

Abstract
In the brain, increased neuronal synaptic activity is accompanied by an increase in local cerebral blood flow that serves to satisfy neuronal metabolic demands. This linkage between neuronal activity and local blood flow has been appreciated for over 100 years. Although this process has been exploited clinically in the form of functional imaging techniques to map brain function, the mechanisms by which increased synaptic activity is communicated to the cerebral microcirculation to generate a vasodilatory response are poorly understood. Recent studies, however, have illuminated a central role for astrocytic calcium (Ca2+) signals as mediators of this process of neurovascular coupling. This review highlights recent evidence implicating astrocytes in the regulation of intracerebral arteriolar diameter, with particular emphasis on the putative signaling molecules and pathways proposed to exert changes on arteriolar physiology.
Introduction
Dynamic regulation of oxygen and glucose supply to match changes in metabolic demand is critical to the maintenance of cerebral homeostasis (Roy and Sherrington 1890). Specifically, increases in neuronal activity must be accompanied by rapid (within 1–2 seconds), spatially localized elevations of oxygen and glucose delivery such that ischemic conditions are averted (Anderson and Nedergaard 2003, Iadecola 2004). This coupling of neuronal activity to increased oxygen and glucose supply occurs as a result of enhanced cerebral blood flow at the level of the cerebral microcirculation and appears to depend on the generation of vasoactive substances in response to neuronal activity. Penetrating (parenchymal) cerebral arterioles receive neuronal innervation primarily from intrinsic neurons (Edvinsson and Hamel 2002, Hamel 2006). While the release of vasoactive substances from such neurons is clearly capable of inducing changes in arteriolar diameter through direct stimulation of vascular smooth muscle cells (SMCs) (Cauli et al. 2004, Rancillac et al. 2006), recent evidence suggests that Ca2+-dependent signaling events in astrocytes also play a critical role in communicating neuronal activity to the cerebral microcirculation, resulting in profound changes in arteriolar diameter and local cerebral blood flow (Anderson and Nedergaard 2003, Filosa et al. 2004, Filosa et al. 2006, Iadecola 2004, Metea and Newman 2006, Mulligan and MacVicar 2004, Simard et al. 2003, Straub et al. 2006, Zonta et al. 2003a).
An individual astrocyte possesses a multitude of processes that surround thousands of neuronal synapses, forming a tripartite synapse with pre- and post-synaptic neurons (Araque et al. 1999). These same astrocytes send additional processes (endfeet) to encase the SMCs of intracerebral arterioles and endothelial cells of capillaries (Grosche et al. 1999, Kacem et al. 1998, Ventura and Harris 1999) (Figure 1). This astrocytic architecture suggests that an individual astrocyte may be capable of integrating the activity of multiple neurons and translating this information into physiological signals, including those that regulate cerebral blood flow.
Figure 1.
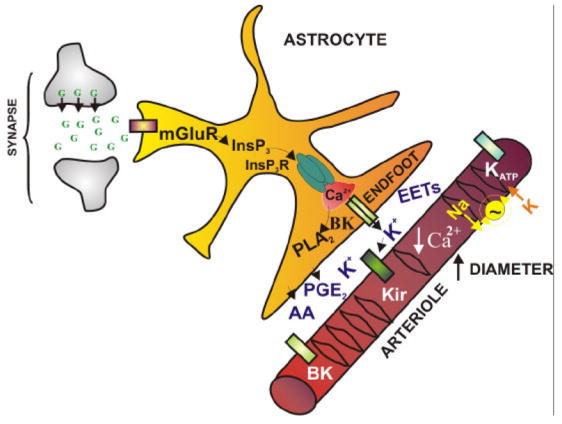
Astrocytic Ca2+ signals couple neuronal activity to changes in vascular function. Neuronally-released glutamate (G) binds metabotropic glutamate receptors (mGluR) on astrocytes inducing production of inositol trisphosphate (InsP3) and increasing astrocytic cytosolic Ca2+ concentration. Ca2+ released from inositol trisphosphate receptors (InsP3R) in astrocytic endfeet, which encase cerebral arterioles, activates Ca2+ dependent signaling mechanisms leading to the generation and/or release of several putative vasoactive factors including potassium ions (K+) and arachidonic acid (AA) metabolites such as prostaglandin E2 (PGE2). These vasoactive factors interact with vascular targets, leading to a decrease in SMC [Ca2+]i and vasodilation. PLA2: phospholipase A2, BK: Ca2+ sensitive large conductance K+ channel, EETs: epoxyeicosatrienoic acids, Kir: strong inwardly rectifying K+ channel (Kir2.1), KATP: ATP sensitive K+ channel, Na+~K+: sodium-potassium ATPase.
Mechanisms Underlying Ca2+ Signals in Astrocytes
Astrocytes act as vital regulators of neuronal function, serving to modulate extracellular potassium concentration ([K+]o) and extracellular volume, and remove neurotransmitters from synapses (Diamond 2005, Kofuji and Newman 2004). These cells possess receptors for a range of neurotransmitters and neuropeptides (Porter and McCarthy 1997). Glutamate-dependent generation of astrocytic Ca2+ signals is a common mechanism by hich neuronal information is transmitted to the cerebral vasculature. Specifically, activation of metabotropic glutamate receptors (mGluRs) located on astrocytic projections that surround synapses of glutamatergic neurons results in an increase in astrocytic cytosolic Ca2+ concentration ([Ca2+]i) that propagates through astrocytic processes, ultimately resulting in a [Ca2+]i increase in the endfoot (Cornell-Bell et al. 1990, Zonta et al. 2003a). These [Ca2+]i increases likely occur through activation of the phospholipase C/inositol trisphosphate (InsP3) cascade, and release of Ca2+ from InsP3 sensitive channels located on the endoplasmic reticulum (Beck et al. 2004, Golovina and Blaustein 1997, 2000, Perea and Araque 2005, Verkhratsky et al. 1998). Several studies using cultured astrocytes suggest that Ca2+ release from ryanodine receptors may also be involved in generating the astrocytic Ca2+ signal (Golovina and Blaustein 1997, 2000). In addition to glutamate, neurotransmitters such as GABA, somatostatin, and vasoactive intestinal peptide released from interneurons may likewise generate vasoactive effects through generation of astrocytic Ca2+ signals, in addition to direct innervation of vessels (Cauli et al. 2004, Kang et al. 1998, Straub et al. 2006).
To understand the mechanisms that underlie communication between perivascular endfeet and cerebral arterioles, the spatiotemporal properties of Ca2+ signals within endfeet in cortical brain slices were recently characterized (Straub et al. 2006). Endfeet were found to possess InsP3 receptors (InsP3Rs) that could be activated by local photorelease of InsP3 within the endfoot or by signals arising from activated neurons. Ryanodine receptors do not appear to play a role in generating Ca2+ increases in endfeet, as determined by a lack of effect of ryanodine and caffeine on endfoot Ca2+ signals. InsP3R in endfeet and perivascular astrocytic processes were found to generate heterogeneous Ca2+ signals, with spatially restricted regions of elevated Ca2+ generated throughout the endfoot and perivascular processes in response to neuronal activity. These findings suggest that Ca2+ signaling within endfeet is highly dynamic in nature, and therefore likely to exert local control over Ca2+-dependent vasoactive signaling pathways involved in neurovascular coupling.
Astrocytic Ca2+ Increases are Involved in Dynamic Regulation of the Cerebral Vasculature
While much effort has been focused on understanding the role of astrocytic Ca2+ signals in communicating information between adjacent astrocytes and neurons, only recently has astrocytic Ca2+ signaling been implicated in the dynamic regulation of the cerebral vasculature. In a seminal study, Zonta and colleagues (Zonta et al. 2003a) linked astrocytic Ca2+ changes to alterations in arteriolar diameter. In this study, astrocytic Ca2+ changes induced by neuronal activity or application of an mGluR agonist were measured in rat cortical brain slices and correlated with alterations in vessel diameter. Electrical stimulation of glutamatergic neurons induced consistent increases in astrocytic Ca2+ that were significantly attenuated, though not completely abrogated, in the presence of the mGluR antagonists MPEP and LY367385. Separate experiments showed that vasodilation occurred in a similar time frame to the increases in astrocytic Ca2+, suggesting that astrocytic Ca2+ changes are coupled to changes in vessel diameter. In addition, utilizing laser Doppler flowmetry to measure cerebral blood flow in vivo, Zonta et al. found that systemic administration of MPEP and LY367385 significantly decreased the hyperemic response to somatosensory stimulation. These findings suggest that astrocyte-derived factors, generated in response to elevations in astrocytic Ca2+, are capable of engaging cerebral arterioles, modulating arteriolar diameter, and dynamically regulating local cerebral blood flow.
In addition to vasodilation, astrocytic Ca2+ increases have also been shown to induce vasoconstriction. Mulligan and MacVicar (2004) found that elevation of Ca2+ within an individual astrocyte, induced through photolysis of the caged divalent ion chelator DMNP-EDTA, resulted in constriction of arterioles in brain slices. Cerebral arteriolar diameter was affected only when a large Ca2+ increase (approximately 185% increase in fractional fluorescence of rhod-2) propagated into an endfoot. No change in the state of the arterioles was seen when a Ca2+ increase was selectively induced within an astrocytic cell body, or when a small Ca2+ increase was generated in an endfoot. Similar to the effects of photoreleasing Ca2+, application of norepinephrine to slices induced an increase in astrocytic Ca2+ that was accompanied by vasoconstriction. Although this study and that of Zonta et al. draw different conclusions about the vasodynamic consequences of astrocytic signaling, they nevertheless strongly support the assertion that Ca2+-dependent, vasoactive signaling processes are localized within astrocytic endfeet.
The Importance of Vascular Tone
While the studies of Zonta et al. (2003a) and Mulligan and MacVicar (2004) illustrate a central role for astrocytic Ca2+ signals in neurovascular coupling, at first glance it is unclear how these studies could generate such disparate results. One likely explanation for these divergent findings lies in the resting state of the arterioles within the brain slices. Cerebral arteries and arterioles in vivo exhibit myogenic tone in response to intravascular pressure (Bayliss 1902). This tone results from SMC depolarization (Harder 1984) and subsequent Ca2+ influx through voltage-gated Ca2+ channels (VDCC) (Knot and Nelson 1995, 1998). This partially constricted state of cerebral arteries and arterioles in vivo allows for vasodilation or vasoconstriction, as required, to maintain a constant level of cerebral perfusion in the face of changing intravascular pressure. Thus, an experimental preparation that recapitulates the dynamic range of the vessel in its native setting – that is, one in which both vasodilation and vasoconstriction can occur – is a prerequisite. Although not attributed to differences in arteriolar diameter by the authors, an elegant example of the effects of vascular tone on the outcome of astrocytic stimulation can be seen in the study of Metea and Newman (2006), who investigated neurovascular coupling mechanisms in whole retinal mounts. When levels of the vasomodulator nitric oxide (NO) were manipulated with NO donors or scavengers, vasodilatory responses could be converted into vasoconstrictions, and vice versa. Similarly, in the study of Mulligan and MacVicar, when vascular tone was induced (by inhibition of nitric oxide synthase), vasodilation was observed in response to endfoot Ca2+ increases. These findings highlight the profound influence of smooth muscle membrane potential, [Ca2+]i, and vascular tone on the outcome of activity-induced vascular responses.
The effect of Ca2+ release through endfoot-resident InsP3R on the diameter of pre-constricted cerebral arterioles has been directly probed by generating endfoot-delimited Ca2+ increases through spatially restricted photorelease of InsP3 within an individual endfoot (Straub et al. 2006). These endfoot-delimited Ca2+ signals induced rapid, local vasodilation of adjacent arterioles that reached 50% of maximum within 1.6 seconds of the uncaging event, and was limited to a short segment of the arteriole (~ 30 μm) centered on the endfoot. These findings suggest that endfeet serve as individual vasoregulatory units in the brain, with the spatial extent of vasodilation dictated by the number of endfeet activated in response to neuronal stimulation. Consistent with these findings, Takano et al. (2006) observed vasodilation of arterioles in vivo in response to endfoot-delimited photorelease of caged Ca2+.
Astrocytic Ca2+ Signals are Linked to Changes in SMC Calcium
If astrocytic Ca2+ signals play a critical role in facilitating changes in cerebral arteriolar diameter, obvious questions arise: what are the chemical mediators of this process, and what alterations in the resting arteriolar signaling processes are brought about by these mediators? Recent evidence suggests that astrocytic Ca2+ signals induce alterations in arteriolar SMC Ca2+ signaling processes (Filosa et al. 2004). Utilizing fluorescence confocal microscopy to provide high temporal and spatial resolution, Filosa et al. (2004) simultaneously measured changes in astrocytic and SMC Ca2+ in cortical slices loaded with the Ca2+ sensitive dye fluo-4 AM. Since arteriolar tone is regulated by SMC membrane potential (Knot and Nelson 1995), which in turn dictates SMC Ca2+ levels (Knot and Nelson 1998), these measurements allow for investigation of the dynamic interplay between astrocytic and SMC Ca2+ signals, with the hypothesis that any astrocytic signal that modulates arteriolar diameter must do so, at least in part, through a manipulation of SMC Ca2+ levels.
With the use of the thromboxane A2 receptor agonist U46619 (100 nM) to preconstrict arterioles, a condition that mimics the vascular tone found in vivo (Brown et al. 2002) and allows both vasodilation and vasoconstriction to be measured, it was observed that arterioles in rat cortical slices exhibit regular oscillations in SMC Ca2+, which are accompanied by synchronized vasomotion (Filosa et al. 2004). This phenomenon of vasomotion has been well documented in vivo (Mayhew et al. 1996, Nilsson and Aalkjaer 2003) and is a fundamental physiological property of mature blood vessels (Filosa et al. 2004, Gokina et al. 1996, Haddock and Hill 2002, Lefer et al. 1990, Lovick et al. 2005, Nilsson and Aalkjaer 2003) that is typically not observed in young, immature vessels, which possess few circumferential SMCs. Importantly, the extent of vasoconstriction induced by U46619 was comparable to that seen in isolated arterioles under physiological pressure. Induction of neuronal synaptic activity via electrical stimulation routinely resulted in a rapid (within ~ 1 second) inhibition of SMC Ca2+ oscillations that was prevented by tetrodotoxin, indicating a requirement for neuronal activity in this process. Simultaneous measurements of astrocytic and SMC Ca2+ signals illustrated that the inhibition of SMC Ca2+ oscillations was preceded by a rise in astrocytic Ca2+, providing further evidence that Ca2+ dependent astrocytic signaling processes play a role in altering SMC Ca2+ levels. Importantly, inhibition of SMC Ca2+ oscillations was accompanied by vasodilation of cerebral arterioles. Thus, a dynamic interplay exists between astrocytic and SMC Ca2+ signals, in which a Ca2+ dependent astrocytic signal alters SMC Ca2+ dynamics to generate a change in vascular diameter.
Potential Mediators of Neurovascular Coupling – Signals Linked to Arachidonic Acid
Several signals based on arachidonic acid (AA) metabolism have been proposed to modulate vessel diameter in response to neuronal activity (Harder et al. 1998, Mulligan and MacVicar 2004, Takano et al. 2006, Zonta et al. 2003a). Specifically, elevation of astrocytic Ca2+ can lead to the Ca2+-dependent activation of phospholipase A 2 (PLA2) and the resultant production of AA, which can be metabolized by a host of different enzymes both within astrocytes and SMCs to generate vasoactive substances (Alkayed et al. 1996, Harder et al. 1998).
Zonta et al. (2003a) provided evidence that the vasodilatory factor released from astrocytes following the astrocytic Ca2+ increase was prostaglandin E2 (PGE2), a product of cyclooxygenase (COX) metabolism of AA (Figure 2). This proposal was based primarily on a decrease in the number of vessels that dilated to neuronal stimulation in the presence of the COX inhibitor acetylsalicylic acid. Consistent with this finding, direct application of PGE2 to brain slices resulted in vasodilation similar to that induced by the mGluR agonist t-ACPD. Additionally, this group has previously shown that PGE2 is released in a pulsatile manner from cultured astrocytes (Zonta et al. 2003b). In support of this mechanism, Takano et al. (2006) found that the non-selective COX inhibitor, indomethacin, and the COX-1-selective inhibitor SC-560 applied to the surface of the intact brain reduced vasodilation associated with elevation of endfoot Ca2+ in vivo. In contrast, Iadecola and colleagues found that selective inhibition or ablation of COX-1 did not alter whisker stimulation-induced elevations in CBF (Niwa et al. 2001). Importantly, in the studies of Zonta et al. and Takano et al., COX inhibition only partially suppressed vasodilation, an observation consistent with the view that multiple mechanisms contribute to the neuronal regulation of arteriolar diameter.
Figure 2.
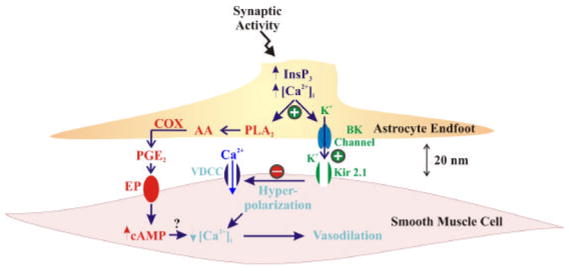
Putative vasoactive signaling pathways involved in neurovascular coupling. Neuronal activity generates elevation of [Ca2+]i in the astrocytic endfoot, through activation of InsP3R, which activates astrocytic BK channels. BK channel activation leads to the local release of K+ ions from endfeet surrounding cortical blood vessels, which activates Kir channels in arteriolar SMCs. Local concentrations of K+ ions between 3–15 mM increase SMC Kir channel conductance, causing SMC membrane potential hyperpolarization, closing voltage-dependent Ca2+ channels (VDCC), and leading to vasodilation. Higher local concentrations of K+ (e.g. >20 mM) would cause membrane potential depolarization, and vasoconstriction. Through an alternate pathway, increased astrocytic [Ca2+]i is postulated to activate phospholipase A2 (PLA2) to generate arachidonic acid (AA) and subsequently prostaglandin E2 (PGE2) through a cyclooxygenase (COX)-dependent mechanism. Activation of EP receptors by PGE2 would lead to increased cAMP in SMCs, ultimately decreasing SMC [Ca2+]i and generating vasodilation through an as yet unidentified mechanism. Modified from (Filosa et al. 2006).
AA can also be converted to epoxyeicosatrienoic acids (EETs) by the action of cytochrome P450 (CYP450) epoxygenases, to produce the vasoactive components 11,12 and 14,15 EET, among others (Alkayed et al. 1996). Harder and colleagues have shown that EETs, which are potent vasodilators (Harder et al. 1998, Roman 2002), activate Ca2+-sensitive, large conductance potassium channels (BK channels) in smooth muscle as well as astrocytes (Gebremedhin et al. 2003). Interestingly, Earley et al. (2005) have recently shown that EETs activate SMC BK channels indirectly through a novel mechanism involving Ca2+ influx mediated by the vanilloid TRP cation channel family member TRPV4, which activates RyRs (Ca2+ sparks), and then subsequently stimulates BK channels (Earley et al. 2005). These findings are consistent with a previous observation that TRPV4 channels are a target of EETs (Vriens et al. 2005). In support of a potential role for EETs in neurovascular coupling, Lovick et al. (2005) recently implicated epoxygenase products in maintaining vasodilation of cortical arterioles following astrocytic AMPA receptor stimulation.
In concert with the theme of BK channel regulation by AA-derived vasoactive substances, Mulligan and MacVicar (2004) postulated a role for 20-hydroxyeicosatetraenoic acid (20-HETE) in generating vasoconstriction of cerebral arterioles in response to neuronal activity. It was suggested that 20-HETE is synthesized by CYP450 hydroxylases in SMCs following diffusion of AA from astrocytic endfeet to SMCs. This proposal was based on the ability of PLA2 and CYP450 hydroxylase inhibitors to prevent vasoconstriction induced by photolysis of caged Ca2+ in astrocytes. In this scenario, 20-HETE is suggested to inhibit BK channels in SMCs, leading to depolarization and constriction. However, as previously noted, the arterioles measured in this study were not pre-constricted, likely existing in a maximally dilated state. Under such conditions SMC BK channels are not engaged in the regulation of vessel diameter (Brayden and Nelson 1992).
It should be noted that blockers of PLA2 and AA metabolism may have direct effects on SMC potassium channels. For example, in isolated SMCs, miconazole and quinacrine have been shown to inhibit KV and BK currents, respectively (Vanheel et al. 1999, Yuan et al. 1995). In addition, since the endothelium of cerebral arteries and arterioles is a rich source of AA products that are involved in setting the basal level of vascular tone and generating vasodilation (Andresen et al. 2006), drug-induced alterations in endothelial function will have profound effects on vascular reactivity. Thus, interpretation of pharmacological interventions aimed at inhibiting AA metabolism in astrocytes is significantly complicated by the potential disruption of endothelium-SMC signaling. Therefore, an attenuation of neurovascular coupling in the presence of inhibitors of AA pathways does not necessarily indicate that the AA metabolites involved are astrocyte derived.
Potential Mediators of Neurovascular Coupling – A Role for Extracellular Potassium
Potassium ions (K+) are among the most potent vasodilatory signals in the cerebral vasculature. Elevation of [K+]o activates strong inward rectifier potassium (Kir) channels (Knot et al. 1996, McCarron and Halpern 1990), in particular the Kir2.1 channel, and the Na+/K+-ATPase in SMCs of extracerebral (pial) arteries (Bradley et al. 1999, Knot et al. 1996, Zaritsky et al. 2000). Activation of SMC Kir channels results in SMC hyperpolarization (Bradley et al. 1999, Zaritsky et al. 2000), which closes voltage-dependent Ca2+ channels in the SMCs and leads to decreased [Ca2+]i and vasodilation (Knot and Nelson 1998, Knot et al. 1996). Interestingly, astrocytes have been proposed to play a critical role in the spatial buffering of K+ within the brain (Newman 1986). Specifically, the K+ siphoning hypothesis put forth by Newman and colleagues suggests that astrocytes function to move K+ from regions of elevated [K+]o around synapses to regions of lower [K+]o around the vasculature.
Consistent with a potential role for K+ in regulating cerebrovascular function, recent evidence suggests that K+ release from astrocytic endfeet plays a critical role in generating rapid vasodilation of cerebral arterioles in response to neuronal activity (Filosa et al. 2006). Specifically, elevation of astrocytic Ca2+ leads to a vasodilation of pre-constricted arterioles in cortical brain slices that is significantly reduced (~70 % reduction) by treatment with the BK channel blockers iberiotoxin or TEA, or by block of Kir channels with barium. This vasodilation is rapid, typically occurring within 2 seconds following onset of electrical stimulation to induce neuronal activity, consistent with the functional hyperemic response observed in vivo. Approximately 40% of the astrocytic K+ current (at 0 mV) in the absence of neuronal stimulation is carried through BK channels, which are preferentially expressed in endfeet (Price et al. 2002). In addition, neuronal stimulation dramatically increases single channel activity consistent with BK channels in “on-endfoot” patch clamp recordings from endfeet within cortical slices. SMCs isolated from parenchymal arterioles exhibit a high density of Kir currents, suggesting that Kir channels in parenchymal arterioles may be a target of K+ released from endfeet through BK channels. Furthermore, neurovascular coupling was diminished in cortical slices from mice in which BK channels were genetically ablated, but not from slices preincubated with the COX inhibitor indomethacin (10 μM). Interestingly, in the presence of indomethacin, treatment with barium or TEA completely abolished vasodilation. Consistent with these in situ data, Gerrits et al. (2002) showed a decrease in whisker stimulation-evoked blood flow changes following block of BK channels with topical application of TEA or iberiotoxin or intravenous administration of TEA, suggesting a critical role for BK channel activity in the functional hyperemic response. Since neither TEA nor iberiotoxin affected neuronal activity, these findings are consistent with the assertion that BK channels in astrocytes and/or SMCs have a critical role in functional hyperemia.
These observations support a model of rapid neurovascular coupling in which neuronal activity is communicated to the vasculature through the Ca2+ dependent activation of endfoot BK channels and release of K+ into the perivascular space, which induces hyperpolarization of SMCs through activation of SMC Kir channels, to promote vasodilation (Figure 2). An interesting feature of this model is that extracellular K+ is capable of accounting for both rapid vasodilation, as described above, and vasoconstriction, depending upon its concentration in the perivascular space and the SMC membrane potential. Specifically, elevation of [K+]o above ~15 mM, as might occur with excessive neuronal stimulation associated with pathological conditions such as spreading depression (Chuquet et al. 2007, Quayle et al. 1997), results in constriction of extracerebral arteries due to depolarization-induced activation of voltage-dependent Ca2+ channels (VDCCs) (Knot et al. 1996).
Limitations of Commonly Utilized Experimental Preparations
Investigations of the mechanisms underlying neurovascular coupling typically utilize either in situ tissue slices or in vivo imaging of astrocytes and cortical arterioles of anesthetized animals. While each technique has its advantages, each also has disadvantages that can limit interpretability of experimental findings. One commonly-cited limitation of brain slice studies is that blood vessels in these preparations lack intraluminal pressure and flow, therefore limiting the activation of autoregulatory mechanisms. Under these conditions, vessels lack normal vascular tone and exist in a maximally dilated state. For this reason, studies employing this preparation typically utilize a vasoconstricting agonist to recapitulate the level of vascular tone generated by physiological pressure, allowing for vasodilation and vasoconstriction as occurs in vivo. Vasoconstricting agonists produce similar levels of tone and vasomotion as are observed when intraluminal pressure and flow are applied to arterioles within slices through cannulation of individual vessels (Lovick et al. 2005). Furthermore, pre-constricted arterioles in brain slices retain their characteristic responsiveness to extracellular K+ (Nakahata et al. 2006), and possess a functional complement of SMC K+ channels (Filosa et al. 2006, Nakahata et al. 2003, Nakahata et al. 2006). One obvious caveat to using this technique is that the level of vascular tone induced by the vasoconstricting agonist should mimic the level of tone generated in vivo in response to physiological pressure.
In vivo imaging of cerebral parenchymal arterioles and astrocytes allows for the study of neurovascular coupling in the presence of physiological levels of intraluminal pressure and flow, suggesting that all physiologically relevant vascular signaling mechanisms should be functional. However, such experimental techniques require the use of anesthetics throughout the course of the experiment, which have been shown to alter vascular tone and other cardiovascular parameters (Jong et al. 2002, Kim et al. 2007, Ueki et al. 1992). In addition, since imaging is performed through a cranial window, cellular access is limited and precludes the use of this preparation with electrophysiological methodologies which are required to understand the ionic basis of vascular function. Furthermore, pharmacological agents must be applied to the exposed surface of the brain rather than at the desired site of action, requiring the application of potentially toxic drug concentrations and yielding unknown concentrations at the recording site. Despite the limitations associated with both techniques, in vivo imaging studies have been largely in concert with findings made in the in situ condition which suggest that astrocytic Ca2+ signals generate vasodilation of arterioles with physiological levels of tone (Filosa et al. 2006, Takano et al. 2006, Zonta et al. 2003a). Furthermore, findings from both approaches support the central role of endfoot Ca2+ signals in the induction of vasodilation through the Ca2+-dependent generation of COX products and K+ signals (Filosa et al. 2006, Gerrits et al. 2002, Takano et al. 2006, Zonta et al. 2003a) (Figure 2).
Since intracerebral blood vessels are the targets of neurovascular coupling, it is critical that both brain slice and in vivo approaches are complemented with appropriate studies on isolated intracerebral arterioles and SMCs. Such studies are required for elucidation of the cellular mechanisms that underlie neurovascular coupling. Furthermore, the judicious use of transgenic mouse models, especially those incorporating tissue-specific genetic manipulations, represents a powerful tool for mechanism formulation, elucidation of potential therapeutic targets, and testing drug specificity.
Future Directions
To define the neuronally-induced signaling events that occur within astrocytes, particularly within astrocytic endfeet, is paramount. These include, but are certainly not limited to, investigations of Ca2+-dependent signaling domains within endfeet and investigation of endfoot ion channel expression and activity in situ and in vivo. Since the process of neurovascular coupling involves numerous cell types, an integrative approach should be utilized, and should include the investigation of as many cellular constituents as possible. This will most certainly require utilization of multiple experimental approaches and tissue/cellular preparations to elucidate biologically relevant findings. Going forward, it will be especially important that putative messengers and mechanisms be validated at the SMC and arteriole level, both within the intact (slice/in vivo) preparation and in isolated arterioles and SMCs. Ideally this will involve the use of animal models in which the putative arteriolar target or signaling mechanism is genetically crippled.
In conclusion, recent evidence clearly supports a central role for astrocytic Ca2+ signals in the generation of vasoactive factors involved in neurovascular coupling. While the identity of the astrocyte derived vasoactive factors involved in this process is still the subject of intense debate, any mechanism proposed to play a role in neurovascular coupling must be capable of inducing rapid vasodilation as is observed in vivo. In addition, each mechanism must have a clearly defined site of action within the vasculature that can account for the effects of neuronal activity on the cerebral circulation. One such mechanism utilizes K+ release from astrocytes through BK channels to promote vasodilation via activation of SMC Kir channels. This mechanism satisfies the temporal requirements of functional hyperemia observed in vivo, and in addition is capable of inducing both vasodilation and vasoconstriction, seemingly accounting for contradictory findings reported in the literature. Importantly, this mechanism is distinct from the previously reported COX-dependent pathway, suggesting that multiple vasoactive pathways likely act in concert to dynamically regulate cerebrovascular responses in vivo.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL83768 to S.V.S. and HL44455 to M.T.N.), and by the Totman Trust for Medical Research. We thank Drs. D. Hill-Eubanks and S. Earley for critical evaluation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996;27:971–9. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- Anderson CM, Nedergaard M. Astrocyte-mediated control of cerebral microcirculation. Trends Neurosci. 2003;26:340–4. doi: 10.1016/S0166-2236(03)00141-3. author reply 344–5. [DOI] [PubMed] [Google Scholar]
- Andresen J, Shafi NI, Bryan RM., Jr Endothelial influences on cerebrovascular tone. J Appl Physiol. 2006;100:318–27. doi: 10.1152/japplphysiol.00937.2005. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–15. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Bayliss W. On the local reactions of the arterial wall to changes of internal pressure. J Physiol. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A, Nieden RZ, Schneider HP, Deitmer JW. Calcium release from intracellular stores in rodent astrocytes and neurons in situ. Cell Calcium. 2004;35:47–58. doi: 10.1016/s0143-4160(03)00171-4. [DOI] [PubMed] [Google Scholar]
- Bradley KK, Jaggar JH, Bonev AD, Heppner TJ, Flynn ER, Nelson MT, et al. Kir2.1 encodes the inward rectifier potassium channel in rat arterial smooth muscle cells. J Physiol. 1999;515 (Pt 3):639–51. doi: 10.1111/j.1469-7793.1999.639ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–5. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- Brown LA, Key BJ, Lovick TA. Inhibition of vasomotion in hippocampal cerebral arterioles during increases in neuronal activity. Auton Neurosci. 2002;95:137–40. doi: 10.1016/s1566-0702(01)00395-2. [DOI] [PubMed] [Google Scholar]
- Cauli B, Tong XK, Rancillac A, Serluca N, Lambolez B, Rossier J, et al. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci. 2004;24:8940–9. doi: 10.1523/JNEUROSCI.3065-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci. 2007;27:4036–44. doi: 10.1523/JNEUROSCI.0721-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–3. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–16. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97:1270–9. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Hamel E. Perivascular nerves in brain vessels. In: Edvinsson L, Krause DN, editors. Cerebral Blood Flow and Metabolism. New York: Lippincott Williams and Wilkins; 2002. [Google Scholar]
- Filosa JA, Bonev AD, Nelson MT. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ Res. 2004;95:e73–81. doi: 10.1161/01.RES.0000148636.60732.2e. [DOI] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- Gebremedhin D, Yamaura K, Zhang C, Bylund J, Koehler RC, Harder DR. Metabotropic glutamate receptor activation enhances the activities of two types of Ca2+-activated K+ channels in rat hippocampal astrocytes. J Neurosci. 2003;23:1678–87. doi: 10.1523/JNEUROSCI.23-05-01678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits RJ, Stein EA, Greene AS. Ca(2+)-activated potassium (K(Ca)) channel inhibition decreases neuronal activity-blood flow coupling. Brain Res. 2002;948:108–16. doi: 10.1016/s0006-8993(02)02957-8. [DOI] [PubMed] [Google Scholar]
- Gokina NI, Bevan RD, Walters CL, Bevan JA. Electrical activity underlying rhythmic contraction in human pial arteries. Circ Res. 1996;78:148–53. doi: 10.1161/01.res.78.1.148. [DOI] [PubMed] [Google Scholar]
- Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–8. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- Golovina VA, Blaustein MP. Unloading and refilling of two classes of spatially resolved endoplasmic reticulum Ca(2+) stores in astrocytes. Glia. 2000;31:15–28. doi: 10.1002/(sici)1098-1136(200007)31:1<15::aid-glia20>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999;2:139–43. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- Haddock RE, Hill CE. Differential activation of ion channels by inositol 1,4,5-trisphosphate (IP3)- and ryanodine-sensitive calcium stores in rat basilar artery vasomotion. J Physiol. 2002;545:615–27. doi: 10.1113/jphysiol.2002.027904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100:1059–64. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- Harder DR. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circ Res. 1984;55:197–202. doi: 10.1161/01.res.55.2.197. [DOI] [PubMed] [Google Scholar]
- Harder DR, Alkayed NJ, Lange AR, Gebremedhin D, Roman RJ. Functional hyperemia in the brain: hypothesis for astrocyte-derived vasodilator metabolites. Stroke. 1998;29:229–34. doi: 10.1161/01.str.29.1.229. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–60. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Jong WM, Zuurbier CJ, De Winter RJ, Van Den Heuvel DA, Reitsma PH, Ten Cate H, et al. Fentanyl-fluanisone-midazolam combination results in more stable hemodynamics than does urethane alpha-chloralose and 2,2,2-tribromoethanol in mice. Contemp Top Lab Anim Sci. 2002;41:28–32. [PubMed] [Google Scholar]
- Kacem K, Lacombe P, Seylaz J, Bonvento G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia. 1998;23:1–10. [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–92. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kim SH, Bae YM, Sung DJ, Park SW, Woo NS, Kim B, et al. Pflugers Arch. 2007. Ketamine blocks voltage-gated K(+) channels and causes membrane depolarization in rat mesenteric artery myocytes. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol. 1995;269:H348–55. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508 (Pt 1):199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. 1996;492 (Pt 2):419–30. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–56. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefer DJ, Lynch CD, Lapinski KC, Hutchins PM. Enhanced vasomotion of cerebral arterioles in spontaneously hypertensive rats. Microvasc Res. 1990;39:129–39. doi: 10.1016/0026-2862(90)90065-y. [DOI] [PubMed] [Google Scholar]
- Lovick TA, Brown LA, Key BJ. Neuronal activity-related coupling in cortical arterioles: involvement of astrocyte-derived factors. Exp Physiol. 2005;90:131–40. doi: 10.1113/expphysiol.2004.028811. [DOI] [PubMed] [Google Scholar]
- Mayhew JE, Askew S, Zheng Y, Porrill J, Westby GW, Redgrave P, et al. Cerebral vasomotion: a 0.1-Hz oscillation in reflected light imaging of neural activity. Neuroimage. 1996;4:183–93. doi: 10.1006/nimg.1996.0069. [DOI] [PubMed] [Google Scholar]
- McCarron JG, Halpern W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am J Physiol. 1990;259:H902–8. doi: 10.1152/ajpheart.1990.259.3.H902. [DOI] [PubMed] [Google Scholar]
- Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci. 2006;26:2862–70. doi: 10.1523/JNEUROSCI.4048-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–9. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nakahata K, Kinoshita H, Hirano Y, Kimoto Y, Iranami H, Hatano Y. Mild hypercapnia induces vasodilation via adenosine triphosphate-sensitive K+ channels in parenchymal microvessels of the rat cerebral cortex. Anesthesiology. 2003;99:1333–9. doi: 10.1097/00000542-200312000-00014. [DOI] [PubMed] [Google Scholar]
- Nakahata K, Kinoshita H, Tokinaga Y, Ishida Y, Kimoto Y, Dojo M, et al. Vasodilation mediated by inward rectifier K+ channels in cerebral microvessels of hypertensive and normotensive rats. Anesth Analg. 2006;102:571–6. doi: 10.1213/01.ane.0000194303.00844.5e. [DOI] [PubMed] [Google Scholar]
- Newman EA. High potassium conductance in astrocyte endfeet. Science. 1986;233:453–4. doi: 10.1126/science.3726539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson H, Aalkjaer C. Vasomotion: mechanisms and physiological importance. Mol Interv. 2003;3:79–89. 51. doi: 10.1124/mi.3.2.79. [DOI] [PubMed] [Google Scholar]
- Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001;88:600–8. doi: 10.1161/01.res.88.6.600. [DOI] [PubMed] [Google Scholar]
- Perea G, Araque A. Glial calcium signaling and neuron-glia communication. Cell Calcium. 2005;38:375–82. doi: 10.1016/j.ceca.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Astrocytic neurotransmitter receptors in situ and in vivo. Progress in Neurobiology. 1997;51:439–55. doi: 10.1016/s0301-0082(96)00068-8. [DOI] [PubMed] [Google Scholar]
- Price DL, Ludwig JW, Mi H, Schwarz TL, Ellisman MH. Distribution of rSlo Ca2+-activated K+ channels in rat astrocyte perivascular endfeet. Brain Res. 2002;956:183–93. doi: 10.1016/s0006-8993(02)03266-3. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Rancillac A, Rossier J, Guille M, Tong XK, Geoffroy H, Amatore C, et al. Glutamatergic Control of Microvascular Tone by Distinct GABA Neurons in the Cerebellum. J Neurosci. 2006;26:6997–7006. doi: 10.1523/JNEUROSCI.5515-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Roy CS, Sherrington C. On the regulation of the blood supply of the brain. J Physiol. 1890;11:85–108. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–62. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub SV, Bonev AD, Wilkerson MK, Nelson MT. Dynamic inositol trisphosphate-mediated calcium signals within astrocytic endfeet underlie vasodilation of cerebral arterioles. J Gen Physiol. 2006;128:659–69. doi: 10.1085/jgp.200609650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, et al. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–7. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Ueki M, Mies G, Hossmann KA. Effect of alpha-chloralose, halothane, pentobarbital and nitrous oxide anesthesia on metabolic coupling in somatosensory cortex of rat. Acta Anaesthesiol Scand. 1992;36:318–22. doi: 10.1111/j.1399-6576.1992.tb03474.x. [DOI] [PubMed] [Google Scholar]
- Vanheel B, Calders P, Van den Bossche I, Van de Voorde J. Influence of some phospholipase A2 and cytochrome P450 inhibitors on rat arterial smooth muscle K+ currents. Can J Physiol Pharmacol. 1999;77:481–9. [PubMed] [Google Scholar]
- Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiol Rev. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, et al. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res. 2005;97:908–15. doi: 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. Inhibition of cytochrome P-450 reduces voltage-gated K+ currents in pulmonary arterial myocytes. Am J Physiol. 1995;268:C259–70. doi: 10.1152/ajpcell.1995.268.1.C259. [DOI] [PubMed] [Google Scholar]
- Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res. 2000;87:160–6. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003a;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- Zonta M, Sebelin A, Gobbo S, Fellin T, Pozzan T, Carmignoto G. Glutamate-mediated cytosolic calcium oscillations regulate a pulsatile prostaglandin release from cultured rat astrocytes. J Physiol. 2003b;553:407–14. doi: 10.1113/jphysiol.2003.046706. [DOI] [PMC free article] [PubMed] [Google Scholar]