

Abstract
Demyelination, a pathological hallmark of multiple sclerosis, may be a necessary but not a sufficient condition for motor dysfunction associated with this disease. We favor a neurodegenerative model of multiple sclerosis and suggest that demyelination creates a permissive environment wherein the denuded axon becomes susceptible to immune-mediated injury. Unfortunately, the cellular effectors responsible for eliciting such axonal injury are currently unknown. Based on previous observations implicating cytotoxic T cells in this injury, we assessed motor function, axon dropout, and axon injury following peptide depletion of the immunodominant CD8+ antiviral T cell response in the IFNγ receptor-deficient mouse model of acute demyelination. We found that the targeted removal of this population of cytotoxic effector cells prior to infection with the Theiler’s murine encephalomyelitis virus caused a substantial preservation of motor function at 45 days postinfection that was associated with preservation of retrograde axonal transport in a subpopulation of surviving axons within the spinal cord. We conclude that cytotoxic T cells may be responsible for the initiation of axon injury following demyelination.
Keywords: Cytotoxic T Cells, Multiple Sclerosis, Axon Injury, Interferon-gamma, Retrograde Transport, MHC Class I
1. INTRODUCTION
Multiple sclerosis (MS) has historically been considered a disease of myelin and the primary focus of therapeutic investigation has centered on the oligodendrocyte and mechanisms of preventing demyelination or inducing remyelination. However, while demyelination is obviously a pathological hallmark of MS, and while demyelination is certainly an underlying etiological event, recent evidence has started to suggest that the clinically relevant locus of functional disability is injury to the axon (Howe and Rodriguez, 2005). Such a neurodegenerative model of MS posits that demyelination is a permissive factor that creates an environment in which the axon becomes susceptible to injury mediated either by loss of axoglial trophic interactions (Compston, 2004; Wilkins and Compston, 2005) or by immune-mediated attack of the denuded axon (Howe and Rodriguez, 2005). Our recent observations of axonal and functional preservation in chronically demyelinated mice genetically deficient in perforin (Howe et al., 2006) and our previous finding that genetic disruption of MHC class I function preserves motor function (Rivera-Quinones et al., 1998) and axon integrity (Ure and Rodriguez, 2002) during chronic demyelination argue strongly in favor of the immune-mediated injury model.
The cellular effector(s) responsible for injuring demyelinated axons is currently unidentified. However, circumstantial evidence, including the roles of perforin and MHC class I indicated above and the fact that CD8+ T cells are the most abundant lymphocytes within MS lesions (Babbe et al., 2000), suggests that class I-restricted cytotoxic T cells may be the culprit. Indeed, the presence of CD8+ T cells within MS lesions correlates with axon injury (Bitsch et al., 2000), and immunostaining indicates that functional MHC class I is substantially upregulated on neurons and axons within active lesions in patients with acute MS (Hoftberger et al., 2004). Unfortunately, the majority of experiments characterizing CD8+ T cells have focused on their role in injuring oligodendrocytes and it is unknown whether cytotoxic T cells are directly interacting with MHC class I on axons within demyelinated lesions in a manner that elicits injury.
In an effort to mechanistically probe such an interaction we chose to analyze axonal injury in mice specifically depleted of an immunodominant population of cytotoxic T cells (Johnson et al., 2001). To do so we utilized a peptide treatment strategy to clonally delete cytotoxic T cells specific for the VP2121–130 epitope of the Theiler’s murine encephalomyelitis virus (TMEV) (Buenz and Howe, 2006). We have previously shown that these T cells comprise the dominant cytotoxic immune response in mice of the H-2b MHC haplotype (Mendez-Fernandez et al., 2003). In order to permit analysis of axon injury within the context of demyelination we chose to use H-2b haplotype mice with a genetic deletion of the interferon gamma receptor (IFNγR−/−). We have previously shown that these mice develop persistent virus infection and a rapidly evolving demyelinating disease that is characterized by a vigorous antiviral cytotoxic T cell response (Johnson et al., 2001). We also previously observed preservation of motor function in these mice following depletion of VP2121–130-specific T cells, a finding which suggested that CD8+ cytotoxic T cells are mediators of axon injury in this viral model of demyelination. In the present study we provide evidence that peptide-mediated depletion of VP2121–130-specific T cells does not prevent the absolute loss of spinal axons associated with the rapidly progressing disease characteristic of TMEV-infected IFNγR−/− mice, but does partially preserve axonal function in the extant population of spinal axons as assessed by retrograde transport of FluoroGold.
2. MATERIALS AND METHODS
Virus
The Daniel’s strain of TMEV was used for all experiments (Lipton, 1975). The virus was grown in BHK-21 cells and titrated via plaque assay on L2 cells, as previously described (Rodriguez et al., 1983). At four to eight weeks of age, mice were intracerebrally infected with 2 × 105 PFU of TMEV in a total volume of 10 μL.
Mice
Mice were either obtained from Jackson Laboratories (Bar Harbor, Maine) (B6 wildtype controls) or were bred in our animal facility (IFNγR−/− mice). The original 129-Ifngrtm1 IFNγR−/− mice were obtained from Michel Aguet (Swiss Institute for Experimental Cancer Research, Epalinges, Switzerland). Handling of all mice conformed to the National Institutes of Health and the Mayo Clinic College of Medicine institutional guidelines.
Peptide depletion
Mice were treated intravenously with three injections of 0.1 mg of the VP2121–130 peptide (FHAGSLLVFM) or the irrelevant Db-restricted E7 control peptide (RAHYNIVTF), as described (Johnson et al., 2001). Each injection was separated by four hours. For experiments lasting longer than 7 days postinfection, mice were boosted with weekly intravenous injections of 0.1 mg of the appropriate peptide.
Brain infiltrating leukocytes and flow cytometry
Lymphocytes were extracted from dounce homogenized brain tissue by centrifugation through a 30% Percoll gradient at 7800 gave for 30 min in a Beckman JA-20 rotor. After centrifugation the myelin debris layer that floated at the top of the gradient was removed by aspiration and the lymphocyte-enriched layer floating just above the red blood cell pellet was diluted to 50 mL in RPMI and centrifuged at 1500 rpm for 5 min in a clinical centrifuge to collect the cell pellet. The lymphocyte-enriched pellet was resuspended in FACS buffer (PBS, 1% BSA, 0.2% sodium azide) and blocked for 20 min in FACS buffer containing 10% FBS and 50% supernatant from the 2.4G2 Fc blocking hybridoma. PerCP-conjugated anti-CD45 antibody and APC-conjugated anti-CD8 antibody were added to the cells at 1:400. R-PE-conjugated Db/VP2121–130 tetramer (Beckman Coulter Immunomics, San Diego, CA) was added at 1:100. Following incubation for 1 hr at 4°C the cells were washed 3 times in FACS buffer, fixed in 2% paraformaldehyde, and analyzed on a FACScan instrument (BD Bioscience, Mountain View, CA).
Rotarod analysis
The Rotamex rotarod (Columbus Instruments, Columbus, OH) was used to assess motor function. Mice were trained under a constant speed protocol and tested in an accelerating rod protocol, as described (McGavern et al., 1999b). Mice were tested at 0, 21, and 45 days postinfection. Rotarod velocity and time at fall from the rod were recorded for each mouse without knowledge of the experimental group.
Axon counting
Calculation of axon numbers was performed essentially as reported (Howe et al., 2006; McGavern et al., 2000; McGavern et al., 1999a). Sections of spinal cord were cut at 1 micron thickness and stained for exactly 20 min with the same batch of 4% para-phenylenediamine to ensure an identical intensity of myelin labeling. Digitized sample images from the smallest thoracic spinal cord section (T6) were collected at 60x magnification from each animal according to a sampling scheme developed previously (Howe et al., 2006; McGavern et al., 2000; McGavern et al., 1999a). Images were captured from regions free of demyelination to ensure the quantitation of only myelinated fibers. Each field measured 17675 μm2, and we collected a total of 2.01 mm2 from the E7-treated mice, 1.91 mm2 from the VP2-treated mice, and 4.14 mm2 from B6 mice. Myelinated axon diameters were calculated following segmentation of the gray values corresponding to the axoplasm from each image. Batch algorithms were generated in Matlab (The Mathworks, Nattick, MA) to automatically calculate the diameter of each axon in the field from the segmented binary image after regions corresponding to the vasculature, cell bodies, longitudinal axons, and demyelination were excluded on the basis of circularity thresholding. Diameters less than 1 μm were excluded from the analysis to eliminate small regions that did not correspond to axons. Axon area measurements were binned for analysis: (i) 1–4 μm2 (small axons); (ii) 4–10 μm2 (medium axons); and (iii) >10 μm2 (large axons). The number of axons in each bin was normalized to the area of spinal cord analyzed to yield axons per mm2. These values were averaged across all animals per group.
Demyelination assessment
Mice were perfused via intracardiac puncture with 50 mL of Trump’s fixative. Spinal cords and brains were removed and post-fixed for 24–48 hours in Trump’s fixative. Cords were removed from spinal columns and cut into one millimeter coronal blocks, and every third block was osmicated and embedded in glycol methacrylate (Rodriguez, 1991). Two μm-thick sections were stained with a modified erichrome/cresyl violet stain (Pierce and Rodriguez, 1989) and morphological analysis was performed on 12 to 15 sections per mouse as previously described (Rodriguez, 1991). Each quadrant from every coronal section from each mouse was graded for the presence or absence of demyelination without knowledge of the experimental group.
Bielschowsky staining
At 45 days postinfection mice were perfused via intracardiac puncture with 50 mL of 4% paraformaldehyde. Paraffin-processed spinal cord was sectioned at 10 μm, deparaffinized, rehydrated, and stained in 15% silver nitrate for 20 min at 37°C in the dark. Following washing in running distilled water, sections were incubated in ammoniacal silver (15% silver nitrate solution plus dropwise ammonium hydroxide until clarified) for 20 min at 37°C in the dark. Sections were then briefly rinsed in weak ammonium hydroxide, developed for 1 to 3 min in Bielschowky developer (6% formalin, 0.4% citric acid (w/v), acidified with nitric acid) diluted in ammoniacal silver, rinsed again in weak ammonium hydroxide, toned in 0.2% gold chloride until gray, incubated in 5% sodium thiosulfate for 1 min at RT, washed in running tap water for 10 min, dehydrated in alcohol, cleared in xylene, and mounted under Permount.
Retrograde labeling
Mice were anaesthetized with sodium pentobarbital (0.08 mg/g) one week prior to endpoint analysis (45 dpi). After dorsal laminectomy of the lower thoracic vertebrae, the spinal cord was hemisected on the right side using microdissecting blades (Harvard Apparatus, South Natick, MA) and the site was packed with Gelfoam (Upjohn Company, Kalamazoo, MI) soaked in a solution of 4% (w/v) FluoroGold (Fluorochrome Inc., Englewood, CO) and 2% (w/v) Fast Blue (Sigma) in sterile PBS. A combination of tracers was used to maximize the number of labeled descending axons. After overlaying the site with untreated Gelfoam, the axial musculature and skin were individually sutured. Analgesic and antiseptic ointments were applied to the sutured skin. Postoperatively, cages were regularly replenished with food pellets and water supplemented with acetaminophen and terramycin antibiotic. Bladder dysfunction occurred in approximately half of the hemisected mice, so manual bladder expression was performed 2–3 times daily. Seven days following the surgery mice were transcardially perfused with 4% paraformaldehyde, and brain and spinal cord were collected. Serial vibratome (Lancer Series 1200) sections (30 μm thick) of the brain were then made and mounted under Vectashield (Vector Laboratories Inc., Burlingame, CA). Cell bodies containing retrograde tracer, visualized by UV illumination (360–370 nm excitation, 420–460 nm emission) with an Olympus AX70 microscope, were counted under 400x magnification. The cells were predominantly labeled with large, brightly fluorescent granules characteristic of FluoroGold. A cell body was counted if a nucleus was visible (excluded tracer) or if, in the absence of a visible nucleus, a large cross-section of the cell body was labeled, as previously described (Ure and Rodriguez, 2002). Neurons in the raphe-reticular complexes were counted together due to the difficulty in discriminating the groups from each other (Ure and Rodriguez, 2002). The brainstem region with the highest density of retrogradely labeled neurons, encompassing 1.1 mm along the rostrocaudal axis, was used for quantitation. Cell bodies were counted in 10 sections, at intervals of every fourth section. In each section all of the labeled cell bodies on the right half of the brainstem (ipsilateral to site of dye application) were counted.
Statistics
All data are presented as mean values ± standard error of the mean. Retrograde labeling and axon numbers were analyzed by one-way ANOVA using the Holm-Sidak pairwise comparison or Dunn’s method to assess intra- and inter-group differences. Flow cytometry data, percent demyelinated quadrants, and rotarod measurements were analyzed by t-test. SigmaStat was utilized for all statistical analysis and for guidance regarding power and sample size.
3. RESULTS
VP2121–130 peptide treatment depletes virus-specific CD8+ T cells from the CNS
It has previously been shown that treatment of H-2b mice with the VP2121–130 peptide prior to TMEV infection and throughout the disease course deletes an immunodominant population of virus-specific CD8+ T cells (Johnson et al., 2001). We used the same depletion strategy in IFNγR−/− mice and assessed the quality of depletion by flow cytometric analysis of VP2121–130-tetramer-positive CD8-positive T cells in the brain-infiltrating leukocyte (BILs) population at 7 days postinfection (dpi). As a control we treated mice with the irrelevant E7 peptide derived from human papillomavirus (Johnson et al., 2001). As shown in Figure 1A, E7-treated mice exhibited a robust population of VP2-tetramer-positive CD8+ cells in the BILs preparation, with approximately 40% of CD45hi cells expressing CD8 (Figure 1C) and approximately 40% of these CD8+ cells expressing a class I molecule specific for the VP2121–130 peptide (Figure 1D). In contrast, VP2 peptide-treated mice exhibited a loss of VP2-tetramer-positive CD8+ cells in the BILs preparation (Figure 1B), and an overall reduction in CD8+ cells in the CD45hi population within the CNS infiltrate (Figure 1C). In fact, CD8+ cells were reduced from 38.3 ± 1.5% of CD45hi cells in the E7-treated mice (n=5) to 17.0 ± 1.7% in the VP2-treated mice (n=5; P<0.001 by t-test), and VP2-tetramer-positive cells were reduced from 40.1 ± 1.9% of CD8+ cells in the E7 mice to 3.1 ± 0.3% in the VP2-treated mice (P<0.001 by t-test). The overall reduction in the total population of CD45hiCD8+ cells in the VP2 peptide-treated mice suggests that the immunodominant VP2121–130-specific population of cytotoxic T cells was not replaced by cells responsive to a secondary viral epitope, as previously reported (Mendez-Fernandez et al., 2003).
Figure 1.
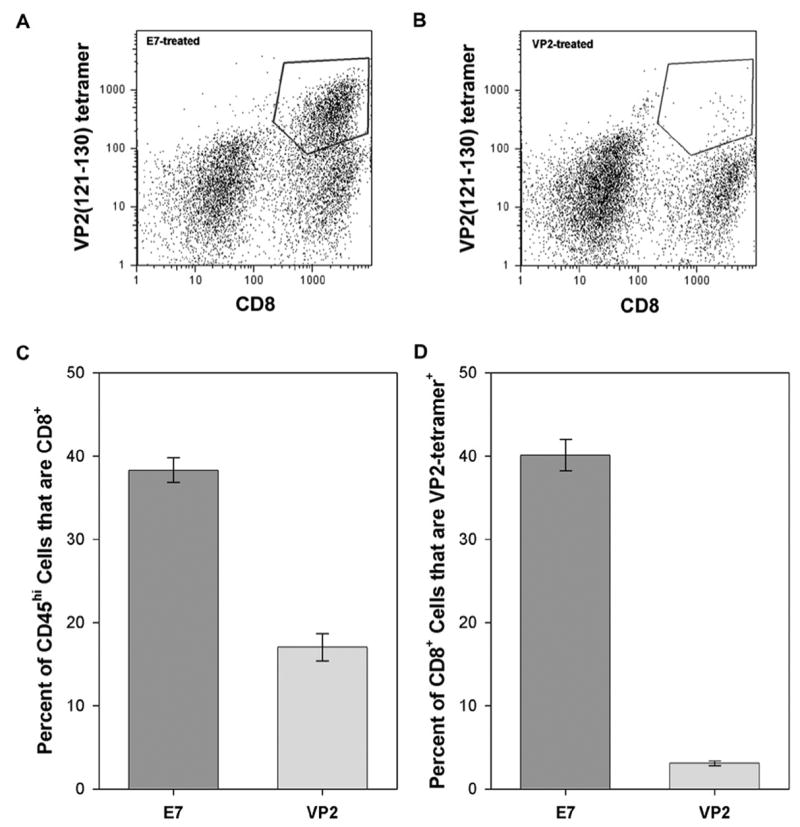
VP2121–130 peptide treatment depletes virus-specific CD8+ T cells from the CNS. IFNγR−/− mice were treated prior to infection and throughout the course of infection with a VP2121–130 peptide to deplete the immunodominant population of CD8+ T cells or with an irrelevant peptide derived from the E7 protein of HPV. Brain infiltrating lymphocytes were collected at 7 days postinfection, stained with anti-CD8 and a tetramer that recognizes VP2121–130-specific T cells, and analyzed by flow cytometry. As shown in panel A and quantified in panel D, approximately 40% of CD8+ T cells were VP2 tetramer-positive in E7-treated mice. In contrast, as shown in panel B and quanitfied in panel D, the population of VP2 tetramer-positive T cells was almost completely depleted in VP2 peptide-treated mice (P<0.001 vs. E7-treated by t-test). Likewise, as shown in panel C, since VP2121–130-specific T cells represent the immunodominant population of T cells in the brain at 7 days postinfection, VP2 peptide depletion effectively reduced the overall population of CD8+ CD45hi cells present in the brain. The polygon in panel A and panel B indicates CD8+ VP2 tetramer-positive cells used for the analysis.
The absence of VP2-specific CD8+ T cells confers preservation of motor function
Next we asked whether depletion of the immunodominant CD8+ T cell population would alter motor function as assessed by rotarod. For this experiment we treated IFNγR−/− mice with either VP2 peptide or E7 peptide throughout 45 days of infection and assessed their motor function at days 0 (uninfected), 21, and 45 postinfection. As shown in Figure 2, we found that deletion of the immunodominant CD8+ population resulted in a substantial preservation of motor function at 45 dpi. While E7-treated mice stayed on the rotarod for only 56 ± 18 seconds (n=6), VP2-treated mice were able to run for 206 ± 49 seconds (n=7; P=0.035 by t-test). We conclude that the absence of VP2121–130-specific cytotoxic T cells throughout the course of disease in IFNγR−/− mice preserved motor function.
Figure 2.
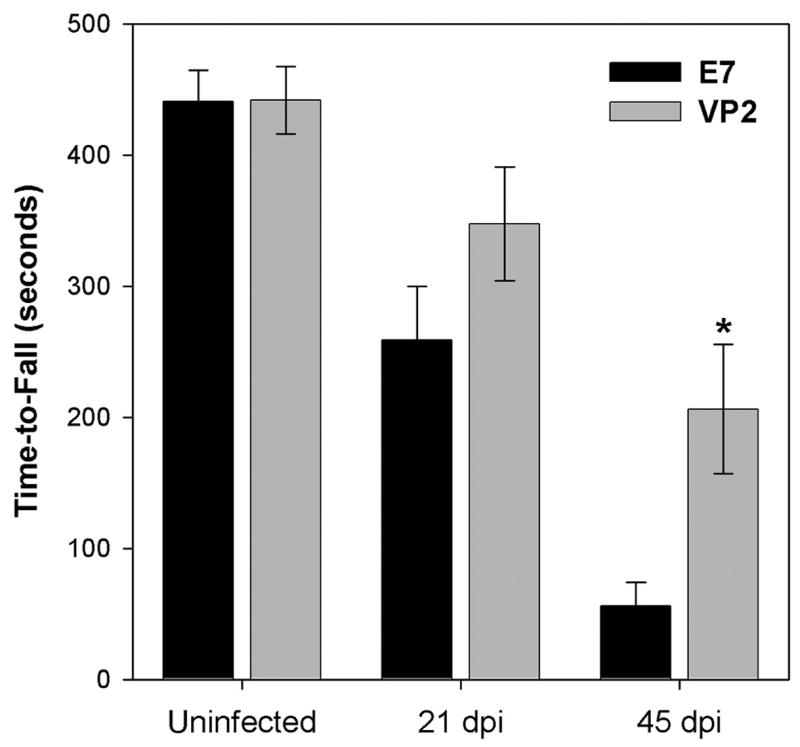
The absence of VP2-specific CD8+ T cells confers preservation of motor function. Function was assessed in VP2- or E7-treated mice prior to infection and at 21 and 45 days postinfection by rotarod analysis. At 45 days postinfection E7-treated mice performed at less than 13% of baseline function, while VP2 peptide-treated mice functioned at almost 50% of baseline (P=0.035 E7 vs VP2 by t-test).
Loss of VP2-specific CD8+ T cells does not change axon injury or demyelination in the spinal cord but does preserve retrograde axonal transport
We have previously found that genetic deletion of the cytotoxic T cell effector molecule perforin confers both preservation of motor function and preservation of medium and large diameter axons within the spinal cord of chronically infected, demyelinated H-2b mice (Howe et al., 2006). Therefore, we predicted that axons would also be spared in the VP2 peptide-treated IFNγR−/− mice and that this protection would correlate with functional preservation. To our surprise, depletion of VP2121–130-specific T cells had no effect on the number of axons as compared to E7-treated mice (Figure 3). While small axons were clearly lost in the infected mice (E7 = 36266 ± 1510 axons per mm2, n=6; VP2 = 37133 ± 2172 axons per mm2, n=7; uninfected = 65637 ± 2227 axons per mm2, n=12; P<0.001 by Kruskal-Wallis ANOVA on ranks), there was no difference between the E7- and VP2-treated groups (P>0.05 by Dunn’s method). Likewise, there was no difference in medium axons between any of the groups (P=0.173 by one-way ANOVA). Finally, while the number of large axons was significantly reduced in the infected mice (E7 = 20736 ± 1118 axons per mm2, n=6; VP2 = 21630 ± 1268 axons per mm2, n=7; uninfected = 26766 ± 1120 axons per mm2, n=12; P<0.001 by Kruskal-Wallis ANOVA on ranks), there again was no difference between the E7- and VP2-treated groups (P>0.05 by Dunn’s method).
Figure 3.
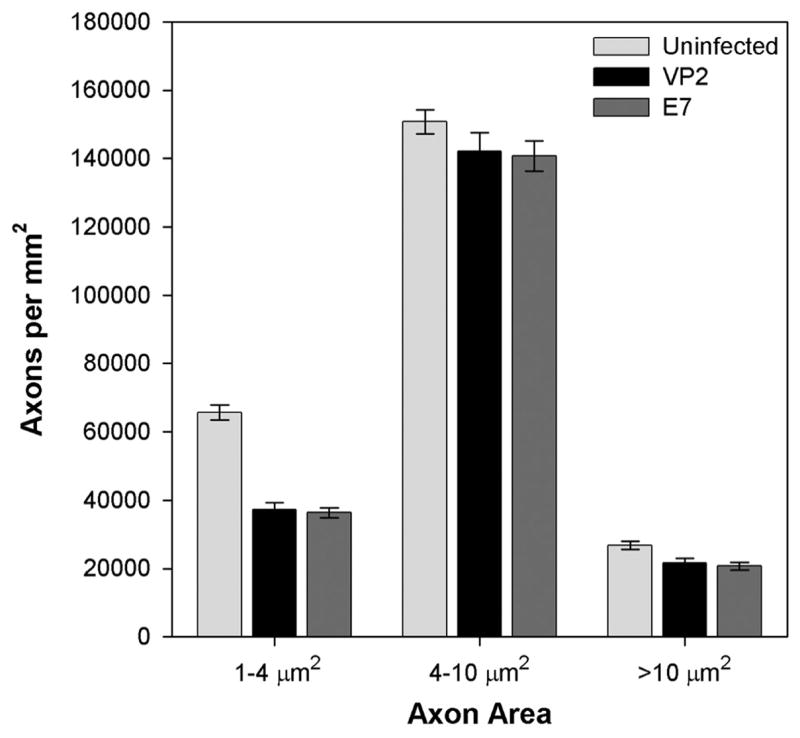
Peptide depletion of VP2-specific CD8+ T cells does not prevent the loss of spinal cord axons. Spinal axons were counted and binned by cross-sectional area in uninfected mice (B6) and in E7 or VP2 peptide-treated mice at 45 days postinfection. Both large (>10 μm2) and small (1–4 μm2) axons were lost in infected mice and the extent of loss did not differ between treatment groups. While medium-sized axons also trended toward reduction in the infected mice this difference was not statistically significant.
Because there was no change in the number of spinal axons following VP2 peptide-treatment, we predicted that demyelination was reduced in the functionally preserved mice. However, we observed no difference in the percent of spinal cord quadrants exhibiting demyelination (E7 = 28.6 ± 2.7% of quadrants; VP2 = 25.8 ± 2.4% of quadrants; P=0.450 by t-test). Likewise, we did not find any differences in the quality or extent of demyelination between the treated groups, as shown in Figure 4. We conclude that neither axon loss nor a difference in demyelination of extant axons explains the preservation of function in IFNγR−/− mice depleted of VP2-specific T cells.
FIgure 4.
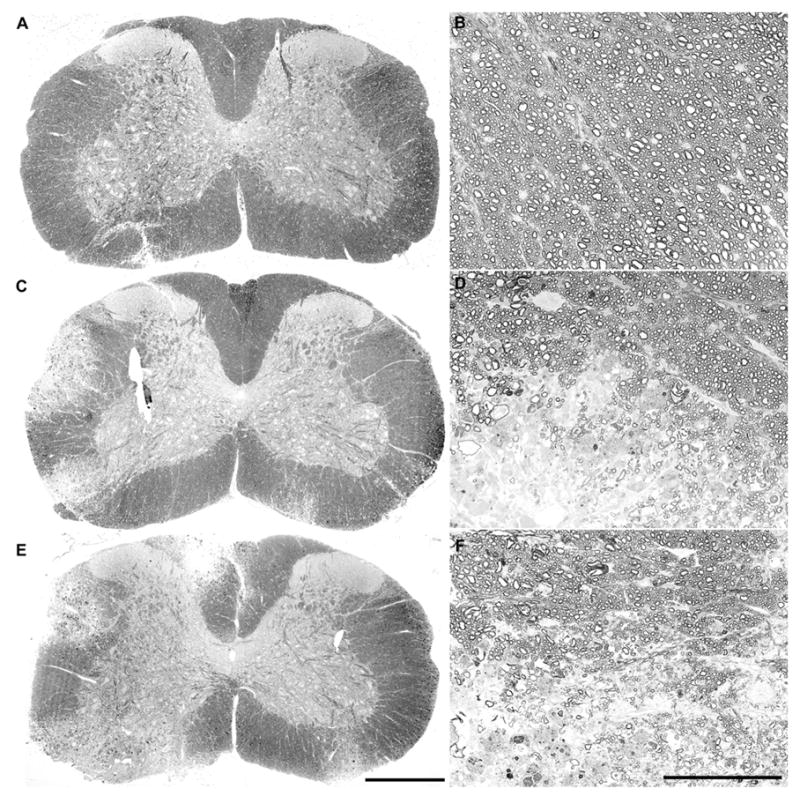
Peptide depletion of VP2-specific CD8+ T cells does not alter the extent or quality of demyelination in the spinal cord. Photomontages of 10x fields collected from uninfected (A) and E7-treated (C) or VP2-treated (E) mice at 45 days postinfection show that the amount and distribution of demyelination in the spinal cord was not grossly changed by peptide treatment. Likewise, higher magnification images of normal myelin in an uninfected mouse (B) and border areas at the junction of demyelinated and normally myelinated areas in E7-treated (D) and VP2-treated (F) mice show that the quality of demyelination in the infected mice was not changed by peptide treatment. The scale bar in E is 500 μm and refers to A, C, and E. The scale bar in F is 100 μm and refers to B, D, and F.
Finally, we asked whether the overall level or pattern of axon injury was reduced in VP2 peptide-treated mice. To do so we analyzed Bielschowsky stained spinal cord sections from VP2-treated and E7-treated mice at 45 dpi. As shown in Figure 5, we were unable to find any apparent difference in the distribution of injured axons, suggesting that, at least grossly, deletion of VP2-specific cytotoxic T cells does not impact the ongoing process of axonal injury during acute, fulminant demyelination. However, in an effort to more carefully probe axon function we assessed the retrograde transport of FluoroGold from the lower thoracic spinal cord to the reticular and raphe nuclear complex in the brainstem. As shown in Figure 6, we found a small but significant preservation of retrogradely labeled neurons in the brainstem of infected mice treated with VP2 peptide. Specifically, we counted 1224 ± 278 (n=30) FluoroGold-labeled neurons in the raphe-reticular complex in VP2-depleted mice but only 1008 ± 378 (n=27) labeled neurons in the E7-treated mice (P=0.014 by one-way ANOVA and Holm-Sidak pairwise comparison). While both infected groups exhibited substantially fewer labeled neurons as compared to uninfected mice (1789 ± 261 neurons; n=7; P<0.001 vs VP2-treated; P≪0.001 vs E7-treated), we conclude that the relative preservation of retrograde labeling in the VP2-treated mice contributes at least in part to the preservation of motor function.
Figure 5.
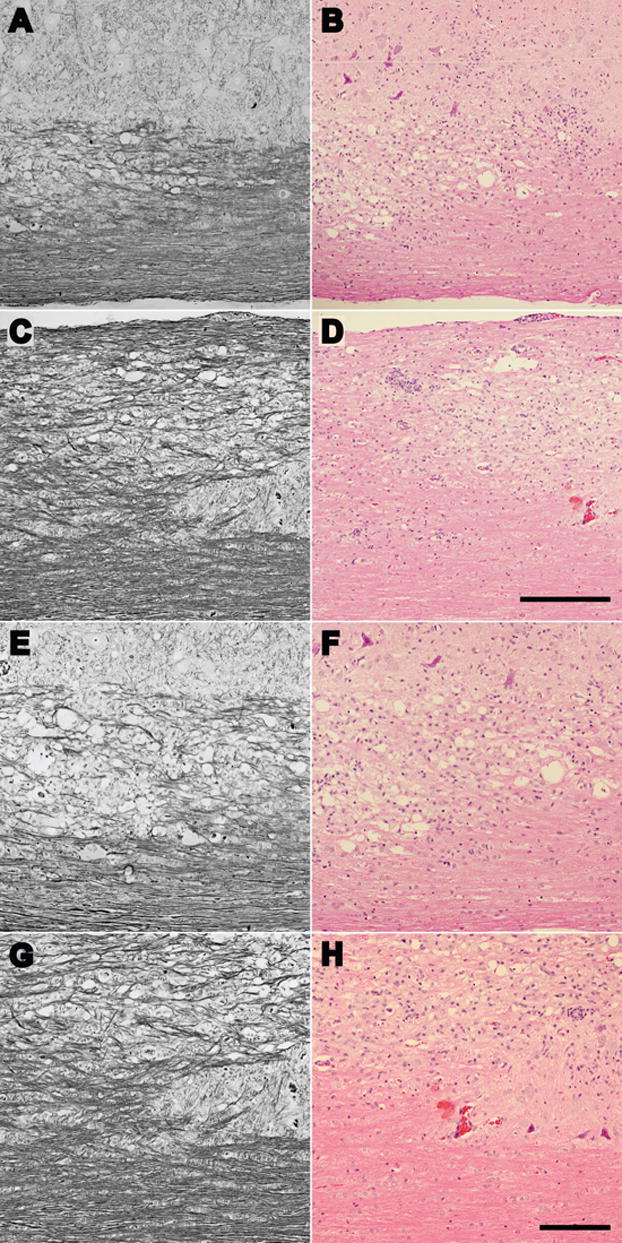
Peptide depletion of VP2-specific CD8+ T cells does not alter gross axonal pathology in the spinal cord. Analysis of Bielschowsky stained (A, C, E, and G) or hematoxylin and eosin stained (B, D, F, and H) spinal cord sections at low magnification (A, B, C, and D) or higher magnification (E, F, G, and H) did not reveal any obvious differences in the extent or pattern of axonal injury between E7-treated mice (A, B, E and F) and VP2-treated mice (C, D, G and H). Images were collected in the ventrolateral region of cervical spinal cord in both treatment groups. The scale bar in D is 500 μm and refers to A–D; the scale bar in H is 100 μm and refers to E–H.
Figure 6.
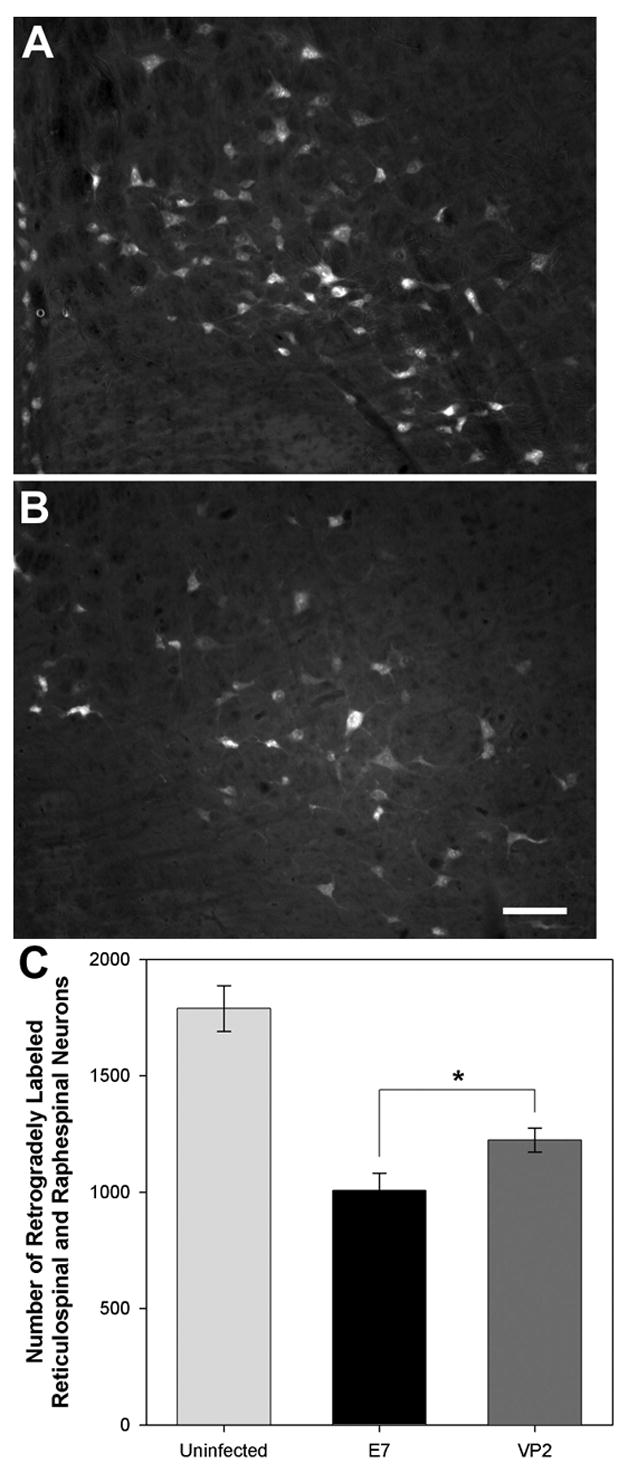
Peptide depletion of VP2-specific CD8+ T cells confers preservation of retrograde axonal transport in reticulospinal and raphespinal axons. Retrograde transport of FluoroGold was assessed in uninfected mice and at 45 days postinfection in VP2 and E7 peptide-treated mice. Representative sections analyzed under UV illumination are shown for VP2-treated (A) and E7-treated (B) mice. As shown in panel C, the number of retrogradely labeled reticular nucleus and raphe nucleus neurons was significantly increased in mice depleted of VP2-specific T cells (P=0.014 by Holm-Sidak one way ANOVA).
4. DISCUSSION
IFNγR−/− mice represent a useful model of fulminant demyelinating disease within the context of an MHC haplotype that is normally resistant to persistent TMEV infection. These animals exhibit rapid onset of demyelination and the development of severe neurologic disease, but because they are of the H-2b MHC haplotype they are amenable to manipulations of the CD8+ antiviral T cell repertoire via treatment with the immunodominant VP2121–130 peptide epitope (Johnson et al., 2001). While we do not consider these mice to necessarily represent a clinically relevant model of human multiple sclerosis, they do permit the exploration of T cell-mediated injury to the nervous system within the context of a rapidly evolving virus-mediated demyelinating insult. Using this model, we have observed a marked preservation of motor function in mice depleted of the immunodominant population of VP2-specific antiviral T cells. To our surprise, we did not find preservation of the number of spinal axons in these mice, despite the fact that we have observed such preservation correlated with protection of motor function in chronically demyelinated mice genetically deficient in perforin (Howe et al., 2006). Because demyelination and overt axonal injury also did not appear to differ between VP2- and E7-treated groups, we hypothesized that the preservation of motor function was due to the retention of some aspect of axonal function that was more subtle than our morphological criteria could detect. Indeed, by testing the health of spinal axons with regard to the retrograde transport of FluoroGold tracer, we found that VP2-depleted mice exhibited approximately 20% more retrogradely labeled neurons as compared to E7-treated mice. While both groups clearly had fewer labeled neurons than uninfected mice, we conclude that the presence of more functional axons in the VP2-depleted mice contributes to the preservation of motor function, despite equivalent overall demyelination and equivalent physical destruction of spinal axons.
We have previously observed that the absence of functional MHC class I due to genetic deletion of β2-microglobulin breaks resistance to persistent TMEV infection in H-2b mice but also promotes preservation of axons as determined by functional assessment (Rivera-Quinones et al., 1998) and retrograde labeling (Ure and Rodriguez, 2002). Likewise, we have recently shown that genetic deletion of perforin confers protection of spinal axon numbers and motor function despite widespread chronic demyelination (Howe et al., 2006). Moreover, others have shown that CD8+ T cells can directly transect neurites (Medana et al., 2001) and injure neurons (Zhu et al., 2006), and CD3+ lymphocytes have been found in contact with demyelinated axons within MS lesions (Neumann et al., 2002). These observations have led us to hypothesize that demyelinated axons become targets for cytotoxic T cell-mediated recognition of axonal MHC class I and are injured via a perforin-dependent mechanism. The findings presented herein confirm this model by showing that depletion of VP2-specific cytotoxic T cells protects motor function and contributes to a relative preservation of functional axons. At the same time, however, these findings are confounded by the absence of an effect on the absolute number of axons and by the relatively small preservation of retrogradely labeled brainstem neurons. Several possible explanations exist. First, our morphological analyses were all performed at 45 days postinfection, a timepoint that approaches the life expectancy of infected IFNγR−/− mice (Fiette et al., 1995; Johnson et al., 2001). It is possible that mice at this timepoint have become so severely debilitated that little anatomical substrate remains for comparison. Second, we only analyzed a subset of brainstem neurons and more detailed assessment of brainstem and cortical motor neuron populations may uncover robust preservation of a critical population of cells. Third, the IFNγR−/− mouse is in many ways a model of fulminant demyelinating disease rather than a model of the chronic, progressive demyelination observed in our previous studies. As such, this model may have additional pathological processes that we have not controlled for in our analysis. The fatal nature of the disease in these mice would argue in favor of this possibility. Indeed, our previous findings that CD4+ and CD8+ T cells produce IFNγ within the spinal cord during infection (Murray et al., 2002) and that this IFNγ is neuroprotective (Rodriguez et al., 2003) suggest that the IFNγR−/− model has additional complexities related to axon preservation.
In parallel, we rule out several possible explanations for the disparities in our observations. First, based on a previous study from our group indicating the absence of an effective immune response to viral epitopes other than VP2121–130 in H-2b mice (Mendez-Fernandez et al., 2003) and based on the data shown in Figure 1, we do not think that an unrecognized population of cytotoxic T cells is responsible for effecting axon injury in a manner that is separable from the induction of functional deficits. Second, we do not think that differences in viral load explain the preservation of motor function in VP2-treated mice, because we have previously observed no statistical difference in the amount or distribution of virus within the spinal cord following depletion of VP2121–130-specific T cells (Johnson et al., 2001). Third, based on both gross histological examination (Figure 4) and quantitative assessment of the extent of demyelination in VP2- or E7-treated mice at 45 days postinfection we do not think that differences in demyelination explain the preservation of motor function that we observed in VP2-treated mice at this timepoint. Thus, we conclude that our current findings support the hypothesis that demyelinated axons and motor function are susceptible to injury mediated by a subset of antiviral cytotoxic T cells. However, it is important to note that our findings still do not clarify whether this injury is mediated by direct recognition of MHC class I molecules presenting VP2121–130 on the surface of infected neurons and axons or whether the injury is indirectly mediated by an axotoxic mileu created when cytotoxic, antiviral T cells attack other cells, such as infected microglia and macrophages (Olson et al., 2001; Olson and Miller, 2004), in the demyelinated lesion.
The disparity between the robust preservation of motor function and the relative lack of axon preservation observed in our present study is compatible with an indirect model in which the absence of a VP2121–130-specific cytotoxic T cell-mediated attack on microglia reduces the axotoxic burden within demyelinated lesions, permitting enhanced motor function without substantially altering the absolute number of axons in the spinal cord (Hoftberger et al., 2004; Linker et al., 2005; Neumann, 2003). Several predictions follow from such an hypothesis. First, treatments that reduce or abrogate microglial activation, such as minocycline, should mimic the effect of VP2 peptide treatment by reducing the production and release of axotoxic cytokines and reactive oxygen species (Giuliani et al., 2005). Second, markers of microglial activation, such as CD45 immunoreactivity and elevated levels of TNFα, should be reduced within demyelinated lesions of mice depleted of the VP2121–130 immunoresponse. Third, preservation of conduction velocity and/or axonal sodium channel distribution should correlate with functional preservation in VP2 peptide-treated mice to a greater degree than either absolute axon number or retrograde transport (Rivera-Quinones et al., 1998; Waxman, 2006). Testing these predictions will help clarify the relationship between CD8+ T cells and axon injury (Howe et al., 2006; Howe and Rodriguez, 2005) and may help rectify the contentious role of MHC class I in demyelinating disease (Begolka et al., 2001; Ercolini and Miller, 2006). Finally, we propose that a cytokine-secreting, reactive oxygen species-producing microglial intermediary positioned between cytotoxic CD8+ T cells and demyelinated axons may provide a highly relevant and highly manipulable therapeutic target for intervention in patients with MS (Craner et al., 2005; Miller et al., 2007; Peterson et al., 2001; Sriram and Rodriguez, 1997; Storch et al., 2002).
Acknowledgments
This work was supported by grants RG3636 (CLH) and CA1011A8 (MR) from the National Multiple Sclerosis Society, by a grant from the Multiple Sclerosis Society of Canada (MR), by funding from Donald and Frances Herdrich (CLH), and by grants P01 NS38468 and R01 NS32129 (MR) from the National Institutes of Health. We thank Dr. Aaron Johnson for experimental guidance, and we thank Laurie Zoecklein, Mabel Pierce, and Louisa Papke for expert technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, Ravid R, Rajewsky K. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begolka WS, Haynes LM, Olson JK, Padilla J, Neville KL, Dal Canto M, Palma J, Kim BS, Miller SD. CD8-deficient SJL mice display enhanced susceptibility to Theiler’s virus infection and increased demyelinating pathology. J Neurovirol. 2001;7:409–420. doi: 10.1080/135502801753170264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123 ( Pt 6):1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Buenz EJ, Howe CL. Picornaviruses and cell death. Trends Microbiol. 2006;14:28–36. doi: 10.1016/j.tim.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Compston A. The pathogenesis and basis for treatment in multiple sclerosis. Clin Neurol Neurosurg. 2004;106:246–248. doi: 10.1016/j.clineuro.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Damarjian TG, Liu S, Hains BC, Lo AC, Black JA, Newcombe J, Cuzner ML, Waxman SG. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia. 2005;49:220–229. doi: 10.1002/glia.20112. [DOI] [PubMed] [Google Scholar]
- Ercolini AM, Miller SD. Mechanisms of immunopathology in murine models of central nervous system demyelinating disease. J Immunol. 2006;176:3293–3298. doi: 10.4049/jimmunol.176.6.3293. [DOI] [PubMed] [Google Scholar]
- Fiette L, Aubert C, Muller U, Huang S, Aguet M, Brahic M, Bureau JF. Theiler’s virus infection of 129Sv mice that lack the interferon alpha/beta or interferon gamma receptors. Vol. 181. 1995. pp. 2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani F, Hader W, Yong VW. Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell-microglia interaction. J Leukoc Biol. 2005;78:135–143. doi: 10.1189/jlb.0804477. [DOI] [PubMed] [Google Scholar]
- Hoftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, Jellinger K, Lassmann H. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol. 2004;14:43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CL, Adelson JD, Rodriguez M. Absence of perforin expression confers axonal protection despite demyelination. Neurobiol Dis. 2006;25:354–359. doi: 10.1016/j.nbd.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CL, Rodriguez M. Remyelination as neuroprotection. In: Waxman SG, editor. Multiple Sclerosis as a Neuronal Disease. Elsevier Academic Press; San Diego: 2005. pp. 389–419. [Google Scholar]
- Johnson AJ, Upshaw J, Pavelko KD, Rodriguez M, Pease LR. Preservation of motor function by inhibition of CD8+ virus peptide-specific T cells in Theiler’s virus infection. Faseb J. 2001;15:2760–2762. doi: 10.1096/fj.01-0373fje. [DOI] [PubMed] [Google Scholar]
- Linker RA, Rott E, Hofstetter HH, Hanke T, Toyka KV, Gold R. EAE in beta-2 microglobulin-deficient mice: axonal damage is not dependent on MHC-I restricted immune responses. Neurobiol Dis. 2005;19:218–228. doi: 10.1016/j.nbd.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Lipton HL. Theiler’s virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun. 1975;11:1147–1155. doi: 10.1128/iai.11.5.1147-1155.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavern DB, Murray PD, Rivera-Quinones C, Schmelzer JD, Low PA, Rodriguez M. Axonal loss results in spinal cord atrophy, electrophysiological abnormalities and neurological deficits following demyelination in a chronic inflammatory model of multiple sclerosis. Brain. 2000;123(Pt 3):519–531. doi: 10.1093/brain/123.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavern DB, Murray PD, Rodriguez M. Quantitation of spinal cord demyelination, remyelination, atrophy, and axonal loss in a model of progressive neurologic injury. J Neurosci Res. 1999a;58:492–504. doi: 10.1002/(sici)1097-4547(19991115)58:4<492::aid-jnr3>3.0.co;2-p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGavern DB, Zoecklein L, Drescher KM, Rodriguez M. Quantitative assessment of neurologic deficits in a chronic progressive murine model of CNS demyelination. Exp Neurol. 1999b;158:171–181. doi: 10.1006/exnr.1999.7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana I, Martinic MA, Wekerle H, Neumann H. Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol. 2001;159:809–815. doi: 10.1016/S0002-9440(10)61755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Fernandez YV, Johnson AJ, Rodriguez M, Pease LR. Clearance of Theiler’s virus infection depends on the ability to generate a CD8+ T cell response against a single immunodominant viral peptide. Eur J Immunol. 2003;33:2501–2510. doi: 10.1002/eji.200324007. [DOI] [PubMed] [Google Scholar]
- Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen Presentation in the CNS by Myeloid Dendritic Cells Drives Progression of Relapsing Experimental Autoimmune Encephalomyelitis. Ann N Y Acad Sci. 2007 doi: 10.1196/annals.1394.023. [DOI] [PubMed] [Google Scholar]
- Murray PD, McGavern DB, Pease LR, Rodriguez M. Cellular sources and targets of IFN-gamma-mediated protection against viral demyelination and neurological deficits. Eur J Immunol. 2002;32:606–615. doi: 10.1002/1521-4141(200203)32:3<606::aid-immu606>3.0.co;2-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H. Molecular mechanisms of axonal damage in inflammatory central nervous system diseases. Curr Opin Neurol. 2003;16:267–273. doi: 10.1097/01.wco.0000073926.19076.29. [DOI] [PubMed] [Google Scholar]
- Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25:313–319. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- Olson JK, Girvin AM, Miller SD. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler’s virus. J Virol. 2001;75:9780–9789. doi: 10.1128/JVI.75.20.9780-9789.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Pierce ML, Rodriguez M. Erichrome stain for myelin on osmicated tissue embedded in glycol methacrylate plastic. J Histotechnol. 1989;12:35–36. [Google Scholar]
- Rivera-Quinones C, McGavern D, Schmelzer JD, Hunter SF, Low PA, Rodriguez M. Absence of neurological deficits following extensive demyelination in a class I-deficient murine model of multiple sclerosis. Nat Med. 1998;4:187–193. doi: 10.1038/nm0298-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. Immunoglobulins stimulate central nervous system remyelination: electron microscopic and morphometric analysis of proliferating cells. Lab Invest. 1991;64:358–370. [PubMed] [Google Scholar]
- Rodriguez M, Leibowitz JL, Powell HC, Lampert PW. Neonatal infection with the Daniels strain of Theiler’s murine encephalomyelitis virus. Lab Invest. 1983;49:672–679. [PubMed] [Google Scholar]
- Rodriguez M, Zoecklein LJ, Howe CL, Pavelko KD, Gamez JD, Nakane S, Papke LM. Gamma interferon is critical for neuronal viral clearance and protection in a susceptible mouse strain following early intracranial Theiler’s murine encephalomyelitis virus infection. J Virol. 2003;77:12252–12265. doi: 10.1128/JVI.77.22.12252-12265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram S, Rodriguez M. Indictment of the microglia as the villain in multiple sclerosis. Neurology. 1997;48:464–470. doi: 10.1212/wnl.48.2.464. [DOI] [PubMed] [Google Scholar]
- Storch MK, Weissert R, Steffer A, Birnbacher R, Wallstrom E, Dahlman I, Ostensson CG, Linington C, Olsson T, Lassmann H. MHC gene related effects on microglia and macrophages in experimental autoimmune encephalomyelitis determine the extent of axonal injury. Brain Pathol. 2002;12:287–299. doi: 10.1111/j.1750-3639.2002.tb00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ure DR, Rodriguez M. Preservation of neurologic function during inflammatory demyelination correlates with axon sparing in a mouse model of multiple sclerosis. Neuroscience. 2002;111:399–411. doi: 10.1016/s0306-4522(02)00012-x. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci. 2006;7:932–941. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- Wilkins A, Compston A. Trophic factors attenuate nitric oxide mediated neuronal and axonal injury in vitro: roles and interactions of mitogen-activated protein kinase signalling pathways. J Neurochem. 2005;92:1487–1496. doi: 10.1111/j.1471-4159.2004.02981.x. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Antony J, Liu S, Martinez JA, Giuliani F, Zochodne D, Power C. CD8+ lymphocyte-mediated injury of dorsal root ganglion neurons during lentivirus infection: CD154-dependent cell contact neurotoxicity. J Neurosci. 2006;26:3396–3403. doi: 10.1523/JNEUROSCI.4767-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]