

Abstract
Skeletal growth is tightly coupled to energy balance via complex and incompletely understood mechanisms. Leptin-deficient ob/ob mice are obese and develop multiple pathologies associated with the metabolic syndrome. Additionally, ob/ob mice have skeletal abnormalities. The objective of this study was to evaluate the effects of leptin deficiency and long-duration selective central leptin repletion via recombinant adeno-associated virus-leptin (rAAV-lep) gene therapy on bone in growing ob/ob mice. The ob/ob mice were injected in the hypothalamus with either rAAV-lep or rAAV-GFP (control vector). Treated ob/ob and untreated wildtype (WT) mice were then maintained on a normal diet for 15 weeks. In a second experiment, similarly treated mice along with a group of pair-fed mice were maintained for 30 weeks. Leptin was not detected in blood of either rAAV-lep or rAAV-GFP-treated mice although rAAV-lep treated mice displayed leptin transgene expression in the hypothalamus. As expected, rAAV-lep normalized body weight and food intake. Compared to WT mice, rAAV-GFP-treated ob/ob mice had decreased femoral length (by 1.6 mm or 10 %, P<0.001), decreased total femur bone volume (by 3.3 mm3 or 19%, P<0.001), but increased cancellous bone volume in the distal femur (by 0.04 mm3 or 60%, P<0.09) and lumbar vertebrae (by 0.26 mm3 or 118%, P<0.001). Treatment with rAAV-lep rescued the ob/ob skeletal phenotype by increasing femoral length and total bone volume, and decreasing femoral and vertebral cancellous bone volume, so that at 15 weeks post-rAAV-lep injection the ob/ob mice no longer differed from WT mice. No further skeletal changes in either the femur or lumbar vertebra were observed at 30 weeks post-rAAV-lep administration. The results suggest that hypothalamic leptin functions as an essential permissive factor for normal bone growth.
Keywords: osteoporosis, peak bone mass, μCT, obesity
I. Introduction
Osteoporosis, with no cure on the horizon, contributes to over 1,500,000 fractures annually in the United States alone [52]. Because a low peak bone mass predisposes individuals to osteoporotic fractures, an understanding of the factors that determine peak bone mass is essential for the prevention of this disease. The acquisition of peak bone mass occurs during childhood and the decade following puberty, and is tightly coupled to energy metabolism. At the extremes, obesity and anorexia each result in skeletal adaptations. Leptin, the protein product of the ob (Lep) gene [55], is a pleiotropic hormone that acts on multiple organs, including bone. Leptin is produced predominantly by fat cells, functions as a sentinel of energy balance, and has been recently identified as an important bone regulatory factor [26]. The obese ob/ob mouse, which cannot produce leptin due to an inactivating mutation in the leptin gene has skeletal abnormalities; compared to wildtype (WT) mice, ob/ob mice have reduced bone length and reduced overall bone mass [27,32,33,36,44]. These findings suggest leptin is required for optimal peak bone growth and quality. However, the mechanisms of action of leptin on the skeleton are not fully understood. In part, this is because the hormone has the potential to affect bone through multiple pathways; an indirect pathway involving a central/hypothalamic relay [3,26,45] and a direct pathway involving the binding of leptin to its receptors directly on cartilage and bone cells [17,20,32,39,46-48].
Some of the indirect effects of leptin on bone may be mediated through the sympathetic arm of the autonomic nervous system; the binding of leptin to its receptors in the hypothalamus stimulates the peripheral cells of the sympathetic nervous system to release noradrenalin which, in turn, could influence bone formation via adrenergic receptors expressed on osteoblasts and other cells [26,42,45]. Additional indirect effects may be mediated through changes in systemic metabolic bone regulatory factors such as IGF-I [10,15,34]. The direct action of leptin on bone and cartilage cells has been amply demonstrated in cell culture [20,36,39,48]. The systemic administration of leptin, via intraperitoneal or subcutaneous injection, has also been shown to increase total body bone mass and bone length in ob/ob mice and increase bone length in calorie-deprived mice, suggesting that bone growth is regulated by the circulating levels of leptin [30,32,44]. Increasing systemic leptin within the physiological range has consistently been shown to increase hypothalamic leptin levels, but the effects of central infusion on circulating leptin levels are less consistent [7,26,29,38,43,46,54]. As a consequence, neither systemic nor central applications of the hormone can unambiguously differentiate between the contributions of systemic versus central leptin in the putative regulation of skeletal metabolism. In addition, all experiments to date investigating the actions of peripheral and central leptin on bone metabolism in rodents have been of short duration (≤ 1 month). Finally, no studies have investigated the effects of leptin on peak bone mass.
We have recently employed central leptin gene transfer technology by non-replicative, non-immunogenic and non-pathogenic virus vector (rAAV) to augment leptin availability selectively in the hypothalamus [21,22,56]. These studies demonstrated that microinjection of rAAV encoding leptin gene (rAAV-lep) into either the third cerebroventricular or other selective hypothalamic sites enhanced leptin transgene expression locally in the hypothalamus and not at distant non-hypothalamic sites for the duration of the experiments [1,2,8,9,11-14,21- 24,34,37,50,51]. The leptin protein transduced in neurons was expressed in amounts sufficient to exert leptin-specific biological effects but insufficient for detection in the hypothalamic tissue surrounding the injection site, cerebroventricular fluid or peripheral circulation [1,2,11,14,21,34,50,51]. Therefore, the objective of this study was to evaluate the effects of leptin deficiency and selective hypothalamic leptin expression of long duration by a single intracerebroventricular (icv) injection of rAAV-lep on bone size and cancellous bone architecture in growing ob/ob mice.
2. Materials and Methods
2.1 Experimental Animals
Male, 8-10 week old leptin-replete WT (C57BL/6) and leptin-deficient ob/ob (weighing 40-50 g) mice, obtained from Jackson Laboratories (Bar Harbor, Maine), were used in the experiments. The mice were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Florida (Gainesville, FL). The mice were housed individually in a temperature (21-23°C)- and light-controlled room (lights on 6am-6pm) under specific pathogen-free conditions. Food and water were available ad libitum.
2.2 Construction and Packaging of rAAV Vectors
The rAAV-lep and rAAV-green fluorescent protein (rAAV-GFP) vectors were constructed and packaged as described elsewhere [9]. In brief, the vector pTR-CBA-Ob EcoRI fragment of pCR-rOb (a gift from Dr. Roger H. Unger, University of Texas Southwestern Medical Center, Dallas, TX) containing rat leptin cDNA was subcloned into rAAV vector plasmid pAAVβGEnh after deleting the EcoRI fragment carrying the β-glucoronidase cDNA sequence [21-23,25,56]. The control vector, rAAV-GFP, was similarly constructed to encode the GFP gene [21,23,25,56].
2.3 Experiment 1
Male WT and ob/ob mice were used in the experiment. The WT mice (n=3) were untreated. The ob/ob mice were anesthetized with sodium pentobarbital (60mg/kg, ip) and placed on a Kopf stereotaxic apparatus with mouse adapter for intracerebroventricular (icv) injection. The coordinates employed for microinjector placement were 0.3 mm posterior to bregma, 0.0 lateral to midline, and 4.2 mm below the dura [11,50,51]. Various procedures to verify icv injection are detailed elsewhere [1,2,8,9,11-14,21-24]. Mice were injected icv with either rAAV-GFP (control vector, 9×107 particles; n=8) or rAAV-lep (9×107 particles; n=9). All mice were then maintained on standard mouse chow fed ad lib for 15 weeks. Food intake and body weight response are detailed elsewhere [11,14]. Also detailed elsewhere are hormone levels (e.g., insulin and leptin) and hypothalamic leptin mRNA expression in response to rAAV-lep therapy in these mice [11,14,50,51].
2.4. Experiment 2
Male ob/ob mice received an icv injection of either rAAV-lep (n=9) or rAAV-GFP (n=9) as described above. Additional controls included untreated (n=7) and pair-fed (mice allowed to consume amounts of food equivalent to that of the rAAV-lep-treated mice, n =7) ob/ob mice. The mice were maintained on standard mouse chow for 30 weeks post-vector administration. Food intake and body weight response during the experimental period, as well as hypothalamic leptin transgene expression and hormonal response to treatment are detailed elsewhere [11,14,50,51].
2.5. Tissue Collection and Analyses
At the end of each experiment, mice were weighed and anesthetized with sodium pentobarbital (60mg/kg; ip). Blood was withdrawn by orbital sinus puncture and serum was stored at -20°C for analysis [11,14,50]. These mice were then sacrificed by decapitation. Femora and 3rd lumbar vertebrae were excised for μCT analysis. The μCT was used for nondestructive three-dimensional evaluation of bone architecture. In all mice, right femora were scanned using a Scanco μCT40 scanner (Scanco Medical AG, Basserdorf, Switzerland) at a voxel size of 12 × 12 × 12 μm and a threshold of 256 (gray scale, 0-1,000). The threshold value was determined empirically. Entire femora (cancellous + cortical bone) were evaluated followed by evaluation of cortical bone at the femoral midshaft and cancellous bone in the distal femoral metaphysis. For the femoral midshaft, 100 slices (1.2 mm) of bone were evaluated and total cross-sectional tissue volume (cortical and marrow volume, mm3), cortical volume/total volume (volume of total tissue occupied by cortical bone, %), marrow volume (mm3) and cortical thickness (μm) were measured. For the femoral metaphysis, ~200 slices (2.4 mm) of bone were measured and included secondary spongiosa only. Analysis of the lumbar vertebra included the entire region of secondary spongiosa between the cranial and caudal growth plates. Direct cancellous bone measurements in the femur and lumbar vertebra included: 1) cancellous bone volume (bone volume unadjusted for tissue volume, mm3), 2) cancellous bone volume/tissue volume (volume of total tissue occupied by cancellous bone, %), 3) trabecular thickness (mean thickness of individual trabeculae, μm), 4) trabecular number (number of trabeculae within the samples tissue, 1/mm), and 4) trabecular separation (the distance between trabeculae, μm) [49]. The coefficient of variation (based on 6 repeat measurements by the same individual) is 0% for total femur bone volume, 2.7% for distal femur cancellous bone volume, 0.1% for midshaft femur total volume, and 0.8% for vertebral cancellous bone volume.
2.6 Statistical Analysis
A one-way ANOVA followed by a Bonferroni post-hoc test was used to evaluate differences among treatment groups. Differences were considered significant at p<0.05. All data are presented as mean±SE.
3. Results
3.1 Experiment 1
rAAV-GFP-treated ob/ob mice weighed significantly more than WT mice (Table 1). At 15 weeks post-vector administration, body weight in rAAV-lep-treated ob/ob mice was decreased compared to their starting weights, 50% lower than in the rAAV-GFP-treated mice, and not different from that of WT mice. Daily food consumption was suppressed in these rAAV-lep injected mice [11,14]. These biological effects resulted from increased leptin supply at targets selectively in the hypothalamus as indicated by analysis of leptin mRNA expression specifically in the hypothalamic neurons and by localization of GFP by immunohistochemistry [9,11,14,21,23,24,50,51]. Leptin protein transduced in the hypothalamus was below the detectable range of radioimmunoassay and immunoblotting assays [1,2,11,14].
Table 1.
Effects of central rAAV-lep therapy on bone in leptin-deficient ob/ob mice at 15 weeks post vector administration. Vectors were administered when the ob/ob mice weighed 45 grams (8-10 weeks old)
| Endpoint | WT
Untreated (n=3) |
ob/ob
rAAV-GFP (n=8) |
ob/ob
rAAV-lep (n=9) |
ANOVA (P<) |
|---|---|---|---|---|
| Body weight at necropsy (g) | 29 ± 1* | 56 ± 2a,b | 28 ± 3 | 0.000 |
| Total femur (cortical + cancellous bone) | ||||
| Length (mm) | 15.3 ± 0.1 | 13.7 ± 0.2a,b | 15.1 ± 0.1 | 0.000 |
| Bone volume (mm3) | 17.6 ± 0.2 | 14.3 ± 0.6a,b | 16.2 ± 0.4 | 0.004 |
| Midshaft femur (cortical bone) | ||||
| Total volume (mm3) | 2.34 ± 0.01 | 2.22 ± 0.051 | 2.19 ± 0.03 | 0.161 |
| Cortical volume/Total volume (%) | 38 ± 1 | 35 ± 1 | 37 ± 1 | 0.202 |
| Marrow volume (mm3) | 1.44 ± 0.02 | 1.45 ± 0.05 | 1.39 ± 0.03 | 0.478 |
| Cortical thickness (μm) | 167 ± 3 | 148 ± 4 | 156 ± 5 | 0.151 |
| Distal femur (cancellous bone) | ||||
| Bone volume (mm3) | 0.07 ±0.003 | 0.11 ± 0.01a*,b | 0.05 ± 0.01 | 0.001 |
| Bone volume/Total volume (%) | 3.2 ± .6 | 4.2 ± .4b | 2.2 ± .3 | 0.003 |
| Trabecular thickness (μm) | 34 ± 1 | 39 ± 1a*,b | 30 ± 1 | 0.000 |
| Trabecular number (1/mm) | 3.4 ± .1 | 2.8 ± .1a,b | 3.2 ± .1 | 0.012 |
| Trabecular spacing (μm) | 297 ± 5 | 359 ± 14a,b | 317 ± 8 | 0.012 |
| Lumbar vertebra (cancellous bone) | ||||
| Bone volume (mm3) | 0.22 ± .01 | 0.48 ± .03a,b | 0.23 ± .01 | 0.000 |
| Bone volume/Total volume (%) | 10.7 ± .7 | 23.4 ± 1.4a,b | 9.8 ± 1.0 | 0.000 |
| Trabecular thickness (μm) | 33 ± 1 | 40 ± 1a,b | 32 ± 1 | 0.000 |
| Trabecular number (1/mm) | 4.4 ± .02 | 6.4 ± .1a,b | 4.8 ± .2 | 0.000 |
| Trabecular spacing (μm) | 227 ± 1 | 153 ± 4a,b | 211 ± 6 | 0.000 |
Data are mean ± SE
Different from WT, P<0.05
P<0.1
Different from rAAV-lep, P<0.05
P<0.1
Compared to WT mice, rAAV-GFP-treated ob/ob mice had lower femoral length (by 1.6 mm or 10 %, P<0.001) and lower total femur bone volume (by 3.3 mm3 or 19%, P<0.001). Treatment with rAAV-lep increased femur length and total femur bone volume, so that at 15 weeks post-rAAV-lep injection the ob/ob mice no longer differed from WT mice in either bone parameter (Table 1 and Figure 1). Significant differences in midshaft cortical measurements were not detected between WT and rAAV-GFP mice (Table 1). Total cancellous bone volume in the distal femur tended to be higher (by 0.04 mm3 or 60%, P<0.09) in the rAAV-GFP-treated ob/ob mice than in WT mice (Table 1 and Figure 2). Cancellous bone architecture in the distal femur was abnormal in the ob/ob mice; trabecular thickness tended to be higher and there was a decrease in trabecular number and an increase in trabecular spacing in the GFP-treated ob/ob mice compared to WT mice. rAAV-lep gene therapy restored femur cancellous architecture in ob/ob mice to values that did not differ from that of WT mice (Table 1). Cancellous bone volume in the lumbar vertebrae, both unadjusted and adjusted for total tissue volume evaluated, was higher in rAAV-GFP-treated ob/ob mice than in WT mice (Table 1). The increased cancellous bone volume in the rAAV-GFP group was due to increases in trabecular thickness and trabecular number. Administration of rAAV-lep to ob/ob mice restored all vertebral measurements to values that did not differ from WT mice.
Figure 1.
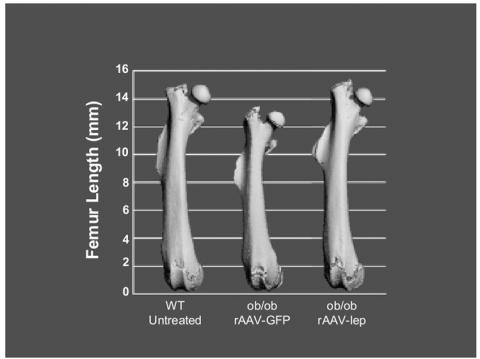
Central rAAV-lep therapy restores femoral length and total bone volume in ob/ob mice.
Figure 2.
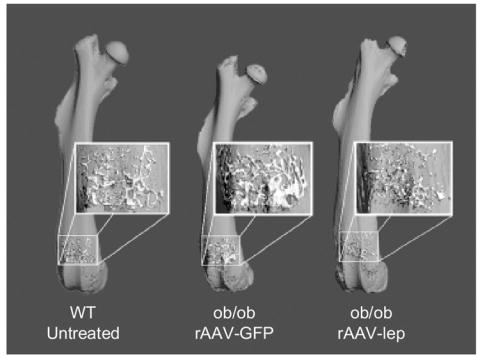
Effects of central rAAV-lep therapy on cancellous bone volume in the distal femur in ob/ob mice.
3.2. Experiment 2
As in the 15-week study, hypothalamic rAAV-lep administration to ob/ob mice decreased weight at 30 weeks post-vector administration (Table 2), along with a sustained decrease in daily food consumption soon after rAAV-lep injection [11,14,34,50,51]. Pair-feeding did not affect bodyweight at week 30 (Table 2), presumably due to an adaptive decrease in energy expenditure [1,2,9]. We have shown previously that pair-fed rodents lose weight early on but regain weight due to homeostatic adaptation [1,2,9,12,13,21-23,25,34,37].
Table 2.
Effects of central rAAV-lep therapy on bone in leptin-deficient ob/ob mice at 30 weeks post vector administration. Vectors were administered when the ob/ob mice weighed 45 grams (8-10 weeks old)
| Endpoint | ob/ob
Untreated (n=7) |
ob/ob
rAAV-GFP (n=9) |
ob/ob
rAAV-lep (n=9) |
ob/ob
Pair-fed (n=7) |
ANOVA P< |
|---|---|---|---|---|---|
| Body weight at necropsy (g) | 70 ± 2* | 68 ± 2 | 29 ± 3a,b,c | 63 ± 2 | 0.000 |
| Total femur (cortical + cancellous bone) | |||||
| Length (mm) | 14.3 ± 0.2 | 14.2 ± 0.2 | 15.3 ± 0.1a,b,c | 14.2 ± 0.2 | 0.000 |
| Bone volume (mm3) | 17.1 ± 0.4 | 15.8 ± 0.5 | 16.8 ± 0.6 | 16.1 ± 0.5 | 0.347 |
| Midshaft femur (cortical bone) | |||||
| Total volume (mm3) | 2.50 ± 0.03 | 2.44 ± 0.04 | 2.39 ± 0.05 | 2.43 ± 0.06 | 0.440 |
| Cortical volume/Total volume (%) | 37.2 ± 0.6 | 35.5 ± 0.9 | 37.6 ± .9 | 35.4 ± .9 | 0.175 |
| Marrow volume (mm3) | 1.57 ± 0.03 | 1.57 ± 0.03 | 1.49 ± 0.03 | 1.57 ± 0.04 | 0.145 |
| Cortical thickness (μm) | 167 ± 3 | 158 ± 5 | 165 ± 5 | 157 ± 5 | 0.404 |
| Distal femur (cancellous bone) | |||||
| Bone volume (mm3) | 0.15 ± .01 | 0.16 ± .02 | 0.08 ± .02a,b,c* | 0.14 ± .02 | 0.004 |
| Bone volume/Total volume (%) | 5.2 ± .5 | 4.9 ± .6 | 3.0 ± .6a*,b* | 4.6 ± .3 | 0.030 |
| Trabecular thickness (mm) | 45 ± 1 | 44 ± 1 | 36 ± 1a,b,c | 42 ± 1 | 0.000 |
| Trabecular number (1/mm) | 2.8 ± .2 | 2.7 ± .1 | 3.1 ± .1 | 2.7 ± .1 | 0.111 |
| Trabecular spacing (mm) | 362 ± 18 | 374 ± 12 | 332 ± 13 | 366 ± 12 | 0.151 |
| Lumbar vertebra (cancellous bone) | |||||
| Bone volume (mm3) | 0.50 ± .02 | 0.56 ± .05 | 0.23 ± .04a,b,c | 0.49 ± .04 | 0.000 |
| Bone volume/Total volume (%) | 21.4 ± 0.8 | 22.7 ± 1.8 | 9.3 ± 1.5a,b,c | 21.0 ± 1.8 | 0.000 |
| Trabecular thickness (mm) | 40 ± 1 | 40 ± 1 | 32 ± 1a,b,c | 40 ± 1 | 0.000 |
| Trabecular number (1/mm) | 5.9 ± .1 | 6.1 ± .2 | 4.5 ± .3a,b,c | 5.9 ± .2 | 0.000 |
| Trabecular spacing (mm) | 166 ± 3 | 159 ± 6 | 236 ± 27a,b,c | 163 ± 4 | 0.005 |
Data are mean ± SE
Different from untreated control, P<0.05
P<0.1
Different from rAAV-GFP control, P<0.05
P<0.1
Different from pair-fed control, P<0.05
P<0.1
Treatment with rAAV-lep resulted in greater femoral length and lower distal femur cancellous bone volume relative to all other treatment groups. Significant inter-group differences were not detected in total femur bone volume or midshaft cortical measurements. Vertebral cancellous bone volume, both unadjusted and adjusted for total volume evaluated, was lower in rAAV-lep-treated mice in comparison to all other groups evaluated. This was due to lower trabecular number and thickness. Pair-feeding of ob/ob mice had no significant effect on any of the skeletal endpoints evaluated.
4. Discussion
Leptin-deficient ob/ob mice have skeletal abnormalities, including decreased bone length, decreased overall bone mass, but site-specific increases in cancellous bone volume. Increasing hypothalamic leptin transgene expression via rAAV-lep gene therapy to levels sufficient to reduce body weight and food intake to normal, as reported here and detailed elsewhere [11,14,34,35,50,51], abolished the skeletal abnormalities. Circulating leptin was not detected in the rAAV-lep-treated ob/ob mice [11,14,50,51], indicating that central leptin expression is sufficient to normalize bone mass and architecture in the genetically obese leptin-deficient ob/ob mouse. Further, that decreased energy intake may not play a role in normalizing bone mass in ob/ob mice is shown by the absence of a skeletal response to pair-feeding.
Dietary obesity is thought to occur as a result of either leptin resistance or insufficiency in the hypothalamus due to reduced leptin entry across the blood brain barrier despite chronic systemic hyperleptinemia [6,7,10,13-17,25,29,34,35,43,53]. However, central leptin therapy designed to circumvent central leptin insufficiency, maintained reduced weight in wild type and genetically obese ob/ob mice for the duration of studies [11,14,34,50,51]. This replication of the effect of daily systemic and central injections [18,19,31,41] indicates that leptin produced by increased leptin transgene expression [11,14,50,51] was biologically active for the 30 week duration of the experiment described here. This long duration efficacy of central leptin gene therapy in mice is in agreement with studies performed in rats [9,13,34,37].
We did not measure bone formation and longitudinal bone growth rates in this study. Fluorochromes were not administered because of a concern that they might interfere with gene therapy. However, net changes can be inferred based on the large differences that were observed. For example, in order to achieve the 1.6 mm difference in bone length between the rAAV-lep and rAAV-GFP-treated groups during the initial 15 weeks of treatment would require hypothalamic leptin to accelerate longitudinal bone growth by approximately 15 μm/day. This large-scale effect would represent an approximate doubling of the growth rate. During the same interval, rAAV-lep treatment induced a net increase in bone resorption that resulted in a 0.02 %/day decrease in cancellous bone mass. Remarkably, no further changes were observed in the subsequent 15-weeks. This finding indicates that the changes induced by increased hypothalamic leptin supply are: 1) rapid adaptive responses and 2) the increases in growth rate and bone resorption induced by sustained leptin are complete within 15 weeks. The peak rates of change for growth and resorption are likely to be much greater than the mean values for the entire 15-week treatment interval. Thus, it is unlikely that administration of fluorochromes at the end of the study would have been informative because the rate of longitudinal bone growth would have returned to normal and/or would have ceased at time of sacrifice.
Kishida et al. [36] have shown that the reduction in long bone length in ob/ob mice is associated with abnormalities in growth plate morphology. Chondrocytes, in both the proliferative and hypertrophic zones, do not form the columnar, organized structures observed in WT mice, but are instead misaligned. Chondrocyte apoptosis and mineralization are also increased in the hypertrophic zone of ob/ob mice. These data suggest leptin is necessary for normal growth plate architecture and normal longitudinal bone growth. They do not, however, reveal whether the physiological actions of leptin on bone length are mediated by systemic or hypothalamic leptin. As reported for this study [11,14] and other studies [50,51], hypothalamic rAAV-lep injection does not result in measurable serum leptin. Thus, increased central leptin is sufficient to normalize bone length in ob/ob mice.
Within the hypothalamus, neuropeptide Y (NPY) network is a major mediator in the integration of energy homeostasis by leptin [34,35]. We observed that leptin transgene expression readily suppressd hypothalamic NPY signaling as indicated by diminished NPY mRNA expression in wildtype rodents and ob/ob mice [1,2,8,14,21-24,34]. Thus, it is possible that the observed skeletal effects are mediated by the central actions of leptin on hypothalamic NPY signal relay [3-5]. However, changes in sympathetic tone [26] and changes in serum levels of cytokines and growth factors, such as IGF-I [9,34], may contribute to the skeletal response to leptin gene therapy.
ob/ob mice have reduced sex steroid levels and elevated cortisol levels, and are prone to diabetes resulting from an insufficiency of central leptin restrain on insulin secretion and glucose metabolism [10,11,14,50,51]. rAAV-lep treatment has been shown to reverse these abnormalities in mice [11,14,50,51] and reduce serum IGF-I levels in rats, as well [9,34]. Sex steroids, cortisol, insulin and IGF-I are mediators of bone growth and turnover. Thus, it seems unlikely that the observed changes in these hormones would not play a role in the skeletal adaptation to rAAV-lep treatment.
In concordance with our results, Ducy et al. [26] have shown that increasing hypothalamic leptin via short-term (4 weeks) intracerebroventricular infusion results in decreased cancellous bone volume in vertebrae of ob/ob mice. The authors interpret this finding as evidence that hypothalamic leptin is antiosteogenic and induces osteopenia. However, this conclusion is based on measurements limited to vertebrae. As shown in the current and earlier studies [27,30,32,33,36,44], leptin deficiency results in decreased femur length and mass. The results presented in this study indicate that increasing hypothalamic leptin in ob/ob mice via rAAV-lep transfer does not result in osteopenia but leads to a normalization of cortical as well as cancellous bone volume to that of WT mice.
Genetic obesity due to leptin deficiency occurs in humans but is very rare [10,15,28,40]. A direct skeletal comparison of leptin-deficient humans and ob/ob mice is problematic because the skeletal phenotype of leptin-deficient individuals has not been described in detail. Limited data suggest leptin-deficient children have advanced skeletal age [28], which is consistent with the growth abnormalities in ob/ob mice. Severe dietary restriction is a more common cause of leptin insufficiency in humans. Based on our findings in ob/ob mice, we suggest studies should be performed to evaluate the possibility that leptin insufficiency contributes to the low bone mass in adolescents with eating disorders.
Leptin insufficiency results in metabolic changes that promote successful adaptation to starvation. Reduced bone growth during chronic calorie restriction may represent such an adaptation. The adaptive significance of the observed increase in vertebral cancellous bone volume in ob/ob mice is not clear. It is possible that this change represents a pathological rather than an adaptive response marked by the complete loss of leptin signaling caused by the mutation. This possibility is supported by the observation that caloric restriction in normal rodents and humans is associated with cancellous osteopenia.
In conclusion, growing, leptin deficient ob/ob mice have skeletal abnormalities that are reversed by delivery of rAAV-lep into the hypothalamus. As was the case for body weight, the WT skeletal phenotype was maintained in the rAAV-lep-treated ob/ob mice for the duration of the studies. These findings suggest leptin is an important permissive factor for bone growth. In the absence of circulating and hypothalamic leptin, bone length and mass are decreased. Hypothalamic leptin is sufficient to normalize bone growth. These findings are consistent with the hypothesis that hypothalamic leptin plays a role in coupling energy homeostasis and bone growth.
Acknowledgments
This work was supported by grants from the National Institute of Health (DK 37273 and 27372 to S.P. Kalra and AA 011140 to R.T. Turner) and the Center for Healthy Aging Research, Oregon State University (to U.T. Iwaniec).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bagnasco M, Dube MG, Kalra PS, Kalra SP. Evidence for the existence of distinct central appetite and energy expenditure pathways and stimulation of ghrelin as revealed by hypothalamic site-specific leptin gene therapy. Endocrinology. 2002;143:4409–4421. doi: 10.1210/en.2002-220505. [DOI] [PubMed] [Google Scholar]
- 2.Bagnasco M, Dube MG, Katz A, Kalra PS, Kalra SP. Leptin expression in hypothalamic PVN reverses dietary obesity and hyperinsulinemia but stimulates ghrelin. Obesity Research. 2003;11:1463–1470. doi: 10.1038/oby.2003.196. [DOI] [PubMed] [Google Scholar]
- 3.Baldock PA, Allison S, Sainsbury A, Enriquez RF, Herzog H, Gardiner EM, Eisman JA. Central regulation of cortical bone: opposing effects of Y2 receptor and leptin pathways. J Bone Miner Res. 2005;20:S413. doi: 10.1359/jbmr.060705. [DOI] [PubMed] [Google Scholar]
- 4.Baldock PA, Sainsbury A, Allison S, Lin EJ, Couzens M, Boey D, Enriquez R, During M, Herzog H, Gardiner EM. Hypothalamic control of bone formation: distinct actions of leptin and y2 receptor pathways. J Bone Miner Res. 2005;20:1851–1857. doi: 10.1359/JBMR.050523. [DOI] [PubMed] [Google Scholar]
- 5.Baldock PA, Sainsbury A, Couzens M, Enriquez RF, Thomas GP, Gardiner EM, Herzog H. Hypothalamic Y2 receptors regulate bone formation. J Clin Invest. 2002;109:915–921. doi: 10.1172/JCI14588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banks WA, DiPalma CR, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity. Peptides. 1999;20:1341–1345. doi: 10.1016/s0196-9781(99)00139-4. [DOI] [PubMed] [Google Scholar]
- 7.Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. Leptin enters the brain by a saturable system independent of insulin. Peptides. 1996;17:305–311. doi: 10.1016/0196-9781(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 8.Beretta E, Dube MG, Kalra PS, Kalra SP. Central LIF gene therapy suppresses food intake, body weight, serum leptin and insulin for extended periods. Peptides. 2002;23:875–984. doi: 10.1016/s0196-9781(02)00021-9. [DOI] [PubMed] [Google Scholar]
- 9.Beretta E, Dube MG, Kalra PS, Kalra SP. Long-term suppression of weight gain, adiposity, and serum insulin by central leptin gene therapy in prepubertal rats: Effects on serum ghrelin and appetite-regulating genes. Ped Res. 2002;52:189–198. doi: 10.1203/00006450-200208000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Bluher S, Mantzoros CS. The role of leptin in regulating neuroendocrine function in humans. J Nutr. 2004;134:2469S–2474S. doi: 10.1093/jn/134.9.2469S. [DOI] [PubMed] [Google Scholar]
- 11.Boghossian S, Dube MG, Torto R, Kalra PS, Kalra SP. Hypothalamic clamp on insulin release by leptin-transgene expression. Peptides. 2006;27:3245–3254. doi: 10.1016/j.peptides.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 12.Boghossian S, Lecklin A, Dube MG, Kalra PS, Kalra SP. Increased Leptin Expression in the Dorsal Vagal Complex Suppresses Adiposity without Affecting Energy Intake and Metabolic Hormones. Obesity. 2006;14:1003–1009. doi: 10.1038/oby.2006.115. [DOI] [PubMed] [Google Scholar]
- 13.Boghossian S, Lecklin A, Torto R, Kalra PS, Kalra SP. Suppression of fat deposition for the life time with gene therapy. Peptides. 2005;26:1512–9. doi: 10.1016/j.peptides.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 14.Boghossian S, Ueno N, Dube MG, Kalra PS, Kalra SP. Leptin gene transfer in the hypothalamus enhances longevity in adult monogenic mutant mice in the absence of circulating leptin. Neurobiol Aging. 2006;27:3245–3254. doi: 10.1016/j.neurobiolaging.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Bray GA. Medical consequences of obesity. J Clin Endocrinol Metab. 2004;89:2583–9. doi: 10.1210/jc.2004-0535. [DOI] [PubMed] [Google Scholar]
- 16.Burguera B, Couce ME, Curran GL, Jensen MD, Lloyd RV, Cleary MP, Poduslo JF. Obesity is associated with a decreased leptin transport across the blood-brain barrier in rats. Diabetes. 2000;49:1219–1223. doi: 10.2337/diabetes.49.7.1219. [DOI] [PubMed] [Google Scholar]
- 17.Buerguera B, Hofbauer LC, Thomas T, Gori F, Evans GL, Khosla S, Riggs BL, Turner RT. Leptin reduces ovariectomy-induced bone loss in rats. Endocrinology. 2001;142:3546–3553. doi: 10.1210/endo.142.8.8346. [DOI] [PubMed] [Google Scholar]
- 18.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Heiman ML. Chronic leptin administration promotes lipid utilization until fat ass is greatly reduced and preserves lean mass of normal female rats. Regul Pept. 2000;92:113–9. doi: 10.1016/s0167-0115(00)00157-9. [DOI] [PubMed] [Google Scholar]
- 20.Cornish J, Callon KE, Bava U, Lin C, Naot D, Hill B, Grey AB, Broom N, Myers DE, Nicholson GC, Reid IR. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol. 2002;175:405–415. doi: 10.1677/joe.0.1750405. [DOI] [PubMed] [Google Scholar]
- 21.Dhillon H. Effects of recombinant adeno-associated virus encoding leptin on body weight regulation and energy homeostasis. University of Florida; Gainesville, FL: 2000. pp. 1–171. http://purl.fcla.edu/fcla/etd/ane5948. [Google Scholar]
- 22.Dhillon H, Ge Y, Minter RM, Prima V, Moldawer LL, Muzyczka N, Zolotukhin S, Kalra PS, Kalra SP. Long-term differential modulation of genes encoding orexigenic and anorexigenic peptides by leptin delivered by rAAV vector in ob/ob mice. Relationship with body weight change. Regul Pept. 2000;92:97–105. doi: 10.1016/s0167-0115(00)00155-5. [DOI] [PubMed] [Google Scholar]
- 23.Dhillon H, Kalra SP, Kalra PS. Dose-dependent effects of central leptin gene therapy on genes that regulate body weight and appetite in the hypothalamus. Mol Ther. 2001;4:139–145. doi: 10.1006/mthe.2001.0427. [DOI] [PubMed] [Google Scholar]
- 24.Dhillon H, Kalra SP, Prima V, Zolotukhin S, Scarpace PJ, Moldawer LL, Muzyczka N, Kalra PS. Central Leptin Gene Therapy Suppresses Body Weight Gain, Adiposity and Serum Insulin Without Affecting Food Consumption in Normal Rats: A Long-Term Study. Regul Pept. 2001;99:69–77. doi: 10.1016/s0167-0115(01)00237-3. [DOI] [PubMed] [Google Scholar]
- 25.Dube MG, Beretta E, Dhillon H, Ueno N, Kalra PS, Kalra SP. Central leptin gene therapy blocks high-fat diet-induced weight gain, hyperleptinemia, and hyperinsulinemia: Increase in serum ghrelin levels. Diabetes. 2002;51:1729–36. doi: 10.2337/diabetes.51.6.1729. [DOI] [PubMed] [Google Scholar]
- 26.Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: A central control of bone mass. Cell. 2000;100:197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- 27.Ealey KN, Fonseca D, Archer MC, Ward WE. Bone abnormalities in adolescent leptin-deficient mice. Regul Pept. 2006;136:9–13. doi: 10.1016/j.regpep.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 28.Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O’Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999:341. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 29.Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–4. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- 30.Gat-Yablonski G, Ben-Ari T, Shtaif B, Potievsky O, Moran O, Eshet R, Maor G, Segev Y, Phillip M. Leptin reverses the inhibitory effect of caloric restriction on longitudinal growth. Endocrinology. 2004;145:343–50. doi: 10.1210/en.2003-0910. [DOI] [PubMed] [Google Scholar]
- 31.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 32.Hamrick MW, Della-Fera MA, Choi YH, Pennington C, Hartzell D, Baile CA. Leptin treatment induces loss of bone marrow adipocytes and increases bone formation in leptin-deficient ob/ob mice. J Bone Miner Res. 2005;20:994–1001. doi: 10.1359/JBMR.050103. [DOI] [PubMed] [Google Scholar]
- 33.Hamrick MW, Pennington C, Newton D, Xie D, Isales C. Leptin deficiency produces contrasting phenotypes in bones of the limb and spine. Bone. 2004;34:376–83. doi: 10.1016/j.bone.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 34.Kalra SP, Kalra PS. Gene-transfer technology: a preventive neurotherapy to curb obesity, ameliorate metabolic syndrome and extend life expectancy. Trends Pharmacol Sci. 2005;26:488–95. doi: 10.1016/j.tips.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Kalra SP, Kalra PS. Subjugation of hypothalamic NPY and cohorts with central leptin gene therapy alleviates dyslipidemia, insulin resistance, and obesity for life-time. Exs. 2006:157–69. doi: 10.1007/3-7643-7417-9_12. [DOI] [PubMed] [Google Scholar]
- 36.Kishida Y, Hirao M, Tamai N, Nampei A, Fujimoto T, Nakase T, Shimizu N, Yoshikawa H, Myoui A. Leptin regulates chondrocyte differentiation and matrix maturation during endochondral ossification. Bone. 2005;37:607–21. doi: 10.1016/j.bone.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Lecklin A, Dube MG, Torto RN, Kalra PS, Kalra SP. Perigestational suppression of weight gain with central leptin gene therapy results in lower weight F1 generation. Peptides. 2005;26:1176–87. doi: 10.1016/j.peptides.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 38.Maness LM, Kastin AJ, Farrell CL, Banks WA. Fate of leptin after intracerebroventricular injection into the mouse brain. Endocrinology. 1999;139:4556–4562. doi: 10.1210/endo.139.11.6319. [DOI] [PubMed] [Google Scholar]
- 39.Maor G, Rochwerger M, Segev Y, Phillip M. Leptin acts as a growth factor on the chondrocytes of skeletal growth centers. J Bone Miner Res. 2002;17:1034–43. doi: 10.1359/jbmr.2002.17.6.1034. [DOI] [PubMed] [Google Scholar]
- 40.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham JN, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Early AR, Barnett AH, Prins JB, O’Rahilly S. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–907. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 41.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 42.Pierroz DD, Baldock P, Bouxsein ML, Ferrari SL. Low cortical bone mass in mice lacking Beta 1 and Beta 2 adrenergic receptors is associated with low bone formation and circulating IGF-1. J Bone Miner Res. 2006;21(1):S26. [Google Scholar]
- 43.Sahu A. Resistance to the satiety action of leptin following chronic central leptin infusion is associated with the development of leptin resistance in neuropeptide Y neurones. J Neuroendocrinol. 2002;14:796–804. doi: 10.1046/j.1365-2826.2002.00840.x. [DOI] [PubMed] [Google Scholar]
- 44.Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept. 2000;92:73–78. doi: 10.1016/s0167-0115(00)00152-x. [DOI] [PubMed] [Google Scholar]
- 45.Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, Karsenty G. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–317. doi: 10.1016/s0092-8674(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 46.Thomas T. The complex effects of leptin on bone metabolism through multiple pathways. Curr Opin Pharmacol. 2004;4:295–300. doi: 10.1016/j.coph.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 47.Thomas T, De Vittoris R, David VN, Lafage-Proust M, Alexandre C. Leptin prevents disuse-induced bone loss in tail-suspended female rats. J Bone Miner Res. 2001;16:S143. doi: 10.1210/en.2004-1509. [DOI] [PubMed] [Google Scholar]
- 48.Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. 1999;140:1630–1638. doi: 10.1210/endo.140.4.6637. [DOI] [PubMed] [Google Scholar]
- 49.Thomsen JS, Laib A, Koller B, Prohaska S, Mosekilde L, Gowin W. Stereological measures of trabecular bone structure: comparison of 3D micro computed tomography with 2D histological sections in human proximal tibial bone biopsies. J Microsc. 2005;218:171–9. doi: 10.1111/j.1365-2818.2005.01469.x. [DOI] [PubMed] [Google Scholar]
- 50.Ueno N, Dube MG, Inui A, Kalra PS, Kalra SP. Leptin modulates orexigenic effects of ghrelin and attenuates adiponectin and insulin levels and selectively the dark-phase feeding as revealed by central leptin gene therapy. Endocrinology. 2004;145:4176–84. doi: 10.1210/en.2004-0262. [DOI] [PubMed] [Google Scholar]
- 51.Ueno N, Inui A, Kalra PS, Kalra SP. Leptin transgene expression in the hypothalamus enforces euglycemia in diabetic, insulin-deficient nonobese Akita mice and leptin-deficient obese ob/ob mice. Peptides. 2006;27:2332–2342. doi: 10.1016/j.peptides.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 52.U.S. Department of Health and Human Services. Bone Health and Osteoporosis: A Report of the Surgeon General. U. S. Department of Health and Human Services, Office of the Surgeon General; 2004. [Google Scholar]
- 53.Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, Sybertz EJ, Strader CD, Davis HR., Jr Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J Clin Invest. 1997;99:385–390. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z-W, Zhou Y-T, Kakuma T, Lee Y, Higa M, Kalra SP, Dube MG, Kalra PS, Unger RH. Comparing the hypothalamic and extrahypothalamic actions of endogenous hyperleptemia. Proc Natl Acad Sci. 1999;96:10373–10378. doi: 10.1073/pnas.96.18.10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 56.Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–85. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]