

SUMMARY
X-linked inhibitor of apoptosis (XIAP), traditionally known as an anti-apoptotic protein, has recently been shown to be involved in copper homeostasis. XIAP promotes the ubiquitination and degradation of COMMD1, a protein that promotes the efflux of copper from the cell. Through its effects on COMMD1, XIAP can regulate copper export from the cell and potentially represents an additional intracellular sensor for copper levels. XIAP binds copper directly and undergoes a substantial conformational change in the copper bound state. This in turn destabilizes XIAP, resulting in lowered steady-state levels of the protein. Furthermore, copper-bound XIAP is unable to inhibit caspases and cells that express this form of the protein exhibit increased rates of cell death in response to apoptotic stimuli. These events take place in the setting of excess intracellular copper accumulation as seen in copper toxicosis disorders such as Wilson's disease and establish a new relationship between copper levels and the regulation of cell death via XIAP. These findings raise important questions about the role of XIAP in the development of copper toxicosis disorders and may point to XIAP as a potential therapeutic target in these disease states.
INTRODUCTION
Apoptosis, or programmed cell death, is a fundamental process that is required for normal development and homeostasis of multicellular organisms. This process is tightly regulated at various levels and deregulated apoptosis has been implicated in a wide variety of human diseases [1-3]. Activation of caspases, a family of cysteine-aspartic-acid specific proteases is a critical event in the induction of apoptosis and occurs in response to a variety of stimuli [4, 5]. Caspase activation ultimately leads to the hallmarks of apoptosis including chromatin condensation, DNA fragmentation and plasma membrane blebbing. Inhibitor of apoptosis proteins (IAPs) provide protection from programmed cell death by binding to and inhibiting specific caspases [6, 7]. All IAPs contain between one and three tandem copies of a ∼70 amino acid motif known as a baculovirus IAP repeat (BIR) domain [8-10]. Of the eight human IAPs identified so far, five also contain a carboxy-terminal RING finger, which possesses E3 ubiquitin ligase activity and directs proteasomal-mediated degradation of target proteins (Figure 1). The BIR domains resemble the structure of zinc fingers and are capable of coordinating one atom of zinc to three cysteines and one histidine while the RING finger can chelate two zinc atoms utilizing cysteine and histidine moieties.
Figure 1.
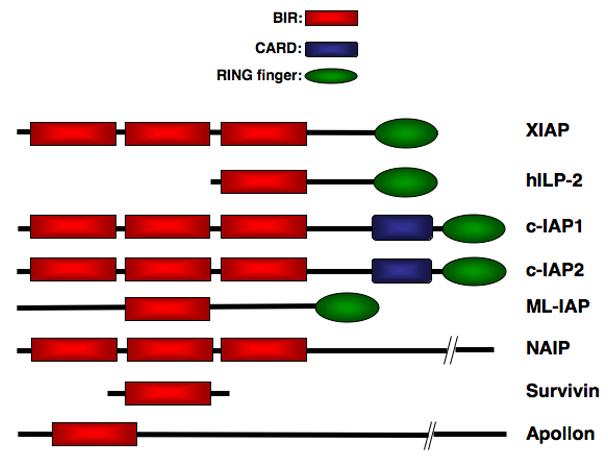
The IAP Family Members. Eight mammalian IAPs have been described so far and all are characterized the presence of one or more baculovirus IAP repeat (BIR) domains.
X-linked inhibitor of apoptosis (XIAP), a 57 kDa protein, is the best characterized member of the IAP family. It contains three amino-terminal BIR domains and a carboxy-terminal RING finger (Figure 1). The role of XIAP as a potent anti-apoptotic protein has been attributed to its ability to directly bind to and inhibit specific caspases [6]. The BIR3 domain of XIAP binds to the amino-terminus of processed caspase-9, an initiator caspase, which is activated following mitochondrial permeabilization and cytochrome c release [11, 12](Figure 2). XIAP binding to caspase-9 prevents its dimerization, and inhibits its protease activity. XIAP-mediated inhibition of the executioner caspases-3 and -7 is the result of binding to BIR2 and a segment immediately amino-terminal to BIR2 in XIAP, which blocks the active site of these caspases [13-16]. Whether the activation of apoptosis is initiated by events that perturb the mitochondria (via caspase-9) or progress directly from cell surface receptors (via caspase-8), the ability of XIAP to inhibit the downstream executioner caspases-3 and -7 makes it a potent and broad inhibitor of cell death. Initiation of the apoptotic cascade involves the inactivation of XIAP through the release of the mitochondrial proteins Smac/DIABLO and Omi/HtrA2 into the cytoplasm where they bind to XIAP at precisely the same domains that mediate the interactions of XIAP with the caspases [17-21]. Acting as competitive inhibitors of caspase binding and through other mechanisms, these factors inhibit XIAP function, thereby facilitating propagation of the apoptotic cascade via the executioner caspases (Figure 2). In addition to its anti-apoptotic properties, XIAP has also been implicated in a variety of intracellular signaling events including the NF-κB pathway [22, 23], the c-Jun-N-terminal kinase pathway [24, 25] and the TGF-β pathway [26-28].
Figure 2.
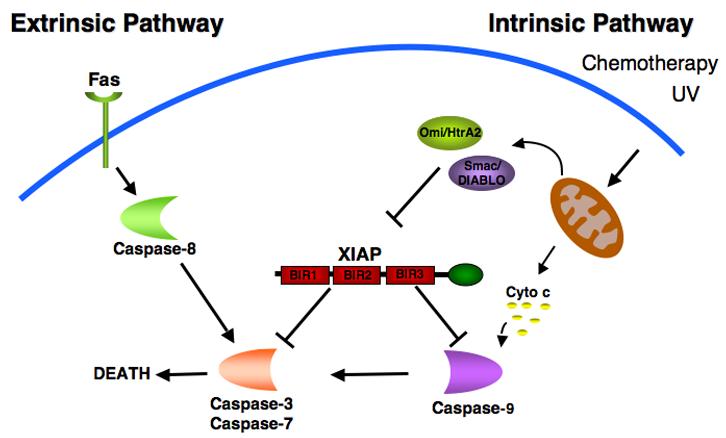
Role of XIAP in the extrinsic and intrinsic cell death signaling pathways. Two different pathways can initiate apoptosis. The death-receptor mediated (extrinsic) pathway involves the activation of caspase-8 following stimulation of the receptor by ligands such as Fas. The mitochondrial (intrinsic) pathway is initiated by a variety of stimuli including DNA damaging agents such as UV and chemotherapy resulting in the mitochondrial release of cytochrome c and subsequent activation of caspase-9. Both pathways converge to activate caspases- 3 and -7, effector caspases responsible for the death of the cell. XIAP regulates both pathways by its ability to directly bind and inhibit caspase-3, -7 and -9. The mitochondrial proteins, Smac and Omi, antagonize the caspase inhibitory properties of XIAP.
More recently, XIAP has been found to play an important role in intracellular copper homeostasis. It had previously been known that a mutation in COMMD1 (Copper Metabolism gene MURR1 domain containing 1) results in an autosomal recessive form of a non-Wilsonian copper toxicosis disorder affecting Bedlington terriers characterized by marked hepatic copper accumulation. A role for XIAP was first suggested in copper metabolism when COMMD1 and XIAP were found to bind each other in vivo. XIAP regulates COMMD1 levels by acting as an E3 ubiquitin ligase for the protein and promoting its proteosomal degradation [29]. By directing COMMD1 for proteasomal degradation, XIAP promotes copper retention in various models including cultured cells and Xiap deficient mice [29]. In addition to this role in copper homeostasis, XIAP turns out to be a copper binding protein whose intracellular levels and anti-apoptotic actions are negatively regulated by intracellular copper [30]. Therefore, by this model, copper itself provides negative feedback to the actions of XIAP within the intracellular environment.
COPPER METABOLISM IN EUKARYOTIC CELLS
Copper, a transition metal with two redox states, is an essential trace element that acts as a catalytic cofactor for a number of enzymes involved in critical biological processes including protection against free radicals (superoxide dismutase) [31], oxidative phosphorylation (cytochrome c oxidase) [32], pigmentation (tyrosinase) [33], collagen maturation (lysyl oxidase) [34] and neuropeptide and peptide hormone production (peptidylglycine alpha-amidating mono-oxygenase, PAM) [35]. However, free copper is very toxic due to its ability to react with hydrogen peroxide and generate highly reactive hydroxyl radicals. Consequently, free copper in the cell is almost undetectable and complex homeostatic mechanisms have evolved to regulate intracellular copper distribution and excretion [36].
Copper is transported into the cell via the high affinity Copper transporter (Ctr) proteins, which are highly conserved from yeast to humans [37, 38] (Figure 3). All Ctr proteins contain three transmembrane domains, and most possess extracellular, methionine rich motifs (MxxM, MxM) at the amino terminal and an additional MxxxM motif in the second transmembrane domain [39]. These domains are essential for binding extracellular copper and facilitating transport into the cell. In yeast, two functionally redundant high affinity copper transporters (yeast Ctr1 and Ctr3) are predominantly responsible for copper import in the form of Cu(I). Metalloreductases, Fre1 and Fre2, bound to the plasma membrane, are responsible for reducing extracellular Cu (II) to Cu (I) prior to transport into the cell [38]. Two Ctr homologs, Ctr1 and Ctr2, have also been identified in vertebrates, and of these, Ctr1 is the main protein responsible for intracellular copper transport. It is ubiquitously expressed and is absolutely required for embryonic development, as Ctr1-deficient mice die halfway through gestation due to growth retardation and impaired development of the neuroectoderm and mesoderm [37]. The exact function of Ctr2 in vertebrates remains unclear although the same protein in yeast has been shown to play an important role in mobilizing vacuolar copper stores [40].
Figure 3.
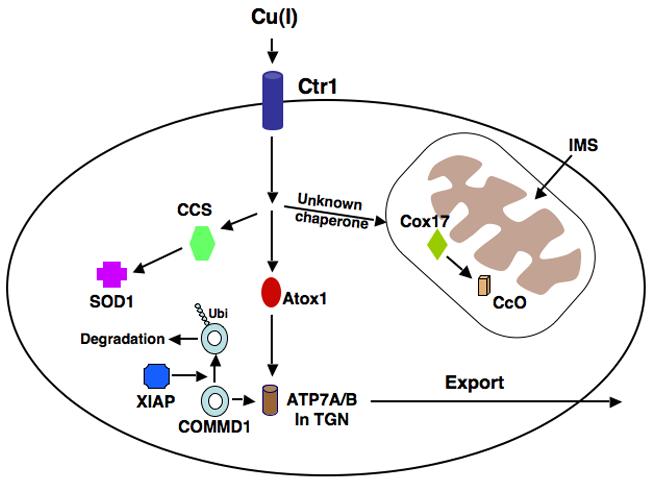
Model of copper uptake and metabolism. Copper enters the cell via the high affinity copper transporter, Ctr1. Atox1 is the chaperone responsible for transporting copper to the P-type ATPases 7A and 7B in the trans Golgi network (TGN). The chaperone, CCS, delivers Cu to cytosolic Cu/Zn superoxide dismutase (SOD1), and Cox17, in the mitochondria, delivers Cu to Cytochrome c Oxidase (CcO). The manner in which copper is transported from the cytosol to Cox 17 in the mitochondria has not been fully elucidated. COMMD1 promotes the efflux of copper from the cell, probably via its interaction with ATP7B. XIAP may reduce copper export by acting as an E3 Ubiquitin ligase and promoting the degradation of COMMD1.
Due to its toxicity, intracellular copper trafficking is very tightly regulated and evolutionarily conserved pathways have developed for the handling of copper upon entry into the cell involving proteins, known as chaperones, whose function is the delivery of copper to specific targets [41] (Figure 3). Delivery of copper to superoxide dismutase (SOD1), a zinc and copper containing protein, occurs via the Copper Chaperone for SOD (CCS), which is found in both the cytoplasm and the mitochondrial intermembrane space (IMS). SOD1 is an important enzyme, which catalyzes the conversion of superoxide anion to hydrogen peroxide and oxygen [42, 43]. Mutations in SOD1 are associated with amyotrophic lateral sclerosis (ALS), a neurodegenerative disorder characterized by the loss of upper and lower motor neurons and muscle atrophy [44, 45]. CCS-mediated delivery of copper to SOD1 is important for the stability of this protein as Ccs-deficient mice exhibit severe reductions in SOD1 levels and activity in various organs despite normal mRNA expression [46].
Another example of intracellular copper handling involves copper delivery to Cytochrome c Oxidase (CcO), the terminal enzyme in the respiratory chain (Figure 3). CcO has two catalytic subunits, Cox1 and Cox2, both encoded by the mitochondrial genome. These subunits require copper as a catalytic co-factor and copper insertion takes place within mitochondria. The copper chaperone Cox17 is one of the factors that plays an important role in the metallation of CcO and its deficiency results in the lack of copper delivery to CcO in yeast [47]. Cox17 is found in both the cytosol and mitochondria. It was originally thought that Cox 17 binds copper in the cytosol, translocates into mitochondria and delivers copper to Cox1 and Cox2 [48-50]. However, further studies in yeast showed that in Cox 17 deficient cells, CcO activity can be fully restored by mitochondrial restricted expression of Cox17 [51]. This indicates that a different and as yet unidentified protein is responsible for transporting copper from the cytosol to mitochondria where it eventually binds to Cox17 in the IMS. Copper is then transferred from Cox17 to two co-chaperones, Cox 11 and Sco1, which incorporate copper into the Cox1 and Cox2 subunits of CcO [52].
The delivery of Cu (I) to the P-type ATPases 7A and 7B (Ccc2 in yeast), the main transporters responsible for copper excretion from cells, also involves copper chaperones and has been extensively studied (Figure 3). ATP7A and 7B, which are both found in the trans-Golgi network, contain six MxCxxC metal binding domains (MBDs) at their amino termini and each MBD is capable of binding one atom of copper [53]. Antioxidant protein 1, or Atox1 (Atx1 in yeast) is a cytosolic copper chaperone responsible for the shuttling of Cu (I) to ATP7A and 7B [54-57]. Following binding to copper and under normal or low copper conditions, ATP7A and 7B translocate Cu (I) into the lumen of the secretory vesicles for integration into cuproenzymes such as ceruloplasmin and tyrosinase [58, 59]. The exact manner in which copper is loaded onto cuproenzymes in the secretory compartment is not known. ATP7A is present in most tissues of the body and is highly expressed in intestinal epithelial cells but is absent in hepatic tissue. In the intestinal epithelium, ATP7A is required to mediate delivery of copper from the cell to the systemic circulation and thus it is essential for the systemic absorption of copper from the gastrointestinal tract [60]. In the presence of elevated copper levels, ATP7A translocates to the plasma membrane and pumps excess copper out of the cell [61]. Mutations of ATP7A result in Menkes disease, an X-linked disorder characterized by a lack of intestinal copper absorption. Affected children have severe neurological damage and present with seizures, hypotonia, and failure to thrive [62]. ATP7B is structurally related to ATP7A, and in humans, it is predominantly expressed in the liver, kidney and brain [53]. Like ATP7A, in elevated copper states ATP7B moves from post-Golgi vesicles to the plasma membrane, specifically the biliary canalicular membrane allowing copper excretion into bile [63]. This constitutes the main pathway for copper efflux from the body and mutations in ATP7B in Wilson's disease result in the pathological accumulation of copper in many organs and tissues, particularly the liver and brain [64]. The hallmarks of the disease are the development of liver damage culminating in cirrhosis, and neurological damage, primarily to the basal ganglia, resulting in dysarthria, dyspraxia and ataxia, along with deposition of copper in the eye manifesting as Kayser-Fleischer rings [65].
XIAP AND COMMD1
Copper accumulation resembling Wilson's disease can occur in humans and animals in the absence of mutations in ATP7B. One such example is the copper toxicosis syndrome that affects a high proportion of a specific dog strain, the Bedlington terrier [66-68]. Although ATP7B is not mutated, these animals have severe impairment of biliary copper excretion similar to that observed in Wilson's disease [69]. Using positional cloning, MURR1 (now referred to as COMMD1) was identified as the gene responsible for this disorder [70]. Its protein product binds to the amino-terminus of ATP7B and presumably regulates its function by a mechanism yet to be identified [71]. COMMD1 is the prototypical member of a family of ten homologous and highly conserved proteins known as COMMD1 to 10 [72]. The defining characteristic is the presence of a unique carboxy-terminal domain termed the COMM domain (copper metabolism gene MURR1). In addition to its role in copper homeostasis, COMMD1 and other COMMD proteins also function as negative regulators of the transcription factor NF-κB. The functional link between these two disparate activities remains elusive.
The first clue that XIAP plays a role in copper homeostasis came from studies that identified COMMD1 as an XIAP-associated factor in a yeast two-hybrid screen [29]. XIAP binds to COMMD1 via its BIR3 domain in a manner independent of its binding to caspases. In keeping with this finding, COMMD1 does not affect the ability of XIAP to inhibit caspase-mediated cell death. However, like COMMD1, XIAP was found to affect intracellular and tissue copper levels, implicating this protein-protein interaction in the regulation of copper homeostasis. When COMMD1 levels are decreased in cultured cells by transfection of short interfering RNA (siRNA) oligonucleotides, intracellular copper levels become elevated, similar to the phenotype of COMMD1 deficient dogs which develop copper toxicosis. On the other hand, ectopic expression of XIAP in cell culture systems results in an increase in intracellular copper levels similar to the effect of COMMD1 deficiency. Corresponding to this finding, fibroblasts derived from Xiap-deficient mice have decreased copper levels compared to wild-type controls, and liver tissue copper content in Xiap-deficient mice is lower than in wild-type controls.
Therefore, XIAP and COMMD1 have inverse effects on intracellular copper content. In keeping with this, XIAP was found to act as a negative regulator of COMMD1 protein levels. An increase in cellular XIAP levels by ectopic expression results in a decrease in COMMD1 levels and, conversely, when endogenous XIAP levels are suppressed, COMMD1 levels increase. This effect is mediated by the E3 ubiquitin ligase activity of XIAP, which promotes the degradation of COMMD1 by the proteasome. Transfection of an XIAP point mutant lacking the E3 ubiquitin ligase activity of the wild-type protein results in an elevation in COMMD1 protein levels, and proteasomal blockade with lactacystin also has the same effect. Therefore, these studies demonstrated an unexpected role for XIAP in the regulation of intracellular copper levels through its ability to control COMMD1 protein levels. While many functions have been attributed to this protein, particularly its ability to inhibit caspases, it is noteworthy that Xiap-deficient mice primarily exhibit abnormalities in copper regulation, implying that other cellular mechanisms cannot compensate for this function of XIAP.
XIAP AND COPPER
In addition to playing an important role in copper homeostasis, more recent studies demonstrated that intracellular copper levels in turn regulate XIAP [30]. Besides its known ability to bind zinc [9, 73, 74], XIAP was found to be a strong copper binding protein. Intracellular copper accumulation induced by non-toxic increases of copper in the growth media resulted in the loading of copper onto XIAP, which can be easily detected by a characteristic alteration in its electrophoretic mobility. Purified recombinant XIAP avidly binds to immobilized copper, changing its electrophoretic mobility in a similar manner. Copper binding to XIAP can be reproduced in various cell-free in vitro systems including the addition of copper to cell lysates from various cell lines or to bacterially made, recombinant XIAP and can be reversed by the use of copper-specific chelators, further indicating that this is indeed a direct event. Interestingly, increasing concentrations of zinc did not reverse copper binding suggesting that copper and zinc bind to distinct sites on XIAP or that the affinity of XIAP for copper is greater than that for zinc. Biochemical studies demonstrated that cysteine residues that are distributed across the whole protein mediate this binding between XIAP and copper. Copper binding proteins classically contain domains that include the CxxC motif and this is the case for ATP7B [53] as well as XIAP, which contains CxxC motifs in each BIR domain as well as in the RING finger.
The interaction between XIAP and copper results in a profound conformational change of XIAP, demonstrated by the characteristic electrophoretic mobility change of copper-bound XIAP as well as a different pattern of proteolytic digestion of the copper bound form. Once XIAP undergoes a conformational change in the presence of copper, it is unable to effectively inhibit caspase-3 activity in vitro. Surprisingly, this loss of inhibitory activity is not due to a change in the ability of copper-loaded XIAP to bind caspase-3, as this interaction remains unaffected, but occurs through a yet undefined molecular mechanism. It therefore follows that intracellular copper accumulation and the resultant generation of the copper-bound form of XIAP increases the susceptibility of cells to apoptotic stimuli such as Tumor Necrosis Factor (TNF).
As well as resulting in a conformational change in the protein, copper binding to XIAP is accompanied by a decrease in its half-life in cells. Consistent with this effect, copper overload states such as Wilson's disease in humans result in decreased levels of XIAP protein in affected liver tissue. Other hallmarks of copper binding to XIAP, such as its altered electrophoretic mobility, are also seen in vivo confirming that this is a phenomenon that takes place in the setting of copper overload syndromes. The observation that copper binding to XIAP and its subsequent degradation occurs in disease states raises important questions about the pathogenesis of copper-induced cellular damage. This brings up a scenario in which aberrant copper accumulation as seen in Wilson's disease and other copper toxicosis disorders results in a conformational change in XIAP, where the protein is no longer able to inhibit caspase-3. In addition, copper destabilizes XIAP, thereby lowering its steady-state levels. The net effect of these two changes is to make the cell more susceptible to apoptotic stimuli and this may contribute to the pathophysiology of copper toxicosis syndromes (Figure 4). Furthermore, these findings point to the possibility of stabilizing XIAP, for example through the use of small molecule inhibitors, as part of the treatment of copper toxicosis disorders.
Figure 4.
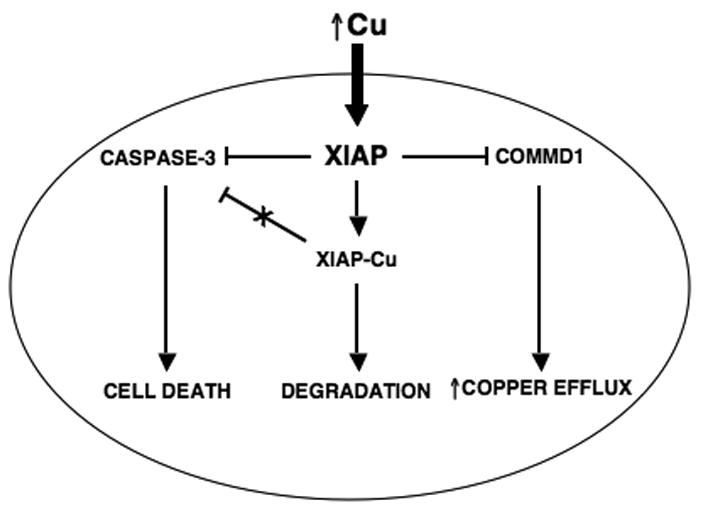
Overview of the relationship between copper, XIAP and the apoptotic threshold. Intracellular copper accumulation induces a conformational change of XIAP, which promotes its degradation and decreases its ability to inhibit caspase-3. The net result of these changes is a lowering of the apoptotic threshold and increased cell death in response to apoptotic stimuli. COMMD1 is involved in the efflux of copper from the cell and XIAP regulates it by promoting its ubiquitination and proteosomal degradation.
The profound effect that increased intracellular copper levels have on XIAP protein levels and function raises important questions that will need further investigation. Studies carried out thus far present the possibility that XIAP may be acting as an intracellular copper sensor. In this role, increases in copper levels result in a lowering of XIAP levels leading to a subsequent elevation of its ubiquitination target, COMMD1, culminating in the efflux of copper from the cell (Figure 4). Another critical question is how copper becomes incorporated into XIAP and whether an unknown protein chaperone is involved. Similar to this, it is also possible that XIAP may act as a copper chaperone, which is responsible for the delivery of copper to another cuproprotein in the intracellular environment. Given that the E3 Ubiquitin ligase activity of XIAP can be directed to the copper regulator COMMD1, it is equally possible that XIAP may function as a regulator of other steps of copper metabolism. For example, the possibility that it may ubiquitinate known copper chaperones, thereby affecting the metallation of well known cuproenzymes such as SOD1 and CcO needs to be examined. Further studies are needed to elucidate the precise role of XIAP in copper metabolism and what role if any this novel function has on the anti-apoptotic and signaling functions of XIAP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Jacobson MD, Weil M, Raff MC. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 2.Kerr JF, Wyllie AH, Currie AR. Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson CB. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 4.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 5.Nicholson DW. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 6.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 7.Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. Embo J. 1998;17:2215–2223. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duckett CS, Nava VE, Gedrich RW, Clem RJ, Van Dongen JL, Gilfillan MC, Shiels H, Hardwick JM, Thompson CB. EMBO J. 1996;15:2685–2694. [PMC free article] [PubMed] [Google Scholar]
- 9.Hinds MG, Norton RS, Vaux DL, Day CL. Nat Struct Biol. 1999;6:648–651. doi: 10.1038/10701. [DOI] [PubMed] [Google Scholar]
- 10.Rothe M, Pan M-G, Henzel WJ, Ayres TM, Goeddel DV. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 11.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 12.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 13.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- 15.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 16.Stennicke HR, Ryan CA, Salvesen GS. Trends Biochem Sci. 2002;27:94–101. doi: 10.1016/s0968-0004(01)02045-x. [DOI] [PubMed] [Google Scholar]
- 17.Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Nature. 2000;406:855–862. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 18.Du C, Fang M, Li Y, Li L, Wang X. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW. Nature. 2000;408:1004–1008. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 20.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 21.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 22.Hofer-Warbinek R, Schmid JA, Stehlik C, Binder BR, Lipp J, de Martin R. J Biol Chem. 2000;275:22064–22068. doi: 10.1074/jbc.M910346199. [DOI] [PubMed] [Google Scholar]
- 23.Levkau B, Garton KJ, Ferri N, Kloke K, Nofer JR, Baba HA, Raines EW, Breithardt G. Circ Res. 2001;88:282–290. doi: 10.1161/01.res.88.3.282. [DOI] [PubMed] [Google Scholar]
- 24.Sanna MG, Da Silva Correia J, Luo Y, Chuang B, Paulson LM, Nguyen B, Deveraux QL, Ulevitch RJ. J Biol Chem. 2002;277:30454–30462. doi: 10.1074/jbc.M203312200. [DOI] [PubMed] [Google Scholar]
- 25.Sanna MG, Duckett CS, Richter BW, Thompson CB, Ulevitch RJ. Proc Natl Acad Sci U S A. 1998;95:6015–6020. doi: 10.1073/pnas.95.11.6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birkey Reffey S, Wurthner JU, Parks WT, Roberts AB, Duckett CS. J Biol.Chem. 2001;276:26542–26549. doi: 10.1074/jbc.M100331200. [DOI] [PubMed] [Google Scholar]
- 27.Lewis J, Burstein E, Birkey Reffey S, Bratton SB, Roberts AB, Duckett CS. J Biol Chem. 2004;279:9023–9029. doi: 10.1074/jbc.M312891200. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi K, Nagai S, Ninomiya-Tsuji J, Nishita M, Tamai K, Irie K, Ueno N, Nishida E, Shibuya H, Matsumoto K. EMBO J. 1999;18:179–187. doi: 10.1093/emboj/18.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burstein E, Ganesh L, Dick RD, van De Sluis B, Wilkinson JC, Klomp LW, Wijmenga C, Brewer GJ, Nabel GJ, Duckett CS. Embo J. 2004;23:244–254. doi: 10.1038/sj.emboj.7600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mufti AR, Burstein E, Csomos RA, Graf PC, Wilkinson JC, Dick RD, Challa M, Son JK, Bratton SB, Su GL, Brewer GJ, Jakob U, Duckett CS. Mol Cell. 2006;21:775–785. doi: 10.1016/j.molcel.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 31.Torres AS, Petri V, Rae TD, O'Halloran TV. J Biol Chem. 2001;276:38410–38416. doi: 10.1074/jbc.M104790200. [DOI] [PubMed] [Google Scholar]
- 32.Hamza I, Gitlin JD. J Bioenerg Biomembr. 2002;34:381–388. doi: 10.1023/a:1021254104012. [DOI] [PubMed] [Google Scholar]
- 33.Prohaska JR, Gybina AA. J Nutr. 2004;134:1003–1006. doi: 10.1093/jn/134.5.1003. [DOI] [PubMed] [Google Scholar]
- 34.Kagan HM, Li W. J Cell Biochem. 2003;88:660–672. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 35.Uauy R, Olivares M, Gonzalez M. Am J Clin Nutr. 1998;67:952S–959S. doi: 10.1093/ajcn/67.5.952S. [DOI] [PubMed] [Google Scholar]
- 36.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 37.Lee J, Prohaska JR, Thiele DJ. Proc Natl Acad Sci U S A. 2001;98:6842–6847. doi: 10.1073/pnas.111058698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puig S, Thiele DJ. Curr Opin Chem Biol. 2002;6:171–180. doi: 10.1016/s1367-5931(02)00298-3. [DOI] [PubMed] [Google Scholar]
- 39.Guo Y, Smith K, Lee J, Thiele DJ, Petris MJ. J Biol Chem. 2004;279:17428–17433. doi: 10.1074/jbc.M401493200. [DOI] [PubMed] [Google Scholar]
- 40.Rees EM, Lee J, Thiele DJ. J Biol Chem. 2004;279:54221–54229. doi: 10.1074/jbc.M411669200. [DOI] [PubMed] [Google Scholar]
- 41.O'Halloran TV, Culotta VC. J Biol Chem. 2000;275:25057–25060. doi: 10.1074/jbc.R000006200. [DOI] [PubMed] [Google Scholar]
- 42.Culotta VC, Klomp LW, Strain J, Casareno RL, Krems B, Gitlin JD. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- 43.Casareno RL, Waggoner D, Gitlin JD. J Biol Chem. 1998;273:23625–23628. doi: 10.1074/jbc.273.37.23625. [DOI] [PubMed] [Google Scholar]
- 44.Rowland LP, Shneider NA. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 45.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 46.Wong PC, Waggoner D, Subramaniam JR, Tessarollo L, Bartnikas TB, Culotta VC, Price DL, Rothstein J, Gitlin JD. Proc Natl Acad Sci U S A. 2000;97:2886–2891. doi: 10.1073/pnas.040461197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glerum DM, Shtanko A, Tzagoloff A. J Biol Chem. 1996;271:14504–14509. doi: 10.1074/jbc.271.24.14504. [DOI] [PubMed] [Google Scholar]
- 48.Beers J, Glerum DM, Tzagoloff A. J Biol Chem. 1997;272:33191–33196. doi: 10.1074/jbc.272.52.33191. [DOI] [PubMed] [Google Scholar]
- 49.Heaton D, Nittis T, Srinivasan C, Winge DR. J Biol Chem. 2000;275:37582–37587. doi: 10.1074/jbc.M006639200. [DOI] [PubMed] [Google Scholar]
- 50.Heaton DN, George GN, Garrison G, Winge DR. Biochemistry. 2001;40:743–751. doi: 10.1021/bi002315x. [DOI] [PubMed] [Google Scholar]
- 51.Cobine PA, Pierrel F, Winge DR. Biochim Biophys Acta. 2006;1763:759–772. doi: 10.1016/j.bbamcr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 52.Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. J Biol Chem. 2004;279:35334–35340. doi: 10.1074/jbc.M404747200. [DOI] [PubMed] [Google Scholar]
- 53.Lutsenko S, Efremov RG, Tsivkovskii R, Walker JM. J Bioenerg Biomembr. 2002;34:351–362. doi: 10.1023/a:1021297919034. [DOI] [PubMed] [Google Scholar]
- 54.Hamza I, Schaefer M, Klomp LW, Gitlin JD. Proc Natl Acad Sci U S A. 1999;96:13363–13368. doi: 10.1073/pnas.96.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larin D, Mekios C, Das K, Ross B, Yang AS, Gilliam TC. J Biol Chem. 1999;274:28497–28504. doi: 10.1074/jbc.274.40.28497. [DOI] [PubMed] [Google Scholar]
- 56.Pufahl RA, Singer CP, Peariso KL, Lin SJ, Schmidt PJ, Fahrni CJ, Culotta VC, Penner-Hahn JE, O'Halloran TV. Science. 1997;278:853–856. doi: 10.1126/science.278.5339.853. [DOI] [PubMed] [Google Scholar]
- 57.Wernimont AK, Huffman DL, Lamb AL, O'Halloran TV, Rosenzweig AC. Nat Struct Biol. 2000;7:766–771. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 58.Dierick HA, Adam AN, Escara-Wilke JF, Glover TW. Hum Mol Genet. 1997;6:409–416. doi: 10.1093/hmg/6.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki M, Gitlin JD. Pediatr Int. 1999;41:436–442. doi: 10.1046/j.1442-200x.1999.01090.x. [DOI] [PubMed] [Google Scholar]
- 60.Voskoboinik I, Camakaris J. J Bioenerg Biomembr. 2002;34:363–371. doi: 10.1023/a:1021250003104. [DOI] [PubMed] [Google Scholar]
- 61.Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J. Embo J. 1996;15:6084–6095. [PMC free article] [PubMed] [Google Scholar]
- 62.Andrews NC. Curr Opin Chem Biol. 2002;6:181–186. doi: 10.1016/s1367-5931(02)00307-1. [DOI] [PubMed] [Google Scholar]
- 63.Roelofsen H, Wolters H, Van Luyn MJ, Miura N, Kuipers F, Vonk RJ. Gastroenterology. 2000;119:782–793. doi: 10.1053/gast.2000.17834. [DOI] [PubMed] [Google Scholar]
- 64.Gitlin JD. Gastroenterology. 2003;125:1868–1877. doi: 10.1053/j.gastro.2003.05.010. [DOI] [PubMed] [Google Scholar]
- 65.Brewer GJ. Proc Soc Exp Biol Med. 2000;223:39–46. doi: 10.1046/j.1525-1373.2000.22305.x. [DOI] [PubMed] [Google Scholar]
- 66.Wijmenga C, Klomp LW. Proc Nutr Soc. 2004;63:31–39. doi: 10.1079/pns2003316. [DOI] [PubMed] [Google Scholar]
- 67.Sternlieb I. J Rheumatol Suppl. 1981;7:94–95. [PubMed] [Google Scholar]
- 68.Haywood S, Fuentealba IC, Foster J, Ross G. Anal Cell Pathol. 1996;10:229–241. [PubMed] [Google Scholar]
- 69.Brewer GJ. Am J Clin Nutr. 1998;67:1087S–1090S. doi: 10.1093/ajcn/67.5.1087S. [DOI] [PubMed] [Google Scholar]
- 70.van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C. Hum Mol Genet. 2002;11:165–173. doi: 10.1093/hmg/11.2.165. [DOI] [PubMed] [Google Scholar]
- 71.Tao TY, Liu F, Klomp L, Wijmenga C, Gitlin JD. J Biol Chem. 2003;278:41593–41596. doi: 10.1074/jbc.C300391200. [DOI] [PubMed] [Google Scholar]
- 72.Burstein E, Hoberg JE, Wilkinson AS, Rumble JM, Csomos RA, Komarck CM, Maine GN, Wilkinson JC, Mayo MW, Duckett CS. J Biol Chem. 2005;280:22222–22232. doi: 10.1074/jbc.M501928200. [DOI] [PubMed] [Google Scholar]
- 73.Miller LK. Trends Cell Biol. 1999;9:323–328. doi: 10.1016/s0962-8924(99)01609-8. [DOI] [PubMed] [Google Scholar]
- 74.Sun C, Cai M, Gunasekera AH, Meadows RP, Wang H, Chen J, Zhang H, Wu W, Xu N, Ng SC, Fesik SW. Nature. 1999;401:818–822. doi: 10.1038/44617. [DOI] [PubMed] [Google Scholar]