

Summary
Directed cell migration involves signaling events that lead to local accumulation of PI(3,4,5)P3 but additional pathways act in parallel. A genetic screen in Dictyostelium discoideum to identify redundant pathways revealed a gene with homology to patatin-like phospholipase A2. Loss of this gene did not alter PI(3,4,5)P3 regulation, but chemotaxis became sensitive to reductions in PI3K activity. Likewise, cells deficient in PI3K activity were more sensitive to inhibition of PLA2 activity. Deletion of the PLA2 homologue and two PI3Ks caused a strong defect in chemotaxis and a reduction in receptor-mediated actin polymerization. In wild type cells, chemoattractants stimulated a rapid burst in an arachidonic acid derivative. This response was absent in cells lacking the PLA2 homologue and exogenous arachidonic acid reduced their dependence on PI3K signaling. We propose that PLA2 and PI3K signaling act in concert to mediate chemotaxis and arachidonic acid metabolites may be important mediators of the response.
Keywords: Chemotaxis; PLA2; PI(3,4,5)P3; arachidonic acid; D. discoideum
Introduction
Chemotaxis, the directed movement of cells along extracellular gradients, is an intriguing and critical cell biological response. A variety of chemoattractants direct cells to proper locations in developing embryos and guide the connections between cells of the nervous system. Following embryogenesis, directed cell movements mediate morphogenetic processes such as remodeling of the vascular system and wound healing. Moreover, leukocytes traffic between the vascular and lymphatic systems and migrate from the blood towards sites of infection. In addition to these roles in normal physiology, inappropriate cell migration is the basis for pathological conditions including cancer metastasis and chronic inflammatory diseases such as atherosclerosis, asthma, and arthritis (Heldin and Westermark, 1999; Baggiolini, 2001; Park et al., 2002; Rao et al., 2002).
The chemotactic response encompasses cell motility, polarity, and directional sensing, all events that involve local phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3) production and degradation at the plasma membrane (Parent and Devreotes, 1999; Rickert et al., 2000; Chung et al., 2001; Wang et al., 2002; Heit et al., 2002). The levels of PI(3,4,5)P3 are regulated by PI 3-kinases (PI3Ks), the PI 3-phosphatase, PTEN (Buczynski et al., 1997; Hirsch et al., 2000; Li et al., 2000; Sasaki et al., 2000; Funamato et al., 2001 and 2002; Iijima and Devreotes, 2002; Stocker et al., 2002) and PI 5-phosphatases, or SHIPs. When D. discoideum cells are exposed to a gradient of chemoattractant, PI3Ks and PTEN bind to the membrane at the front and rear, respectively, PI(3,4,5)P3 selectively accumulates at the leading edge, and new F-actin filled pseudopodia are extended at corresponding sites. Unable to degrade PI(3,4,5)P3, pten- cells fail to sharply localize the phosphoinositide and extend projections in multiple directions at once, which impairs polarity and chemotaxis (Iijima and Devreotes, 2002; Iijima et al., 2004). These observations have established a link between local plasma membrane PI(3,4,5)P3 levels, actin polymerization, and pseudopodia extension. This role for local PI(3,4,5)P3 in polarity and directional sensing appears to be conserved throughout phylogeny in cells such as D. melanogaster hemocytes, human neutrophils and fibroblasts, neurons, and a variety of embryonic cells (Stramer et al., 2005; Wang et al., 2002; Wu et al., 2000; Schneider et al., 2005; Chadborn et al., 2006; Montero, 2003).
In spite of these observations, an essential requirement for local PI(3,4,5)P3 accumulation has been surprisingly difficult to establish. In D. rerio, PI(3,4,5)P3 is uniformly elevated around the entire perimeter of chemotaxing primary germ cells (Dumstrei et al., 2004). Abrogation of PI3K activation has no effect on migration of D. melanogaster border cells. Migration of PI3Kγ−/− mouse macrophages in response to chemokines is only partially impaired, with inhibition ranging from 50–90%, depending on the chemokine examined (Hannigan et al., 2002). T-cells and B-cells have been reported to be sensitive to PI3K inhibitors (Reif et al., 2004), but under more physiological conditions, T-cells are reported to be relatively insensitive to PI3K inhibitors (Sotsios et al., 1999; Smit et al., 2003; Cinamon et al., 2003; Ward, S.G., 2004). In D. discoideum, while over production of PI(3,4,5)P3 disrupts chemotaxis, loss of PI3K activity (deletion of two out of five PI3Ks) leading to more than 95% reduction of PI(3,4,5)P3, hardly affects the ability of cells to sense and orient in chemotactic gradients (Buczynski et al., 1997; Funamoto et al., 2001; Huang et al., 2003; Chen et al., 2003; Loovers et al., 2006). Taken together, these studies in different systems suggest that, although local PI(3,4,5)P3 plays an important role in chemotaxis, there are likely redundant mediators.
Our studies of chemoattractant-mediated actin polymerization, one of the primary biochemical responses underlying pseudopod extension, have also pointed to alternative pathways. We examined the biphasic responses of PI(3,4,5)P3 accumulation and actin polymerization during perturbations that decreased or elevated PI(3,4,5)P3 levels in D. discoideum. In cells with deficient PI3K activity, the second phase of actin polymerization was blocked, but the first phase was unaffected. Similar results were obtained with EGF stimulation of breast carcinoma cells (Mouneimne et al., 2004). Taken together, these observations suggest that the second phase of actin polymerization is mediated by PI(3,4,5)P3, but the first phase is regulated differently (Chen et al., 2003; unpublished data).
Based on these findings, we have speculated that there are additional pathways, not merely additional PI3Ks, downstream of G-protein activation, which act in parallel with the PI3K/PTEN system, controlling actin polymerization, pseudopod extension, and directional movement (Figure 1A). Parallel pathways could explain why elevation of PI(3,4,5)P3 causes a dramatic defect while its removal does not. Lowered PI(3,4,5)P3 would not be expected to cause a significant phenotype unless the parallel pathways are also inhibited. Accordingly, we designed a screen to search for mutants where chemotaxis is selectively impaired when PI3K is inhibited. This procedure uncovered a gene with homology to a patatin-like phospholipase A2 (PLA2), suggesting that this enzyme acts in parallel with PI3K pathways to mediate chemotaxis.
Figure 1.
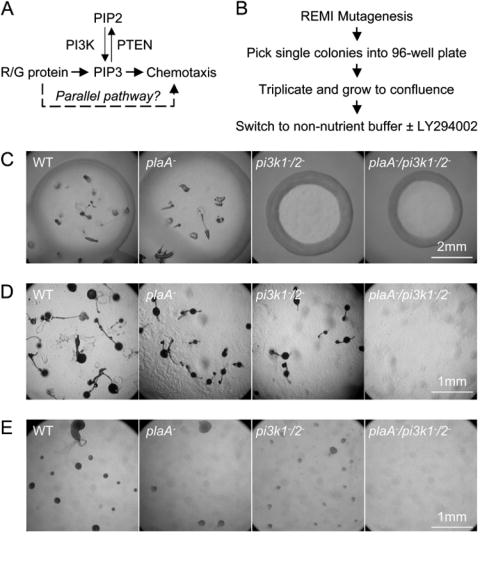
(A) A model for regulation of chemotaxis. The dashed line shows a putative pathway that acts in parallel with the PI3K/PTEN pathway. (B) Scheme of the genetic screen. After mutagenesis, individual phenotypically wild-type colonies were separately plated in 96-well format, replicated and induced to differentiate in non-nutrient buffer containing different concentrations of LY. Mutants with altered sensitivity to LY are scored based on their ability to aggregate. Phenotypes of wild type, plaA−, pi3k1-/2- and plaA−/pi3k1-/2- cells on bacteria lawn (C), non-nutrient agar (D), and in under-buffer assay (E).
Experimental Procedures
Cell culture, development, and mutagenesis
D. discoideum cells were cultured in HL5 medium and allowed to differentiate for 5 hours, unless otherwise indicated, in development buffer (DB) as previous described (Parent et al., 1998). To isolate mutants sensitive or resistant to LY294002, wild type cells were mutagenized and genes identified using restriction enzyme mediated integration (REMI) method (Adachi et al., 1994; Van Es et al., 2001).
Live cell imaging and quantification
Fluorescent images of living cells expressing GFP fusion proteins and chemotactic movements of cells towards cAMP containing micropipettes were performed as previously described (Parent et al., 1998). IP lab, Image J and the Matlab imaging tool box (Mathworks) were used to collect and process data (Chen, et. al, 2003).
Cellular responses to chemoattractant stimulation
PH domain translocation, actin polymerization, and calcium influx assays were performed as previously described (Parent et al., 1998; Iijima and Devreotes., 2002; Zigmond et al., 1997; Milne and Coukell, 1991).
Protein purification and phospholipase A2 assays
Wild type cells expressing PLA2A-FLAG were cultured to a density of 3-8 × 106 cells/ml. Typically, 500 ml of cells were collected and starved at 2 × 107 cells/ml for 2 hours, collected and filter-lysed in 50 mM HEPES (pH= 7.5) at a density of 1 × 108 cells/ml (Parent and Devreotes, 1998). Cell lysates were subjected to two rounds of centrifugation at 15 Krpm for 20 minutes and the supernatant was centrifuged at 55 Krpm for 20 minutes. The final supernatant was loaded on an ion exchange column (Q fast flow, Amersham). The Q column was washed with 0.1 M NaCl with 50 mM HEPES (pH= 7.5) and then eluted with 0.5 M NaCl with 50 mM HEPES (pH= 7.5). The eluted fraction (3-4 ml) was incubated with 200 μl Flag-agarose (Sigma) for 2-3 hours at 4°C. Agarose beads were collected, washed and incubated at 4°C for 10 minutes with 400 μl of 200 ng/μl FLAG-peptide (Sigma) in 100 mM HEPES, 0.1% Triton X-100. After centrifugation, the supernatant was collected and subjected to further analysis. In some experiments, 10 mM sodium phosphate buffer (pH= 7.0) was used instead of 50 mM HEPES. Phospholipase A2 assays were performed as previously described with minor modifications (Ackermann et al., 1994). Extracted products were separated on a Silica gel 60 TLC plate (EMD chemicals) in chloroform: methanol: acetic acid: water (75: 20: 2:1,v/v/v/v). Then TLC plate was sprayed with 3H enhancer (PE) and exposed to HyBlot film (Denville) at -80°C for two days.
3H-arachidonic acid labeling assay
Cells were starved for 3 hours in DB and labeled with 3H-arachidonic acid for another 2 hours. Labeled cells were resuspended at 3 × 107 cells/ml in DB and shaken at 200 rpm at room temperature. At various time points after adding 500 nM cAMP, 300 μl of cells were collected into 1ml of chloroform: methanol: acetic acid (2:4:1, v/v/v) to stop the stimulation. Lipids were extracted and subjected to TLC analyses as described in the previous section.
Results
Isolation of mutants defective in aggregation in the presence of PI3K inhibitors
We screened for components in pathways that act in parallel with PI3K/PTEN, as outlined in Figure 1A and 1B. Restriction enzyme mediated insertional mutagenesis (REMI) was used to generate random insertions in a population of wild type cells (Adachi et al., 1994). Mutagenized cells were clonally plated onto bacteria lawns and cells from phenotypically wild type single colonies were transferred into 96-well plates. These were grown to confluency, triplicated, then switched to non-nutrition buffer containing no, low (30-50 μM), or high (>150 μM) concentrations of the PI3K inhibitor, LY294002 (LY), respectively. During starvation, untreated cells begin to sense and secrete cAMP which directs chemotactic migration into large, tight aggregates containing several million cells. The low concentration of inhibitor does not significantly alter this process, while the high concentration blocks it, causing the cells to remain as a monolayer, or form small, loose aggregates. Phenotypes were visually inspected under a dissection microscope after 24 hours. Of 7043 insertional mutants, we found ten mutants that were unable to aggregate in the presence of the low concentration of inhibitor and six mutants that were able to resist the high concentration. The wild type cells did not show an aggregation defect until 80 μM LY and completely failed at 120 μM. We were able to identify the insertion site on two sensitive and one resistant mutant. Sensitive clones, S1 and S2, formed small aggregates at 20 to 40 μM LY and failed completely at 80 μM while resistant clone R1 aggregated normally up to 160 μM. Consistent behavior was observed when each of the clones was retested with another PI3K inhibitor, wortmannin. In this report, we focus on characterization of mutant S1.
A patatin-like phospholipase A2 homologue is required for aggregation in the presence of LY
We recovered the tagged sequence integrated into the genome of S1, and determined the position of the insertion site. It was found to be 191 bp upstream of the start codon of a predicted ORF that shared sequence homology with the group VI phospholipase A2 (PLA2) family of proteins (Supplemental Figure 1A). These enzymes are found both in plants, where they are known as patatin or patatin-related phospholipase A2 (Holk et al., 2002), and in animals where they are referred to as Ca2+-independent PLA2 (iPLA2, group VI PLA2). The family includes enzymes that use triacylglycerol as well as phospholipids as substrates. We suggest the names plaA and PLA2A for the new gene (DDB0205513) and its product. We found thirteen additional predicted patatin-like or group VI PLA2s homologues in the D. discoideum genome (Supplemental Figure 1B). Based on sequence analyses, there are no secreted or cytosolic PLA2s in this organism (Schaloske and Dennis, 2006). By sequence comparison, the fourteen PLA2s can be roughly divided into three groups (Supplemental Figure 1C). At least two additional ORFs (DDB0219540 and DDB0231758) are closely related to plaA. Some of the ORFs have additional domains, including pleckstrin-like (DDB0184540), protein kinase-like (DDB0185559) and growth factor motifs (DDB0185561).
To confirm that the LY-sensitive phenotype was caused by inactivation of plaA, we targeted it by homologous recombination, thereby replacing part of the gene with a blasticidin resistance marker (Bsr) (Supplemental Figure 2A). The disruption of plaA recapitulated the phenotype of the original insertional mutant. On bacteria lawns, individual plaA− cells proliferated to form wild-type size plaques that differentiated normally to form fruiting bodies (Figure 1C). Similarly, the plaA− cells were able to aggregate normally and form fruiting bodies like wild type cells after 24 hours on non-nutrient agar (Figure 1D). However, in the presence of low LY in the under-buffer aggregation assay, the plaA− cells displayed an aggregation defect while wild type cells were not affected (Figure 2A). Therefore, the phenotypes of the plaA− cells were similar to those of the original mutant, proving that a deficiency in plaA causes the LY-sensitive phenotype.
Figure 2.
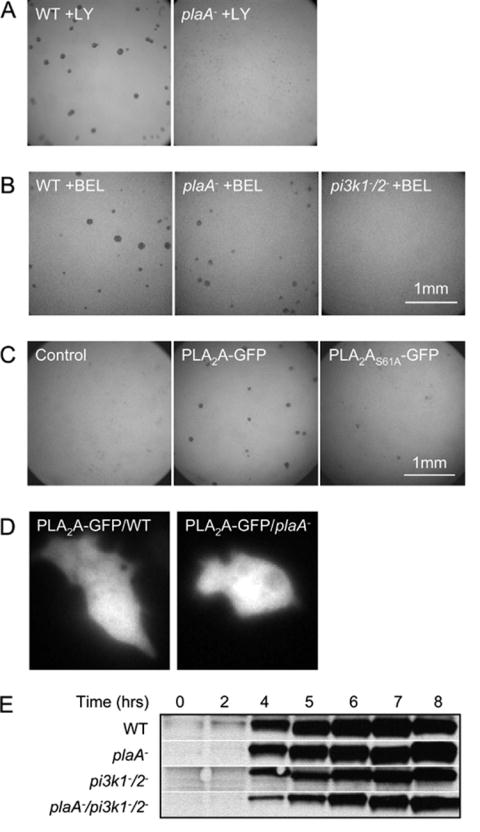
PLA2A and PI3K actin parallel to mediate aggregation. (A, B) Under-buffer assay of indicated cells in the absence or presence of 60 μM LY or 5 μM BEL are shown. (C) Complementation of plaA− cells by GFP tagged wild type and inactive (S61A) version of PLA2A protein. The phenotype of cells in the under-buffer assay in the presence of 20 μM LY is shown. (D) Fluorescence images of wild type and plaA− cells expressing PLA2A-GFP. (E) Immuno-blot shows expression of camp receptor, cAR1, in wild type, plaA−, pi3k1-/2- and plaA−/pi3k1-/2- cells during development. Bands are 40 kD. All experiments were repeated at least 3 times.
When treated for 5 minutes with 5 μM bromoenol lactone (BEL), a iPLA2 inhibitor (Ackermann et al., 1995), the pi3k1-/2- cells failed to aggregate while both treated wild type and plaA− cells still aggregated normally in under buffer assays (Figure 2B). Thus, cells lacking plaA require PI3K activity for aggregation and, conversely, those deficient in PI3K1 and PI3K2 require PLA2 activity.
PLA2A and PI3K act in parallel to mediate aggregation
If PLA2A and PI3K are in redundant pathways as suggested, a combined deficiency would be expected to display a severe defect compare to either single disruption. We disrupted plaA in cells lacking two of the five “class I-like” PI3K (pi3k1-/2- cells) (Buczynski et al., 1997; Funamoto et al., 2001) using a hygromycin resistance marker (data not shown; Egelhoff et al, 1989). As predicted, the plaA−/pi3k1-/2- cells were unable to aggregate on non-nutrient agar while wild type, plaA− and pi3k1-/2- cells developed normally and formed fruiting bodies (Figure 1D). Upon closer examination, we noticed that the plaA−/pi3k1-/2- cells formed loose aggregates on non-nutrient agar after 24 hours, suggesting they were motile. Similar results were observed in the under-buffer assays where wild type and plaA− cells aggregated normally, pi3k1-/2- cells formed small aggregates, but plaA−/pi3k1-/2- cells remained as a monolayer (Figure 1E). Thus whereas neither PLA2A nor PI3K1/PI3K2 are essential, each becomes indispensable in the absence of the other.
To further explore the interdependence of PLA2A and PI3K signaling, we made an expression construct of PLA2A tagged with GFP at the C-terminal (PLA2A-GFP). When expressed in plaA− cells, this construct was able to rescue the LY-sensitive phenotype in the under-buffer aggregation assay (Figure 2C). At 20 μM LY, aggregation of plaA− cells was inhibited, but when PLA2A-GFP was expressed in the plaA− cells, they aggregated normally. The rescue was partial since PLA2A-GFP/plaA− cells failed to aggregate at 60 μM LY, a concentration that did not block the aggregation of wild type cells (data not shown). We also expressed an inactive version of PLA2A-GFP (S61A) in which the catalytic site was mutated. Based on fluorescence and immuno-blot of GFP, S61A was expressed at the same level as wild type PLA2A-GFP, but it completely failed to rescue the LY-sensitive phenotype in the presence of 20 μM LY. When PLA2A-GFP was expressed in plaA−/pi3k1-/2- cells, it restored the capacity of cells to aggregate in the under buffer assay (data not shown). PLA2A-GFP appeared to localize largely in the cytosol in both wild type and plaA− cells and its distribution did not change upon cAMP stimulation (Figure 2D).
The phenotypes observed in the plaA− and plaA−/pi3k1-/2- cells are not due to defects in development. Since the plaA− cells formed fruiting bodies at the same time after starvation as wild type cells both on non-nutrient agar and on bacteria lawns, the developmental program is not altered (Figure 1C and D). Similar sensitivity to LY was observed whether it was added at start or after 5 hours of starvation. As a further indicator of development, the time course of expression of the chemoattractant receptor, cAR1, was examined by immunoblot (Figure 2E). cAR1 levels start to increase at 3 to 4 hours after starvation and reached a peak at 5 to 6 hours, when chemotactic phenotypes are typically assessed. Wild type and plaA− cells had essentially identical patterns of expression of cAR1. Although the pi3k1-/2- and plaA−/pi3k1-/2- cells expressed a slightly lower amount of cAR1, the time course was similar to wild type[1]. Therefore, the non-aggregation phenotype of the plaA−/pi3k1-/2- cells was more likely due to a defect in chemotaxis or cell-cell signaling than a failure to differentiate.
PLA2A is not required for PI(3,4,5)P3 production
To monitor the dynamics of PI(3,4,5)P3 production, PHcrac-GFP was expressed in wild type, plaA−, pi3k1-/2-, and plaA−/pi3k1-/2- cells (Figure 3, supplemental video 1-6). PHcrac-GFP binds to PI(3,4,5)P3 and translocates to the plasma membrane upon cAMP stimulation (Iijima and Devreotes, 2002; Huang et al., 2003; Lemmom, 2003). In response to a uniform cAMP stimulus, PHcrac-GFP in plaA− cells displayed a biphasic response, as previously reported for wild type cells (Chen et al., 2003; Loovers, et al., 2006). PHcrac-GFP was recruited to the entire plasma membrane within seconds and returned to the cytosol after 30 seconds, then a second phase, localized to membrane extensions, ensued for the next several minutes (Figure 3A). When cells were pretreated with 30 μM LY and stimulated with cAMP, the first phase of PI(3,4,5)P3 accumulation was still detectable while the second phase was nearly absent, indicating a similar extent of inhibition in each case. In the pi3k1-/2- and plaA−/pi3k1-/2- cells, PHcrac-GFP did not translocate to membrane upon cAMP stimulation. Quantitative results were obtained when the translocation of the PHcrac-GFP was assessed by pelleting of membranes from cell lysates (Figure 3B,C). The extent of inhibition with 20 and 100 μM LY was essentially identical in the wild type and plaA− cells. Thus, neither the dynamics of PI(3,4,5)P3 production nor its sensitivity to inhibition was altered by the loss of PLA2A, indicating that the enzyme does not regulate the PI3K/PTEN pathway.
Figure 3.
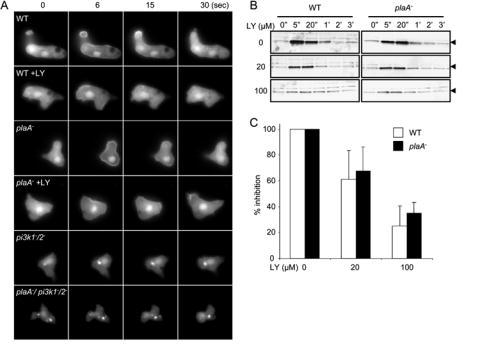
PI(3,4,5)P3 accumulation in wild type, plaA−, pi3k1-/2- and plaA−/pi3k1-/2- cells. (A) Chemoattractant-induced translocation of PHcrac-GFP to the plasma membrane is observed by epifluorescence microscopy. Images were captured at the indicated time points after uniform stimulation of 1 μM cAMP. When indicated, 30 μM of LY was added 10 minutes before stimulation. (B) Immuno-blotting shows amounts of membrane-associated PHcrac-GFP (arrow) at the indicated time points after addition of 1 μM cAMP. (C) Quantification of membrane-bound PHcrac-GFP was accomplished by densitometry and NIH image. The integrated amount of membrane-bound PHcrac-GFP above the prestimulated level was determined. Normalized values relative to controls in the absence of LY are shown. Experiment was repeated 3 times. Means and standard deviations are shown.
PLA2A is required for chemotaxis in the absence of PI3K activity
To directly observe the chemotactic responses, cells were exposed to chemoattractant gradients created by a cAMP-filled micropipette and directed migration was observed by phase contrast microscopy. Both wild type and plaA− cells were examined in the presence and absence of 30 μM of LY (Figure 4A, supplemental video 7-10). During ten minutes of observation, wild type cells polarized in the cAMP gradient and moved towards the tip of the micropipette, forming streams by attaching to each other during the directional movement. When cells were treated with LY, they initially lost polarity, then recovered within a few minutes, extended pseudopods, and started to move towards the source of chemoattractant. The LY-treated cells did not form streams in the course of the experiment. The plaA− cells were able to polarize and carry out chemotaxis, similar to wild type cells. However, addition of 30 μM of LY to the plaA− cells severely inhibited chemotaxis and movement. In the presence of LY, only a fraction of plaA− cells were motile and even fewer moved towards the micropipette. These observations suggest that the migration of the plaA− cells is sensitive to inhibition of PI3K activity.
Figure 4.
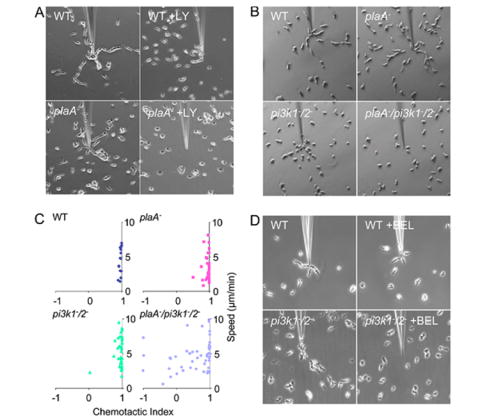
Chemotactic responses towards a micropipette filled with 10 μM cAMP. Cells were observed at 10-second intervals for 10 minutes. There was a random distribution of cells at the onset and images at 10 minutes are shown. (A) Chemotaxis of wild type and plaA− cells was observed in the absence or presence of 30 μM LY using phase-contrast microscopy. LY was added as indicated 10 minutes before the start of the experiments. (B) Chemotactic responses of wild type, plaA−, pi3k1-/2-, plaA−/pi3k1-/2- cells were observed by DIC microscopy. (C) The speed (S), calculated from the distance a cell covered divided by the experimental time, and chemotactic index (CI), defined as the cosine of the angle formed by the line between the cell start point and the micropipette tip and the line from the cell start to ending point (Iijima and Devreotes, 2002; Chen et al., 2003), of wild type (S = 4.38 ± 1.94; CI = 0.96 ± .03), plaA− (S = 3.24 ± 1.82; CI = 0.94 ± 0.10), pi3k1-/2- (S = 5.60 ± 2.58; CI = 0.92 ± 0.17), plaA−/pi3k1-/2- (S = 4.66 ± 2.14; CI =; 0.55 ± 0.57) cells were calculated and plotted (n= 15, 37, 37, 49, respectively). Three independent experiments were quantified. (D) Chemotaxis of wild type and pi3k1-/2- cells was observed in the absence or presence of BEL by phase-contrast microscopy. Cells were treated with BEL (5 μM) for 10 minutes, then the inhibitor was washed away. Cells were allowed to recover for 5 minutes before the start of the experiments.
We next examined the chemotaxis of the plaA−/pi3k1-/2- cells (Figure 4B, supplemental video 11-14). As we observed previously, wild type cells and plaA− cells were well polarized, formed streams and moved towards cAMP. Although the pi3k1-/2- cells were less polarized, they did line up and migrate towards the micropipette efficiently. Furthermore, treatment of pi3k1-/2- cells with LY did not significantly impair their chemotactic response (Supplemental video 15). In contrast, the plaA−/pi3k1-/2- cells showed strong defects in maintaining cell shape and in directed migration. While the polarities of these cells were similar to those of the pi3k1-/2- cells, many of them ignored the direction of the gradient, moved randomly, and changing direction frequently. A few cells very close to the micropipette moved in the correct direction, although sometimes they subsequently turned away. For each experimental condition, we quantified the speed and chemotactic index from three sets of independent experiments (Figure 4C). The mean chemotactic indexes showed differences. In wild type cells, the mean was close to one, indicating that the cells moved straight towards the needle. In plaA− and pi3k1-/2- cells, the indicies were slightly lower, indicating a fraction of cells were unable to chemotax as well as wild type cells. In plaA−/pi3k1-/2- cells, the index was low and standard deviation was large. About 40% of the cells (i.e. those closest to the pipet) appeared to be more or less chemotactic while the others moved randomly. Unlike the plaA− cells treated with LY, the speed of random movement of the plaA−/pi3k1-/2- cells was not significantly reduced compared those of wild type cells. These observations suggest that the chemotaxis of the plaA− cells is sensitive to inhibition of PI3K activity.
Then we asked whether pi3k1-/2- cells were more sensitive to inhibition of PLA2 activity than wild type cells. Cells were treated with different concentration of BEL, for 5 minutes, then washed. In chemotaxis assays, this treatment did not affect the directed movement of wild type cells whereas it caused a strong reduction in chemotaxis of the pi3k1-/2- cells (Figure 4D, supplemental video 16-19).
PLA2A and PI3K act in parallel to regulate cAMP-induced actin polymerization
As outlined in the Introduction, evidence suggests that the first phase of actin polymerization is regulated by events that act in parallel with the PI3K/PTEN pathway (Chen et al., 2003). To characterize the role of PLA2A, we carried out biochemical assays of actin polymerization in the plaA− and plaA−/pi3k1-/2- cells (Figure 5). In the absence of LY, wild type and plaA− cells displayed similar responses upon cAMP stimulation (Figure 5A). An initial peak (approximately two fold above unstimulated cells) occurred at 4 to 8 seconds. Since these were highly polarized cells, a defined second peak was not obvious, but the F-actin level remained above baseline for several minutes as previously described (Chen et al., 2003). Treatment with LY reduced this signal during the prolonged second phase as expected. The initial peak in the plaA− cells appeared to be slightly decreased as well (Figure 5B). As previously determined, the first peak of actin polymerization was identical in wild type and pi3k1-/2- cells (data not shown). Compared to the pi3k1-/2- cells, the first phase of actin polymerization in the plaA−/pi3k1-/2- cells was clearly decreased (Figure 5C). When treated with LY, a further decrease to 50% of the wild type level was observed. Therefore, in the plaA− background, the loss of PI3K activity had a detectable consequence on the initial peak of actin polymerization. These results suggest that either the PI3K or the PLA2 pathway can support the initial actin polymerization response and inhibition is only observed when both pathways are inhibited.
Figure 5.
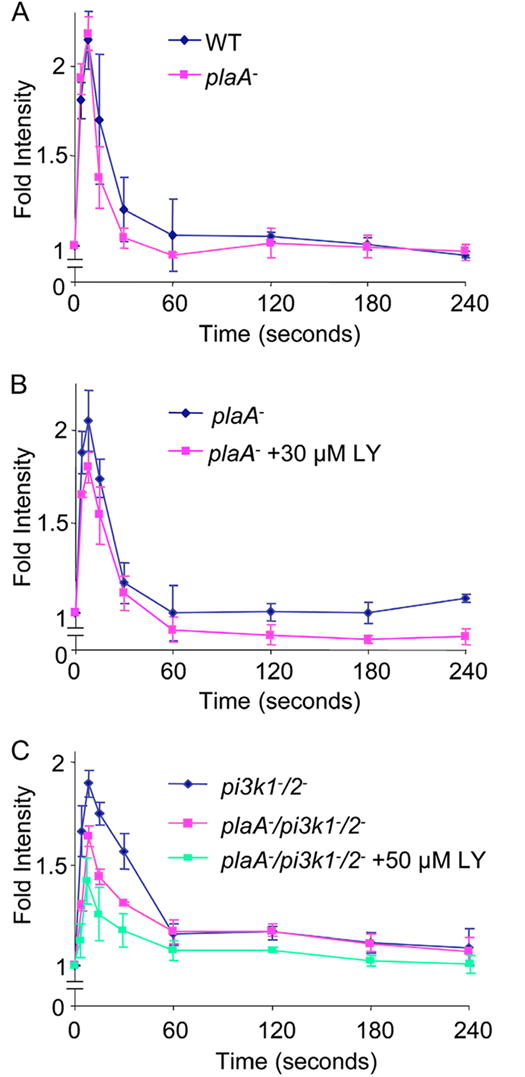
Chemoattractant-induced actin polymerization assay. (A) Wild type (diamonds) and plaA− cells (pink squares). (B) plaA− cells in the absence (blue diamonds) or presence of 30 μM LY (pink squares). (C) pi3k1-/2- (blue diamonds) and plaA−/pi3k1-/2- cells in the absence (pink squares) or presence of 50 μM LY (green squares). For each experiment, all values were normalized to the amount of F-actin at time 0, which was taken before the addition of 100 nM cAMP. When indicated, LY was added 10 minutes before experiments. Experiments were repeated at least 3 times. Values are Means ± standard deviation.
Purified PLA2A converts PC to arachidonic acid and is inhibited by calcium
To verify that PLA2A had PLA2 activity, we tagged it with a FLAG sequence at the C-terminal (PLA2A-FLAG) and purified the protein from D. discoideum cells. In the under-buffer assay, PLA2A-FLAG was able to rescue the LY-sensitive phenotype of the plaA− cells as did PLA2A-GFP (data not shown). We purified the recombinant protein by ion-exchange chromatography followed with affinity chromatography (Figure 6A). The protein was expressed at low levels, but the recovery from the purification was over 50% and we were able to obtain a sufficient amount of purified protein (typically 10 μg/109 cells) for in vitro PLA2 assays. The purified PLA2A-FLAG was able to produe arachidonic acid in buffer containing EGTA or EDTA using phophatidylcholine (PC) as substrate (Figure 6B). The specific activity of the purified PLA2A was similar to that of PLA2 from snake venom (2.7×10-11 μmol/μg-min versus 7.3×10-11 μmol/μg-min, respectively) at a substrate concentration of 10-9 M. The PLA2 activities of previously reported iPLA2s, with the exception of the patatin-related PLA2s, are not activated by Ca2+, and thus they have been called Ca2+-independent PLA2 (Ackermann et al., 1994; Chakraborti, 2002; Holk et al, 2002). The PLA2 activity of PLA2A-FLAG did not require Ca2+, in fact it was inhibited in buffer containing very high (5 mM) CaCl2 (Figure 6B). The purified enzyme bearing the active site S61A mutation displayed no activity under any condition, consistent with its inability to rescue the mutant phenotype. We did not assess whether the enzyme displayed activity against triacylglycerol and this have not ruled out that it can also function as a triacylglycerol lipase.
Figure 6.
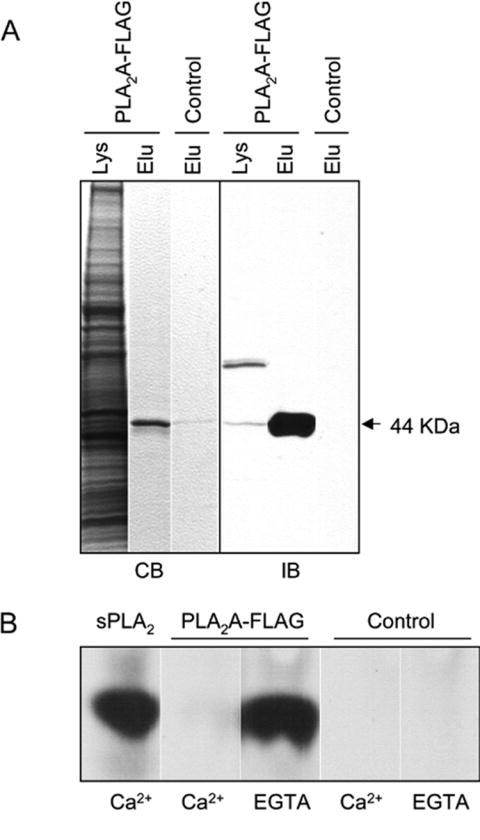
Conversion of phosphatidylcholine to arachidonic acid by purified PLA2A-FLAG. (A) PLA2A-FLAG was affinity purified. Coomassie Blue stained gel (CB) and immuno-blot (IB) shows cell lysate and eluted fractions prepared from PLA2A-FLAG expressing cells or wild type cells. The major eluted band from PLA2A-FLAG expressing cells is at the predicted size of 44 kD, as indicated by arrow. A similar-sized non-specific band was eluted from wild type cells but not recognized by Flag antibodies. Another non-specific band (~ 70 kD) recognized by FLAG antibody is present in the cell lysate, but not in the purified fractions. (B) Thin layer chromatography (TLC) of 3H-arachidonic acid (3H-AA) released from 3H-phosphatidylcholine (3H-PC) by purified PLA2A-FLAG. The eluted fractions were incubated with 3H-PC in buffer containing 5 mM Ca2+ or 5 mM EGTA for 60 minutes at 25°C. 3H-AA generated by a snake venom sPLA2 (Sigma P7778) was used as positive control. The eluted fraction from wild-type cells was used as negative control. The experiment was repeated at least five times.
Altered arachidonic acid metabolism underlies phenotype of cells deficient in PLA2A
To further elucidate the defect in the plaA− cells, we examined the profiles of lipids visualized by steady-state labeling with 3H-arachidonic acid. After several hours of incubation, cells were washed, stimulated with chemoattractant, and lipids were extracted for thin layer chromatography analyses (Figure 7A). Both wild-type and plaA− cells displayed similar levels of the major labeled components, presumably phosphatidylcholine, phosphotidylethanolamine and other lipids. The wild-type cells also displayed a prominently 3H-labeled component which co-chromatographed near arachidonic acid on thin-layer chromatography. Chemoattractants induced a rapid two to three fold increase in the level of this 3H-arachidonic acid derivative that peaked within 30 seconds and declined more slowly (Figure 7B). The levels of the 3H-arachidonic acid derivative were reduced by more than 70% in the plaA− cells and chemoattractant-induced increases were undetectable. The Rf of the compound was somewhat variable (Rf=0.815-0.903), usually running slightly faster than or with arachidonic acid (Rf=0.83 ± 0.02). We excised the spot and chromatographed it in an independent TLC system. In the second system, the compound co-chromatographed with arachidonic acid (Rf=0.31) and was widely separated from the methyl ester (Rf=0.59). These observations suggest that this component is 3H-arachidonic acid or a very closely related, perhaps unstable, derivative.
Figure 7.
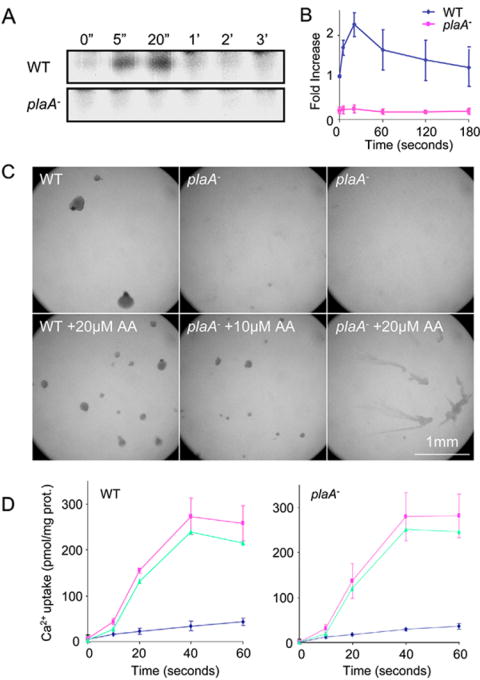
We wondered whether the severely depressed levels of the arachidonic acid derivative in the plaA− cells might underlie the defect in chemotaxis. We tested whether exogenous arachidonic acid might mitigate the defect. Similar to previous under-buffer assays, wild-type cells were unaffected by 10 μM LY whereas plaA− cells were unable to aggregate in the presence of this inhibitor. However, addition of 5 to 20 μM arachidonic acid partially reversed this defect (Figure 7C). Exogenous arachidonic induced the plaA− cells to aggregate at concentrations LY as high as 40 μM whereas wild-type cells were able to aggregate up to 80 μM. Oleic acid did not have the same effect as arachidonic acid. These results suggest that the observed deficit in the arachidonic acid derivative may underline the chemotactic defect.
Published reports have implicated arachidonic acid as a regulator of calcium influx in a variety of cells, including D. discoideum. However, as shown in Figure 7D, basal and chemoattractant-mediated Ca2+ influxes were very similar in wild type and plaA− cells. These observations suggest that the effects of PLA2A on chemotaxis and arachidonic acid metabolism are unlikely to be mediated through effects of calcium uptake.
Discussion
We have found that pathways involving PLA2 and PI3K act in parallel to mediate chemotaxis. Neither loss of PLA2A nor significant lowering of PI(3,4,5)P3 above has a dramatic effect on chemotaxis. However, simultaneous inhibition of both activities caused a severe defect in chemotaxis. The absence of PLA2A does not affect PI(3,4,5)P3 production indicating that PLA2A is not upstream of PI3K. Thus, PLA2A and its metabolites appear to be part of a parallel pathway regulating chemotaxis and actin polymerization.
These signaling events are involved in the first and second phases of actin polymerization (Chen et al., 2003). The second phase is absent in pi3k1-/2- cells, indicating that PI(3,4,5)P3 is the important regulator for the second phase. Our current results suggest that PI3Ks and PLA2A are equally important for the rapid initial peak; when both are inhibited the response is significantly decreased. A portion of the initial phase remains even in LY treated plaA−/pi3k1-/2- cells, possibly due to our inability to completely remove PLA2 or PI3K activity, as there are multiple homologues of each enzyme. Alternatively, yet another pathway is involved. It is likely that the decrease in the first peak of actin polymerization contributes to the defect in chemotaxis (Figure 4D). However, other events which play a role in chemotaxis may also be defective.
PLA2A is more likely to function in the response to chemoattractant gradient than motility per se. The plaA−/pi3k1-/2- cells exhibit poor chemotaxis but actively form pseudopods and move randomly at the same speed as wild type cells. LY treatment is apparently not equivalent to deletion of PI3Ks. When first added, LY causes cells to round up but when exposed to a chemoattractant gradient, wild type cells recover within a few minutes, start to extend pseudopods, and move towards the micropipette. Similar results have been observed in several laboratories (personal communications with Van Haastert, P.J.M. and Firtel, R.A.). However, when treated with LY, the plaA− cells remain rounded for at least 20 minutes and do not respond to the applied gradient. With longer recovery these treated cells regain random motility. Thus PLA2A may also be involved in the mechanism by which the applied gradient overcomes the LY-induced motility block.
There are parallel pathways in the chemotaxis network and each pathway contains multiple redundant components. First, the network is branched after G-protein, with PI3K/PTEN regulating one branch and PLA2A being involved in the other. Inhibition of either pathway does not block chemotaxis since the remaining pathway can compensate. Second, within each pathway, there are several isoforms of the key enzymes. It has been shown that more than two PI3Ks control PI(3,4,5)P3 levels and we might expect several PLA2s also act in concert. These layers of redundancy may in part explain why chemotaxis has been difficult to inhibit specifically.
We have identified a specific PLA2 homologue and have shown that its ability to generate an arachidonic acid derivative may be required for chemotaxis. The observed rapid chemoattractant-induced increase in the levels of the 3H-arachidonic acid derivative does not occur in plaA− cells. The transient burst of this compound in response to uniform stimulation of wild type cells is identical to the behavior of other molecules such as PI(3,4,5)P3 that have been implicated in directional sensing and found at the cell’s leading edge when the stimulus is applied as a gradient. Further studies are required to precisely identify the derivative and determine its location in chemotaxing cells. The strong defect in arachidonic acid metabolism is remarkable considering that there are 14 patatin-like PLA2s in D. discoideum. It is possible that PLA2A targets a specific phospholipid substrate or phospholipids within a specific compartment. The chemoattractant regulation and specific defect in the mutant indicates that PLA2A plays more than a “housekeeping” role.
Exogenous arachidonic acid (and not oleic acid) can compensate for the loss of PLA2A, indicating a specific requirement for an achidonic acid metabolite. Uniform application of arachidonic acid can decrease sensitivity to PI3K inhibition even though it would not be expected to produce an internal gradient within the cell. Perhaps arachidonic acid enhances the activity of another signaling molecule which is asymmetrically localized. While normally a localized increase in arachidonic acid synergizes with the other component to produce optimal response, uniform levels may be able to partially function by enhancing the other activity. Alternatively, other products of PLA2A such as a lyso-phospholipid may also be required for full rescue.
We have as yet no insight as to the target of the PLA2A metabolites. Studies in mammalian cells have suggested that arachidonic acid may regulate store-operated calcium channels (Smani et al., 2004). Furthermore, previous studies in D. discoideum have suggested that products of PLA2, arachidonic acid and platelet activation factor (PAF, synthesized from lyso-phosphatidylcholine), affect chemoattractant-induced Ca2+ influx and can trigger Ca2+ influx directly (Sordano et al., 1993; Schaloske et al., 1995; Schaloske et al., 1997). Ca2+ influx stimulated by cAMP was blocked with PLA2 inhibitors and then bypassed by arachidonic acid and related components (Schaloske et al., 1998). However, we found that specific genetic disruption of PLA2A that leads to a chemotaxis defect and altered arachidonic acid metabolism does not affect Ca2+ uptake. Other studies have also not supported a role for Ca2+ in chemotaxis. Cells lacking a putative IP3 receptor homolog (iplA-), in which Ca2+ influx in response to cAMP is abolished, displayed no chemotaxis defect (Traynor et al., 2000). Consistently we found that iplA- cells were not more sensitive than wild type cells to inhibitors of PI3K (data not shown).
Our studies provide genetic evidence that PLA2 and PI3K activities act in parallel. Similar mechanisms might exist in other organisms. Arachidonic acid releasing phospholipases are enriched in nerve growth cones and have been implicated in neurite outgrowth (Hornfelt, et al, 1999). Studies of chemotaxis in monocytes revealed requirements for cPLA2 and iPLA2 with distinct characteristics (Carnevale and Cathcart, 2001). Inhibition of cPLA2 could be overcome by exogenous arachidonic acid as shown in our studies. It is likely that such parallel pathways are conserved and that PLA2 is a central regulator of chemotaxis along with PI3K/PTEN. Consideration of the parallel roles of these pathways may help to resolve some of the conflicting results observed by inhibition of PI3K in other systems.
Supplementary Material
Acknowledgments
Research was supported by PHS grants 28007 and 34933 to PND, an LLS Fellowship 3374-05 to MI, and an ACS Fellowship PF-02-109-01-DDC to MAL. Sequence data for D. discoideum was obtained from the dictyBase website at http://www.dictybase.org/. We thank Dr. Daniel M. Raben for his technical support and suggestions for lipid analyses. We thank Dr. Richard A. Firtel for the pi3k1-/2- cell line.
Footnotes
An interesting phenomenon was observed in the course of the experiments to determine cAR1 levels. Initially, pla2a-/pi3k1-/2- cells displayed the wild type time course and level of cAR1 during development. However, after culture for four weeks, cAR1expression levels gradually declined in these cells while those in pla2a- and pi3k1-/2- cells remained similar to those in wild type. Similar phenomenon had been observed in gα2- and gβ- cells (unpublished observations).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackermann EJ, Kempner ES, Dennis EA. Ca2+-independent cytosolic phospholipase A2 from Macrophage-like P388D1 cells. J Biol Chem. 1994;269:9227–9233. [PubMed] [Google Scholar]
- Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- Adachi H, Hasebe T, Yoshinaga K, Ohta T, Sutoh K. Isolation of Dictyostelium discoideum cytokinesis mutants by restriction enzyme-mediated integration of the Blasticidin S resistance marker. Biochem Biophys Res Commun. 1994;205:1808–1814. doi: 10.1006/bbrc.1994.2880. [DOI] [PubMed] [Google Scholar]
- Baggiolini M. Chemokines in pathology and medicine. J Intern Med. 2001;250:91–104. doi: 10.1046/j.1365-2796.2001.00867.x. [DOI] [PubMed] [Google Scholar]
- Buczynski G, Grove B, Nomura A, Kleve M, Bush J, Firtel RA, Cardelli J. Inactivation of two Dictyostelium discoideum genes, DdPIK1 and DdPIK2, encoding proteins related to mammalian phosphatidylinositide 3-kinases, results in defects in endocytosis, lysosome to postlysosome transport, and actin cytoskeleton organization. J Cell Biol. 1997;136:1271–1286. doi: 10.1083/jcb.136.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussoloni F, Sordano C, Benfenati E, Bozzaro S. Dictyostelium cells produce platelet-activating factor in response to cAMP. Eur J Biochem. 1991;196:609–615. doi: 10.1111/j.1432-1033.1991.tb15856.x. [DOI] [PubMed] [Google Scholar]
- Carnevale KA, Cathcart MK. Calcium-independent phospholipase A(2) is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. J Immunol. 2001;167:3414–3421. doi: 10.4049/jimmunol.167.6.3414. [DOI] [PubMed] [Google Scholar]
- Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol. 2001;2:515–521. doi: 10.1038/88710. [DOI] [PubMed] [Google Scholar]
- Chadborn NH, Ahmed AI, Holt MR, Prinjha R, Dunn GA, Jones GE, Eickholt BJ. PTEN couples Sema3A signaling to growth cone collapse. J Cell Sci. 2006;119:951–957. doi: 10.1242/jcs.02801. [DOI] [PubMed] [Google Scholar]
- Chakraborti S. Phospholipase A2 isoforms: a perspective. Cell Signal. 2002;15:637–665. doi: 10.1016/s0898-6568(02)00144-4. [DOI] [PubMed] [Google Scholar]
- Chen L, Janetopoulos C, Huang YE, Iijima M, Borleis J, Devreotes PN. Two phases of actin polymerization display different dependencies on PI(3,4,5)P3 accumulation and have unique poles during chemotaxis. Mol Biol Cell. 2003;14:5028–5037. doi: 10.1091/mbc.E03-05-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Funamoto S, Firtel RA. Signaling pathways controlling cell polarity and chemotaxis. Trends Biochem Sci. 2001;26:557–566. doi: 10.1016/s0968-0004(01)01934-x. [DOI] [PubMed] [Google Scholar]
- Dumstrei K, Mennecke R, Raz E. Signaling pathways controlling primordial germ cell migration in zebrafish. J Cell Sci. 2004;117:4787–4795. doi: 10.1242/jcs.01362. [DOI] [PubMed] [Google Scholar]
- Egelhoff TT, Brown SS, Manstein DJ, Spudich JA. Hygromycin resistance as a selectable marker in Dictyostelium discoideum. Mol Cell Biol. 1989;9:1965–1968. doi: 10.1128/mcb.9.5.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Haase I, Simmeth E, Gerisch G, Müller-Taubenberger A. A brilliant monomeric red fluorescent protein to visualize cytoskeleton dynamics in Dictyostelium. FEBS Letters. 2004;577:227–232. doi: 10.1016/j.febslet.2004.09.084. [DOI] [PubMed] [Google Scholar]
- Funamoto S, Milan K, Meili R, Firtel RA. Role of phosphatidylinositol 3’ kinase and a downstream pleckstrin homology domain-containing protein in controlling chemotaxis in Dictyostelium. J Cell Biol. 2001;153:795–810. doi: 10.1083/jcb.153.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan M, Zhan L, Li Z, Ai Y, Wu D, Huang C. Neutrophils lacking phosphoinositide 3-kinase-γ show loss of directionality during N-formyl-Met-Leu-Phe-induced chemotaxis. Proc Natl Acad Sci. 2002;99:3603–3608. doi: 10.1073/pnas.052010699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Hill K, Welti S, Yu J, Murray JT, Yip SC, Condeelis JS, Segall JE, Backer JM. Specific requirement for the p85-p110alpha phosphatidylinositol 3-kinase during epidermal growth factor-stimulated actin nucleation in breast cancer cells. J Biol Chem. 2000;275:3741–3744. doi: 10.1074/jbc.275.6.3741. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Hornfelt M, Ekstrom PA, Edstrom A. Involvement of phospholipase A2 activity in the outgrowth of adult mouse sensory axons in vitro. Neuroscience. 1999;91:1539–1547. doi: 10.1016/s0306-4522(98)00684-8. [DOI] [PubMed] [Google Scholar]
- Huang YE, Iijima M, Devreotes PN. Receptor mediated regulation of PI3Ks confines PI(3,4,5)P3 to the leading edge of chemotaxing cells. Mol Biol Cell. 2003;14:1913–1922. doi: 10.1091/mbc.E02-10-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima M, Devreotes PN. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- Iijima M, Huang YE, Luo HR, Vazquez F, Devreotes PN. Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J Biol Chem. 2004;279:16606–16613. doi: 10.1074/jbc.M312098200. [DOI] [PubMed] [Google Scholar]
- Jenkins CM, Wolf MJ, Mancuso DJ, Gross RW. Identification of the calmodulin-binding domain of recombinant calcium-independent phospholipase A2beta. Implications for structure and function. J Biol Chem. 2001;276:7129–7135. doi: 10.1074/jbc.M010439200. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Phosphoinositide Recognition Domains. Traffic. 2003;4:201–213. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-β2 and -β3 and PI3Kγ in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- Loovers HM, Postma M, Keizer-Gunnink I, Huang YE, Devreotes PN, van Haastert PJM. Distinct roles of PI(3,4,5)P3 during chemoattractant signaling in Dictyostelium: A quantitative in vivo analysis by inhibition of PI3-kinase. Mol Biol Cell. 2006;17:1503–1513. doi: 10.1091/mbc.E05-09-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JL, Coukell MB. A Ca2+ transport system associated with the plasma membrane of Dictyostelium discoideum is activated by different chemoattractant receptors. J Cell Biol. 1991;112:103–110. doi: 10.1083/jcb.112.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero JA, Kilian B, Chan J, Bayliss PE, Heisenberg CP. Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Curr Biol. 2003;13:1279–1289. doi: 10.1016/s0960-9822(03)00505-0. [DOI] [PubMed] [Google Scholar]
- Mouneimne G, Soon L, DesMarais V, Sidani M, Song X, Yip SC, Ghosh M, Eddy R, Backer JM, Condeelis J. Phospholipase C and cofilin are required for carcinoma cell directionality in response to EGF stimulation. J Cell Biol. 2004;166:697–708. doi: 10.1083/jcb.200405156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent CA, Devreotes PN. A cell’s sense of Direction. Science. 1999;284:765–770. doi: 10.1126/science.284.5415.765. [DOI] [PubMed] [Google Scholar]
- Park YH, Kantor L, Wang KK, Gnegy ME. Repeated, intermittent treatment with amphetamine induces neurite outgrowth in rat pheochromocytoma cells (PC12 cells) Brain Res. 2002;951:43–52. doi: 10.1016/s0006-8993(02)03103-7. [DOI] [PubMed] [Google Scholar]
- Rao Y, Wong K, Ward M, Jurgensen C, Wu JY. Neuronal migration and molecular conservation with leukocyte chemotaxis. Genes Dev. 2002;16:2973–2984. doi: 10.1101/gad.1005802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif K, Okkenhaug K, Sasaki T, Penninger JM, Vanhaesebroeck B, Cyster JG. Cutting edge: differential roles for phosphoinositide 3-kinase, p110gamma and p110delta, in lymphocyte chemotaxis and homing. J Immunol. 2004;173:2236–2240. doi: 10.4049/jimmunol.173.4.2236. [DOI] [PubMed] [Google Scholar]
- Rickert P, Weiner OD, Wang F, Boune HR, Servant G. Leukocytes navigate by compass: role of PI3Kγ and its lipid products. Trends Cell Biol. 2000;10:466–473. doi: 10.1016/s0962-8924(00)01841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- Schaloske R, Malchow D. Mechanism of cAMP-induced Ca2+ influx in Dictyostelium: role of phospholipase A2. Biochem J. 1997;327:233–238. doi: 10.1042/bj3270233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaloske R, Sonnemann J, Malchow D, Schlatterer C. Fatty acids induce release of Ca2+ from acidosomal stores and activate capacitative Ca2+ entry in Dictyostelium discoideum. Biochem J. 1998;332:541–548. doi: 10.1042/bj3320541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaloske R, Sordano C, Bozzaro S, Malchow D. Stimulation of calcium influx by platelet activating factor in Dictyostelium. J Cell Sci. 1995;108:1597–1603. doi: 10.1242/jcs.108.4.1597. [DOI] [PubMed] [Google Scholar]
- Schaloske RH, Dennis E. The phospholipase A2 superfamily and its group numbering system. BBA. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Schneider IC, Haugh JM. Quantitative elucidation of a distinct spatial gradient-sensing mechanism in fibroblasts. J Cell Biol. 2005;171:883–892. doi: 10.1083/jcb.200509028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- Smit MJ, Verdijk P, Van der Raaij-Helmer EMH, Navis M, Hensbergen PJ, Leurs R, Tensen CP. CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3-kinase. Blood. 2003;102:1959–1965. doi: 10.1182/blood-2002-12-3945. [DOI] [PubMed] [Google Scholar]
- Sordano C, Cristino E, Bussolino F, Wurster B, Bozzaro S. Platelet activating factor modulates signal transduction in Dictyostelium. J Cell Sci. 1993;104:197–202. doi: 10.1242/jcs.104.1.197. [DOI] [PubMed] [Google Scholar]
- Sotsios Y, Whittaker GC, Westwick J, Ward SG. The CXC chemokine stromal cell-derived factor activates a Gi-coupled phosphoinositide 3-kinase in T lymphocytes. J Immunol. 1999;163:5954–5963. [PubMed] [Google Scholar]
- Stocker H, Andjelkovic M, Oldham S, Laffargue M, Wymann MP, Hemmings BA, Hafen E. Living with lethal PIP3 levels: viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB. Science. 2002;295:2088–2091. doi: 10.1126/science.1068094. [DOI] [PubMed] [Google Scholar]
- Stramer B, Wood W, Galko MJ, Redd MJ, Jacinto A, Parkhurst SM, Martin P. Live imaging of wound inflammation in Drosophila embryos reveals key roles for small GTPases during in vivo cell migration. J Cell Biol. 2005;168:567–573. doi: 10.1083/jcb.200405120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor D, Milne JL, Insall RH, Kay RR. Ca2+ Signaling is not required for chemotaxis in Dictyostelium. EMBO J. 2000;19:4846–4854. doi: 10.1093/emboj/19.17.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Es S, Wessels D, Soll DR, Borleis J, Devreotes PN. Tortoise, a novel mitochondrial protein, is required for directional responses of Dictyostelium in chemotactic gradients. J Cell Biol. 2001;152:1–13. doi: 10.1083/jcb.152.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Bio. 2002;4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- Ward SG. Do phosphoinositide 3-kinases direct lymphocyte navigation? Trends Immunol. 2004;25:67–74. doi: 10.1016/j.it.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Wu D, Huang CK, Jiang H. Roles of phospholipid signaling in chemoattractant-induced responses. J Cell Sci. 2000;113:2935–2940. doi: 10.1242/jcs.113.17.2935. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Sozzani S, Altruda F, Mantovani A, Hirsch E. Lipids on the move: phosphoinositide 3-kinases in leukocyte function. Immunol Today. 2000;21:260–264. doi: 10.1016/s0167-5699(00)01649-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.