdeS (1 μg) for 30 minutes at 37°C. After the incubation, iodoacetate was added to a final concentration of 20 mM to stop the reaction, and the generation of Fc fragments determined as described in the Methods. Percent inhibition was determined by comparing the experimental peak areas to the peak area of a control reaction without human IgG γ chain added. Note the protein G capture step was not included in this assay because of the differences in affinity of protein G for γ chain and intact IgG.
3.5. Detection of IdeS activity in bacterial culture media
The finding that there is a critical ratio of IgG to enzyme required for activity makes the detection of unknown levels of IdeS secretion from bacteria more challenging. For studies to detect an unknown level of IdeS activity it is important to test for activity with different concentrations of IgG as substrate and to include dilutions of the unknown enzyme sample to ensure that IdeS will be detected if present. Using this basic checkerboard approach we tested the bacteria-free supernatant from a stationary culture of S. pyogenes isolate AP1, grown at 37°C. The results, presented in Figure 6, indicate that an Fc product could be detected when the bacterial culture supernatant from as early as late log or early stationary phase was mixed with 12.5 μg of human IgG. This Fc product was not detected when iodoacetate was added at the same time as the bacterial culture media but was unaffected by the addition of the cysteine protease inhibitor E64 (data not shown). Similar activity was detected when a higher IgG substrate concentration was used under identical assay conditions (data not shown). These results indicate that despite the potential problems from substrate inhibition or degradation of Fc fragments at high enzyme concentration, it was practical to detect IdeS activity in crude bacterial supernatants.
Fig. 6.
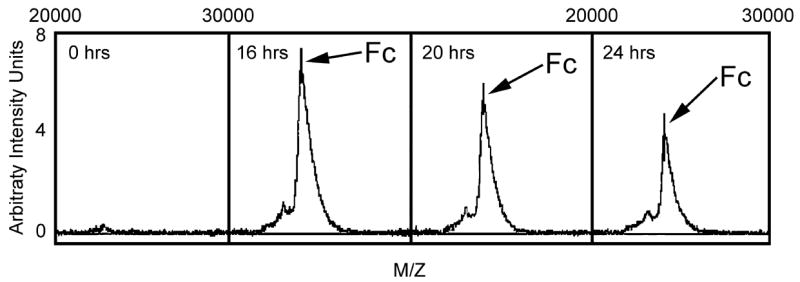
Detection of IdeS activity in bacterial culture media. An aliquot of bacteria-free media harvested from a culture of S. pyogenes isolate AP1 growing in Todd-Hewitt broth at different times was assayed for IdeS activity as described in the Methods. Control samples harvested at the time of inoculation (0 hours) were included in each assay.
3.6. Detection of IdeS activity in plasma
By virtue of the selectivity of the protein G capture step in the assay, we wanted to determine whether the IdeS enzyme could function in plasma or if a naturally occurring inhibitor(s) or competitive substrates would regulate enzyme activity. In preliminary studies, we could demonstrate that IdeS generated fragments artificially added to samples of 10% human plasma could be detected using the bacterial capture step and mass spectral analysis (data not shown). The ability of IdeS to cleave IgG present in different concentrations of human plasma was tested. For these experiments 1% or 10% human plasma was incubated with recombinant IdeS (2 μg) in a final volume of 70 μl PBS for 30 minutes at ambient temperature. At that time, an aliquot of the reaction mixture was subjected to the protein G capture step and subsequent mass spectral analysis. The results presented in Figure 7 upper panel demonstrate the appearance of the characteristic Mr~25,300 Fc fragment in human plasma treated with IdeS. This peak was not observed in samples in which 20 mM iodoacetate was added, see Figure 7 lower panel. Note in the spectra from samples containing human plasma, a Mr~66,000 peak and a Mr~33,000 double charge peak was detected in every sample. These peaks are due to contaminating human serum albumin. These peaks did not however overlap with the Fc fragment peak generated by IdeS activity, see Figure 7. These results indicated that IdeS is highly specific for IgG and that there was no inhibition (competitive, specific or non-specific) of this bacterial cysteine protease by any component in the human plasma.
Fig. 7.
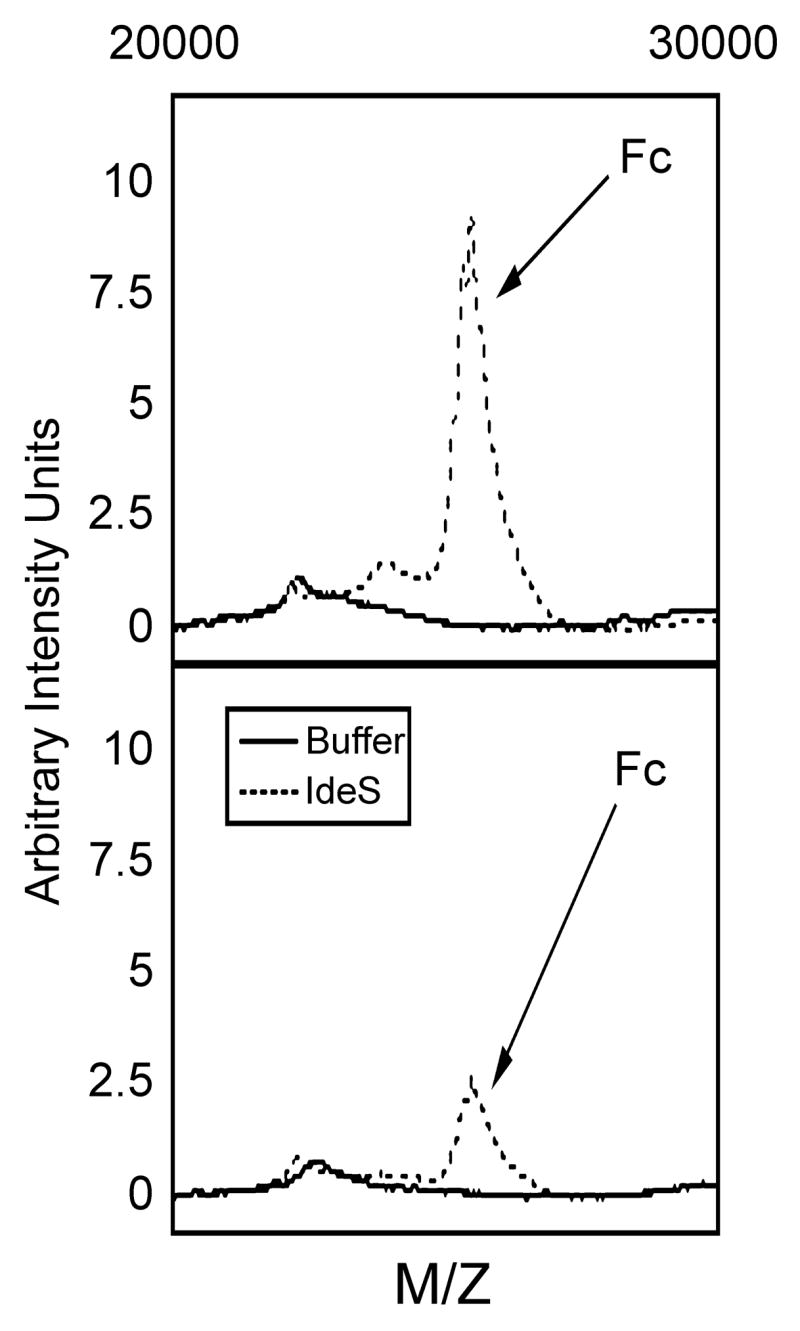
Analysis of human plasma treated with IdeS following capture by bacterial bound protein G. Human plasma (10% or 1%) was incubated with recombinant IdeS (2 μg) in a final volume of 70 μl PBS for 30 minutes at 37°C and the generation of Fc fragments assayed as described in the Methods. The upper panel represents the results obtained with 10% human plasma sample and the lower panel the results for the 1% sample. Not shown in the spectra are peaks at Mr~66,000 representing contaminating human serum albumin and the corresponding double charge peak at Mr~33,000 which were present in all samples containing human plasma.
4. Discussion
IdeS is an unusual bacterial protease that was designated as a cysteine protease based on X-ray crystallographic analysis (von Pawel-Rammingen et al., 2002). This prediction was subsequently validated by site directed mutagenesis confirming the importance of a catalytic triad composed of Cys94, His242 and Asp246 (Lei et al., 2003). Unlike most bacterial cysteine proteases, IdeS is encoded in the streptococcal genome, not as a precursor protein, but as an active enzyme (von Pawel-Rammingen et al., 2002). IdeS has a very limited range of protein substrates and is unable to cleave a single chain synthetic peptide containing the known clip site for the enzyme (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen et al., 2003; Vincents et al., 2004; Wenig et al., 2004).
Currently, IdeS activity is measured by the generation of a unique Mr~ 25,000 Fc fragment of IgG detected using SDS-PAGE, HPLC or Biacore techniques (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen and Bjorck, 2003; Vincents et al., 2004; Wenig et al., 2004; Lei et al., 2004; Lei et al., 2001a; Agniswamy et al., 2004). These assays are time consuming, require high concentrations of purified reagents and are not amenable to analysis of multiple samples. Furthermore it has been reported that the enzymatic activity requires occupancy of an exosite for activity (Vincents et al., 2004), displays declining rate of catalysis at higher IgG concentrations (Vincents et al., 2004) and can dimerize (Agniswamy et al., 2006). All of these unusual properties make the characterization and detailed analysis of the mechanism of action of this enzyme in an infected host difficult to model.
In this paper we describe a protein G capture assay for IdeS activity that incorporates an Fc capture step and a low resolution mass spectral read out. This assay is based on the ability to detect the product of IdeS activity, a Mr~25,300 Fc fragment that retains the ability to bind streptococcal protein G. The distinct difference in size between the IgG substrate (Mr~ 150,000) and the Fc product (Mr~ 23,500) does not require a high resolution mass spectrometer for this particular application. In previous studies in our laboratory, we have used a similar strategy to monitor interaction of bacteria with fragments of fibrinogen (Hess and Boyle, 2006) or as a capture platform for specific antibodies to facilitate detection of different post-translationally modified forms of a targeted antigen (Saouda et al., 2002; Romer and Boyle, 2003; Rezcallah et al., 2004; Hess et al., 2005).
The results presented in Figure 4 suggested some form of multimeric binding of IdeS to IgG was required for enzymatic activity to be observed. This would be consistent with a dimer of IdeS being required (Agniswamy et al., 2006) and potentially suggests that that structure could only be achieved in the native S-S linked dimer of heavy chains present in the Fc region of native IgG. Based on these findings we predicted that isolated γ heavy chain would not be a substrate for IdeS but could potentially bind IdeS and act as a competitive inhibitor of cleavage of the native IgG substrate. The results of the experiments designed to test this prediction confirmed that IdeS would not cleave isolated heavy chain but could inhibit the cleavage of native IgG in the reaction mixture (see Figure 5). The ability to distinguish isolated heavy chain, Fc fragments and intact IgG, using a mass spectral readout, facilitates this type of study.
In addition to being able to distinguish among IgG fragments, the protein G capture step allows this assay procedure to detect IdeS activity in complex reaction mixtures. Unlike the current assays for IdeS that require high concentrations of homogeneous reactants and are slow and technically difficult (von Pawel-Rammingen et al., 2002; von Pawel-Rammingen and Bjorck, 2003; Vincents et al., 2004; Wenig et al., 2004; Lei et al., 2004; Lei et al., 2001a; Agniswamy et al., 2004), the protein G capture mass spectral approach can be carried out in complex mixtures. The results presented in Figures 6 and 7 demonstrate it was possible to detect IdeS activity in the bacterial culture media of S. pyogenes grown in Todd-Hewitt broth culture media or using a complex substrate like human plasma which contained multiple proteins in addition to the specific IdeS substrate, IgG.
The versatility of the assay procedure and the ability to conduct analysis in complex mixtures suggests that this method will be valuable for analysis of IdeS activity in S. pyogenes and other related streptococcal species (Lannergard and Guss, 2006). Currently, characterization of this enzyme is complicated by the absence of a simple synthetic substrate assay and the absence of an efficient, rapid and quantitative method for characterizing the immunoglobulin degrading potential of the protein. The protein G capture assay using a mass spectral read-out described in this study represents an improved procedure to monitor IdeS activity in a rapid format. The protein G capture step coupled with a more sensitive mass spectrometer potentially would allow the approach to be extended to a more detailed analysis of the precise cleavage site of the immunoglobulin. In addition, the assay is amenable to use with complex reaction conditions e.g. in the presence of multiple plasma proteins and acute-phase reactants, which might be anticipated to be present at the site of infection with S. pyogenes and related bacteria that produce immunoglobulin degrading enzymes.
Acknowledgments
This research is supported in part by National Institutes of Health grants # AI 50731 and AI 53718, a AAAS-Merck grant, and by the von Liebig Foundation. The authors would also like to thank Christine Dorman, Nicole Bressler and Emily Coyle for their excellent technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agniswamy J, Lei B, Musser JM, Sun PD. Insight of host immune evasion mediated by two variants of group a streptococcus Mac protein. J Biol Chem. 2004;279:52789–52796. doi: 10.1074/jbc.M410698200. [DOI] [PubMed] [Google Scholar]
- Agniswamy J, Nagiec MJ, Liu M, Schuck P, Musser JM, Sun PD. Crystal structure of group A streptococcus Mac-1: insight into dimer-mediated specificity for recognition of human IgG. Structure. 2006;14:225–235. doi: 10.1016/j.str.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Akesson P, Moritz L, Truedsson M, Christensson B, von Pawel Rammingen U. IdeS, a Highly Specific Immunoglobulin G (IgG)-Cleaving Enzyme from Streptococcus pyogenes, Is Inhibited by Specific IgG Antibodies Generated during Infection. Infect Immun. 2006;74:497–503. doi: 10.1128/IAI.74.1.497-503.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SD. A proteolytic enzyme produced by group A streptococci with special reference to its effect on the type-specific M antigen. J Exp Med. 1945;81:573–592. doi: 10.1084/jem.81.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MM. Annihilating host defense. Nat Med. 2001;7:1285–1286. doi: 10.1038/nm1201-1285. [DOI] [PubMed] [Google Scholar]
- Hess JL, Boyle MD. Fibrinogen fragment D is necessary and sufficient to anchor a surface plasminogen activating complex in Streptococcus pyogenes. Proteomics. 2006;6:375–378. doi: 10.1002/pmic.200500189. [DOI] [PubMed] [Google Scholar]
- Hess JL, Blazer L, Romer T, Faber L, Buller RM, Boyle MD. Immunoproteomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;815:65–75. doi: 10.1016/j.jchromb.2004.07.047. [DOI] [PubMed] [Google Scholar]
- Kagawa TF, Cooney JC, Baker HM, McSweeney S, Liu M, Gubba S, Musser JM, Baker EN. Crystal structure of the zymogen form of the group A streptococcus virulence factor SpeB: an integrin-binding cysteine protease. Proc Natl Acad Sci USA. 2000;97:2235–2240. doi: 10.1073/pnas.040549997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata S, Tamura Y, Murakami J, Terao Y, Nakagawa I, Hamada S. A novel, anchorless streptococcal surface protein that binds to human immunoglobulins. Biochem Biophys Res Commun. 2002;296:1329–1333. doi: 10.1016/s0006-291x(02)02078-8. [DOI] [PubMed] [Google Scholar]
- Lannergard J, Guss B. IdeE, an IgG-endopeptidase of Streptococcus equi sp equi. FEMS Microbiol Lett. 2006;262:230–235. doi: 10.1111/j.1574-6968.2006.00404.x. [DOI] [PubMed] [Google Scholar]
- Lasakosvitsch F, Gentil LG, dos Santos MR, da Silveira JF, Barbieri CL. Cloning and characterisation of a cysteine proteinase gene expressed in amastigotes of Leishmania (L) amazonensis. Int J Parasitol. 2003;33:445–454. doi: 10.1016/s0020-7519(03)00010-9. [DOI] [PubMed] [Google Scholar]
- Lei B, DeLeo FR, Hoe NP, Graham MR, Mackie SM, Cole RL, Liu M, Hill HR, Low DE, Federle MJ, Scott JR, Musser JM. Evasion of human innate and acquired immunity by a bacterial homolog of CD11b that inhibits opsonophagocytosis. Nature Med. 2001a;7:1298–1305. doi: 10.1038/nm1201-1298. [DOI] [PubMed] [Google Scholar]
- Lei B, DeLeo FR, Reid SD, Voyich JM, Magoun L, Liu M, Braughton KR, Ricklefs S, Hoe NP, Cole RL, Leong JM, Musser JM. Opsonophagocytosis-inhibiting Mac protein of group A streptococcus: identification and characteristics of two genetic complexes. Infect Immun. 2002;70:6880–6890. doi: 10.1128/IAI.70.12.6880-6890.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei B, Liu M, Meyers EG, Manning HM, Nagiec MJ, Musser JM. Histidine and aspartic acid residues important for immunoglobulin G endopeptidase activity of the group A streptococcus opsonophagocytosis-inhibiting Mac protein. Infect Immun. 2003;71:2881–2884. doi: 10.1128/IAI.71.5.2881-2884.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei B, DeLeo FR, Musser JM. Author reply to phagocytic killing. Nat Med. 2001b;10:1045–46. [Google Scholar]
- Lei B, Mackie SM, Lukomski S, Musser JM. Identification and immunogenicity of group A streptococcus culture supernatant proteins. Infect Immun. 2000;68:6807–6818. doi: 10.1128/iai.68.12.6807-6818.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto HH, Schirmeister T. Cysteine Proteases and Their Inhibitors. Chem Rev. 1997;97:133–172. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- Rezcallah MS, Boyle MD, Sledjeski DD. Mouse skin passage of Streptococcus pyogenes results in increased streptokinase expression and activity. Microbiology. 2004;150:365–371. doi: 10.1099/mic.0.26826-0. [DOI] [PubMed] [Google Scholar]
- Romer T, Boyle MDP. Application of immunoproteomics to analysis of post-translational processing of the anti-phagocytic M protein of Streptococcus pyogenes. Proteomics. 2003;3:29–35. doi: 10.1002/pmic.200390005. [DOI] [PubMed] [Google Scholar]
- Romer T, Rescellah M, Boyle MDP. Use of surface enhanced laser desorption ionization time-of-flight mass spectrometry to analyze post-translational modification of bacterial virulence factors. Research Signpost. 2002;37:1–18. [Google Scholar]
- Saouda M, Romer T, Boyle MDP. Application of immuno-mass spectrometry to analysis of a bacterial virulence factor. BioTechniques. 2002;32:916–992. doi: 10.2144/02324pt03. [DOI] [PubMed] [Google Scholar]
- Saouda M, Wu W, Conran P, Boyle MDP. Streptococcal pyrogenic exotoxin B (SpeB) enhances tissue damage initiated by other Streptococcus pyogenes products. J Infect Dis. 2001;184:723–731. doi: 10.1086/323083. [DOI] [PubMed] [Google Scholar]
- Vincents B, von Pawel-Rammingen U, Bjorck L, Abrahamson M. Enzymatic characterization of the streptococcal endopeptidase, IdeS, reveals that it is a cysteine protease with strict specificity for IgG cleavage due to exosite binding. Biochemistry. 2004;43:15540–15549. doi: 10.1021/bi048284d. [DOI] [PubMed] [Google Scholar]
- von Pawel-Rammingen U, Bjorck L. IdeS and SpeB: immunoglobulin-degrading cysteine proteinases of Streptococcus pyogenes. Curr Opin Microbiol. 2003;1:50–55. doi: 10.1016/s1369-5274(03)00003-1. [DOI] [PubMed] [Google Scholar]
- von Pawel-Rammingen U, Johansson BP, Bjorck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J. 2002;217:1607–15. doi: 10.1093/emboj/21.7.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Pawel-Rammingen U, Johansson BP, Tapper H, Bjorck L. Streptococcus pyogenes and phagocytic killing. Nat Med. 2001;10:11044–11045. doi: 10.1038/nm1002-1044. Erratum in: Nat Med (2002) 8, 1329. [DOI] [PubMed] [Google Scholar]
- Wenig K, Chatwell L, von Pawel-Rammingen U, Bjorck L, Huber R, Sondermann P. Structure of the streptococcal endopeptidase IdeS, a cysteine proteinase with strict specificity for IgG. Proc Natl Acad Sci USA. 2004;101:17371–17376. doi: 10.1073/pnas.0407965101. [DOI] [PMC free article] [PubMed] [Google Scholar]