

Abstract
By surveying a filtered, high-quality set of SNPs in the human genome, we have found that SNPs positioned 1, 2, 4, 6, or 8 bp apart are more frequent than SNPs positioned 3, 5, 7, or 9 bp apart. The observed pattern is not restricted to genomic regions that are known to cause sequencing or alignment errors, for example, transposable elements (SINE, LINE, and LTR), tandem repeats, and large duplicated regions. However, we found that the pattern is almost entirely confined to what we define as “periodic DNA.” Periodic DNA is a genomic region with a high degree of periodicity in nucleotide usage. It turned out that periodic DNA is mainly small regions (average length 16.9 bp), widely distributed in the genome. Furthermore, periodic DNA has a 1.8 times higher SNP density than the rest of the genome and SNPs inside periodic DNA have a significantly higher genotyping error rate than SNPs outside periodic DNA. Our results suggest that not all SNPs in the human genome are created by independent single nucleotide mutations, and that care should be taken in analysis of SNPs from periodic DNA. The latter may have important consequences for SNP and association studies.
More than 11.5 million single nucleotide polymorphisms (SNPs) are reported in the human genome (dbSNP build 125). These are spread throughout the genome and are not restricted to certain genomic regions or genetic elements such as exons, introns, transposons, or tandem repeat sequences. Most SNPs are believed to be the product of independent single mutational events in the past, or occasionally due to multiple recurrent mutations in the same nucleotide position (Stoneking 2001). After the completion of the human genome (International Human Genome Sequencing Consortium 2001, 2004), many efforts have gone into studying genetic variation in the genome sequence, with SNP variation being the primary focus of the HapMap project. One aim of the HapMap project is to provide a high-resolution haplotype map of the human genome by genotyping 270 individuals from four human populations of African, Asian, and European ancestry in 5.6 million SNP loci (The International HapMap Consortium 2003; The International HapMap Consortium, in prep.). The current HapMap release (#21) contains genotypes of 3.3 million nonredundant, high-quality SNPs.
SNPs are not the only widespread variation in the genome. Insertions and deletions (indels) occur throughout the genome, giving rise to local structural polymorphisms (Tuzun et al. 2005; Conrad et al. 2006). Furthermore, recent large-scale studies have reported widespread occurrence of copy number variations longer than 1000 bp (1 kb) in the human genome (Tuzun et al. 2005; Conrad et al. 2006; Freeman et al. 2006; Redon et al. 2006). These variations point to a very dynamic and plastic genome that undergoes many changes in the transmission from parent to child and possibly throughout the somatic history of an individual.
In this study, we report on a systematic small-scale pattern of SNPs that adds to the complexity of the genome and that cannot be explained by viewing all SNPs as the result of independent single nucleotide mutations. We filtered all known SNPs in the human genome by stringent criteria to obtain a highly reliable set of SNPs, excluding SNPs with ambiguous positions or validation problems. By examining the filtered SNPs, we observed that SNPs positioned 1, 2, 4, 6, or 8 bp apart are more frequent than SNPs positioned 3, 5, 7, or 9 bp apart (see Fig. 1). This holds even when we correct for nucleotide frequencies and site dependencies in nucleotide usage in the genome. If all positions in the genome had the same probability of being an SNP, we would expect equal numbers of SNP pairs in all distances >1. For SNP pairs in distance 1 (direct neighbor SNPs), the high CpG mutation rate is expected to lead to an over-representation compared to distances >1 (Hwang and Green 2004).
Figure 1.

Definitions of distances in SNP pairs and an example of periodic DNA. Distances are calculated between all SNPs, thus the figure shows three pairs with three distances. The distance (d) between any two SNPs is defined as the positive difference between the two genomic SNP positions, for example, d = 1 indicates neighboring SNPs. The distance definition is chosen such that distances are additive between neighboring SNPs. Identical SNP pairs are defined as two SNPs each with identical alleles (here SNP1: A/G, SNP2: A/G, d = 9). Different SNP pairs are defined as two SNPs with different alleles (here SNP1: A/G, SNP2: C/T, d = 4; SNP1: C/T, SNP2: A/G, d = 5). To the right, an example of periodic DNA is shown. The period is 3, and it is shown that SNPs are allowed in the pattern.
One possible and obvious explanation of this 1, 2, 4, 6, 8 pattern is systematic sequencing and/or alignment errors. We ruled out this possibility by using only filtered SNPs (as defined in Methods), and by observing that the pattern is far from restricted to genomic regions associated with sequencing and alignment errors; for example, transposable elements (SINE, LINE, and LTR), tandem repeats, and large duplicated regions. Moreover, the pattern is highly abundant in transcripts.
To further scrutinize the observation, we defined “periodic DNA.” Periodic DNA is (small) sequences of DNA with a high degree of periodicity in nucleotide usage (defined rigorously in Methods), and periodic DNA is thus expected to contain the pattern systematically. Surprisingly, we found that by excluding SNPs in periodic DNA, the pattern virtually disappears. Hence the structure of periodic DNA may hint at the origin of the pattern.
The fundamental observation is that in a segment of periodic DNA, for example, ATATATATAT, a base change, say, A to G, may be observed in several of the A positions and more frequently than by chance. This pattern could be created by copy number alterations in the AT repeat, but we find that the pattern is persistent even when the flanking regions of the SNPs align perfectly to the reference genome sequence and there are no gaps in the alignment. Hence, length polymorphism/variation cannot explain the pattern. This implies that even in a short segment of periodic DNA with period p (in the above example, p = 2), the presence of one SNP increases the probability of a second identical SNP in distances 1p, 2p, . . . bp, in the same segment. This is visible as an excess of identical SNPs in certain distances. For example, periodic DNA with periods 1, 2, or 4 is expected to have an over-representation of identical SNPs in a distance 4 bp, whereas only periodic DNA with periods 1 or 5 are expected to have an over-representation of identical SNPs in distance 5.
In this study, we document this pattern in detail.
Results
General pattern
When surveying the frequency spectrum of all pairs of SNPs in various distances (d), we found that pairs of identical SNPs generally follow a 1, 2, 4, 6, 8 pattern, whereas pairs of different SNPs are almost uniformly distributed for d > 1 (Fig. 2; Supplemental Fig. S1). The CpG effect (Hwang and Green 2004) accounts for the over-representation of pairs of different SNPs with d = 1. The frequency spectrum for pairs of identical SNPs indicates that the pattern might exist for distances up to 15 bp (Supplemental Fig. S1). However, when looking at the frequency spectrum chromosome-wise (Supplemental Fig. S2A,B), the pattern only appears to be persistent for distances up to 9 bp. We therefore restricted our further investigations to pairs of SNPs with d ≤ 9 bp.
Figure 2.

Density spectrum of SNP pairs. (A) SNP pairs from the entire genome. The 2, 4, 6, 8 pattern is visible in all SNP pairs, and caused by the variation in identical pairs. Pairs of different SNPs are almost uniformly distributed for d > 1. Red represents identical pairs of SNPs and blue represents pairs of different SNPs. (B) SNP pairs inside and outside periodic DNA. The 2, 4, 6, 8 pattern is strongest in periodic DNA and nearly disappears outside periodic DNA. Also, there is a strong general over-representation of SNP pairs in periodic DNA when compared to the rest of the genome.
Using only SNPs outside transposable elements (SINE, LINE, and LTR), tandem repeats (as defined by RepeatMasker) and large duplicated regions (>1 kb), respectively, did not remove the pattern (Supplemental Fig. S3A–C).
To further validate the pattern, we analyzed only the random HapMap-ENCODE regions (The ENCODE Project Consortium 2004, 2007). The random HapMap-ENCODE regions consist of seven randomly picked 500-kb regions, which have been resequenced in the HapMap populations to obtain a dense, unbiased map of the variation in the genome. The pattern is less visible in these regions than in the entire genome (Supplemental Fig. S3D), but the number of observations is also much smaller, and noise is likely to disturb the picture.
Periodic DNA
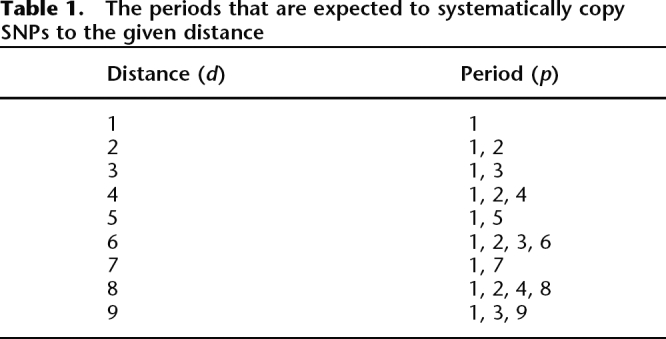
To investigate the frequency pattern for d = 1, . . . , 9, all periodic DNA is identified for these distances (see Methods and Table 1). Periodic DNA makes up 4.3% of the entire genome and has a mean length of 16.9 bp. When comparing SNP pairs in the entire genome (Fig. 2A) and SNP pairs inside and outside periodic DNA (Fig. 2B), it is seen that the 2, 4, 6, 8 pattern is almost entirely confined to periodic DNA. Furthermore, it is clear that pairs of identical SNPs as well as pairs of different SNPs are highly over-represented in periodic DNA, compared to the entire genome (Fig. 2B). To test for an excess of pairs of identical SNPs, we used a test that takes into account the composition of the reference sequence, and the actual frequencies of the six types of SNPs (A/C, A/G, A/T, C/G, C/T, G/T) (see Methods). The expected fraction of identical SNP pairs in the entire genome is 26.7%, whereas 31.9% (39,123) is observed (P < 10−100; 95% CI: 31.6%–32.2%). In periodic DNA, the expected fraction of identical SNP pairs is 37.0%, whereas 56.8% (11,987) is observed (P < 10−100; 95% CI: 56.1%–57.6%).
Table 1.
The periods that are expected to systematically copy SNPs to the given distance
The density of SNPs is higher in periodic DNA than in the rest of the genome. Thus, 7.4% of the SNPs are located in periodic DNA (4.3% of the genome), which is a 1.8 times higher SNP density than in the rest of the genome. Pairs of identical SNPs show the most significant discrepancy, with 28.1% of all pairs of identical SNPs located in periodic DNA. Pairs of different SNPs are less over-represented, with 10.0% of all pairs of different SNPs located in periodic DNA.
The distribution of periodic DNA on the nine different periods is shown in Figure 3. It is seen that sequences with periods 1, 2, or 4 are over-represented compared to the other periods. This implies that we expect pairs of identical SNPs in distances 1, 2, 4, 6, or 8 bp to be more frequent than identical SNP pairs in distances 3, 5, 7, or 9 bp. This is in good concordance with the observed frequency spectrum for pairs of identical SNPs (Fig. 4; Supplemental Fig. S1). Furthermore, Figure 4, A and B, shows that in the entire genome, as well as in periodic DNA, identical SNP pairs in distances d = 2, 4, 6, or 8 are highly over-represented compared to the expected frequency, whereas SNP pairs in distance 3 are less over-represented, and SNP pairs in distances 5, 7, or 9 are only slightly over-represented. The expected frequency of identical SNP pairs cannot be estimated for d = 1 in this way, because of the CpG mutational bias (Hwang and Green 2004).
Figure 3.

Period distribution of periodic DNA. The histogram shows the number of times a pattern of period p has been observed.
Figure 4.

Location of periodic DNA. Estimated and observed frequencies for pairs of identical SNPs in: the entire genome (see Supplemental Fig. S1A for individual chromosomes) (A); periodic DNA (4.3% of the entire genome; see Supplemental Fig. S1B for individual chromosomes) (B); the overlap between exons and periodic DNA (0.06% of the entire genome) (C); the overlap between transcripts and periodic DNA (1.56% of the entire genome) (D); and the overlap between tandem repeats and periodic DNA (0.12% of the entire genome) (E). Because of the over-representation of SNP pairs in distance d = 1, we only estimated the frequencies for d = 2, . . . , 9.
SNPs in periodic DNA have more genotyping problems than SNPs outside periodic DNA. By examining all genotyped SNPs in all individuals from the HapMap project, genotyping failed in 41.1% of the cases for SNPs inside periodic DNA, but only in 19.9% for SNPs outside periodic DNA. This difference is highly significant (P-value < 10−13). If we omit SNPs that failed to be genotyped in any individuals, the error rates are 21.4% inside periodic DNA and 12.3% outside periodic DNA, which is highly significant too (P-value < 10−13).
Location of periodic DNA
Subsequently, we restricted the analysis to the intersection of periodic DNA with various other genomic regions.
Periodic DNA is under-represented in exons. Exons make up 2.1% of the entire genome, but only 1.4% of the periodic DNA is located in exons. The frequency pattern of pairs of identical SNPs in the overlap shows a damped version of the 2, 4, 6, 8 pattern (Fig. 4C), but the pairs of identical SNPs are not significantly over-represented (P = 0.38).
Periodic DNA does not correlate with transcripts. Transcripts (exons + introns) make up 37.5% of the genome, and 36.9% of the periodic DNA is located in transcripts. The 2, 4, 6, 8 pattern is highly abundant in transcripts (Fig. 4D) with an over-representation of pairs of identical SNPs (P < 10−100).
Periodic DNA does not correlate with tandem repeats. Tandem repeats make up 2.80% of the genome, and 2.83% of the periodic DNA is located in tandem repeats. As expected from the periodic nature of tandem repeats, the 2, 4, 6, 8 pattern is abundant in the overlap of the two (Fig. 4E), and pairs of identical SNPs are highly over-represented compared to the expected level (P = 1.7 × 10−27).
Periodic DNA found in tandem repeats is longer (mean length 36.1 bp) than generally in the genome (mean 16.9 bp). The overlap contains 9.3% of all identical SNP pairs and 12.4% of all different SNP pairs found in periodic DNA. A possible explanation is that more SNP pairs are cut by the edges of short sequences.
Periodic DNA does not correlate with transposable elements. Transposable elements make up 46.4% of the genome, and 43.2% of periodic DNA is located in transposable elements.
Discussion
We have observed that identical pairs of SNPs in the human genome are more frequent in distances 2, 4, 6, and 8 bp, than in distances 3, 5, 7, and 9 bp. The immediate explanation of this observation is sequencing errors and/or alignment errors. To rule out this possibility, we first compiled a set of high-quality SNPs, that is, SNPs that map to a unique position in the genome, and with an exact match between the flanking regions and the reference genome sequence. In this way, all SNPs that might be wrongly placed in the genome are excluded. Furthermore, to avoid study-specific ascertainment biases, we used all SNPs reported to dbSNP as a starting point. For this set of filtered SNPs, we observed that the pattern is highly pronounced. Furthermore, we observed that the pattern is persistent even when we ignore SNPs in genomic regions that may cause sequencing and/or alignment problems, for example, transposable elements, tandem repeats, and large duplicated regions (Bailey et al. 2001, 2002; Fredman et al. 2004). We therefore concluded that the pattern is not caused by direct sequencing or alignment errors, and that the pattern is not confined to any known type of genomic elements related to such errors.
Interestingly, the entire pattern is virtually embedded in periodic DNA, which makes up only 4.3% of the genome and has 1.8 times higher SNP density than the rest of the genome. Furthermore, periodic DNA is not correlated with tandem repeats or other repetitive elements, indicating that periodic DNA is different from these types of genomic elements.
In the overlap of periodic DNA and exons, the 2, 4, 6, 8 pattern is damped, which may be because of selective constraints on exons. Oppositely, the pattern is preserved in periodic DNA overlapping with transcripts (exons + introns), consequently suggesting fewer (or no) selective constraints on introns.
Our results indicate that a proportion of all SNPs in the human genome is not created by independent single nucleotide mutations. We speculate that many different mechanisms such as polymerase slippage (Weber and Wong 1993; Walsh et al. 1996), unequal crossover events (Jeffreys et al. 1999), and gene conversion (Holliday 1964; Lewin 2004) could lead to the observed pattern. Polymerase slippage is a mechanism whereby the DNA polymerase jumps backward or forward on the template sequence, leading to two copies of a small fragment of the template sequence, or a deletion of a similar fragment. Unequal crossover occurs when two overcrossing chromosomes do not break in the same position, leading to one product with a deletion and one with a copy of a small fraction of the sequence. Both of these two mechanisms lead to length polymorphisms that are either preserved or repaired by repair mechanisms. Gene conversion, on the other hand, copies small fragments of DNA to new positions in the genome without creating length polymorphisms. In this study, we excluded all SNPs that are positioned in connection to a length polymorphism, implying that if polymerase slippage or unequal crossover is the underlying mechanism, the length polymorphism must have been repaired.
Alternatively, a complex process of context-dependent mutations could potentially create a similar pattern, although such a process may be difficult to envisage. We note, however, that the CpG mutation bias is caused by a context-dependent mutation process, and the possibility of a more elaborate process accounting for the observed pattern is difficult to rule out per se. The exact nature of the molecular mechanism(s) is to be revealed in future studies.
In conclusion, our results show that periodic DNA has some distinctive genomic features: (1) there is an excess of SNPs in periodic DNA compared to non-periodic DNA; (2) SNPs in periodic DNA are distributed according to a 2, 4, 6, 8 pattern; (3) care should be taken in analysis of SNPs from periodic DNA since SNPs in periodic DNA have a higher genotyping error rate than SNPs outside periodic DNA. The latter may have important consequences for SNP and association studies.
Methods
Reference sequence
Reference sequence hg17 (NCBI build 35) was used (2001) (International Human Genome Sequencing Consortium 2004). In the analysis all gaps were omitted, resulting in a reference sequence of length 2,866,216,770 bp (here referred to as the entire genome).
Genomic elements
Exon and transcript regions
Transcripts were downloaded as the “Known Genes” track from the UCSC Table Browser (Karolchik et al. 2004). This track contains start and end positions for all exons inside a transcript, and is used to define exonic regions as well as transcripts.
Tandem repeat regions
Tandem repeat regions were defined by the “Simple Repeats” track in the UCSC Table Browser (Karolchik et al. 2004). This track displays simple tandem repeats (possibly imperfect) identified by Tandem Repeats Finder (Benson 1999), which is specialized for this purpose.
Transposable elements
The transposable elements were found using the “SINE,” “LINE,” and “LTR” regions from the “RepeatMasker” tracks from the UCSC genome browser (Karolchik et al. 2004). The “RepeatMasker” track was created by the RepeatMasker program version 20040130, which screens DNA sequences for interspersed repeats (http://www.repeatmasker.org/). RepeatMasker uses the Repbase library (update 8.12 is used) of repeats from the Genetic Information Research Institute (GIRI) (Jurka 2000).
Large duplicated regions
The large duplicated regions were found using “Segmental Dups” and the “RepeatMasker” tracks from the UCSC genome browser (Karolchik et al. 2004). After downloading these two tracks, they were filtered to contain only duplicated sequences that are at least 95% similar and have a length of at least 100 bp. The “Segmental Dups” track contains duplicated sequences of at least 1000 bp (Bailey et al. 2001).
SNP data
To avoid false patterns due to study specific biases, for example, as discussed in Clark et al. (2003, 2005), Koboldt et al. (2006), and Pe’er et al. (2006), we used SNPs from all projects that reported to dbSNP build 125 and SNPs from HapMap phases I + II (rel21a NCBI build 35) (The International HapMap Consortium 2003; The International HapMap Consortium, in prep.). SNP data from dbSNP were downloaded as the “SNPs” track from the UCSC Genome Browser (Karolchik et al. 2004). The track contains NCBI dbSNP build 125. The data were filtered to contain only two-allele, perfectly mapped SNPs. To select “true” SNPs only, we only kept SNPs that met one of the following filtering criteria: “by-frequency,” “by-2hit-2allele,” or “by-hapmap,” or were validated by HapMap phases I + II, and had the minor allele reported at least twice (rel21a NCBI build 35).
We only selected unambiguously mapped SNPs, where the flanking sequences surrounding a SNP had exactly one hit to the human genome (weight = 1). To avoid SNPs with potential alignment problems on the local scale (<10 bp, e.g., due to indels), we only selected SNPs that were perfectly mapped on the local scale, i.e, where the alignment of the flanking sequences and the reference genome were exactly 1 bp apart (location type = ‘exact’). To ensure that our automated filtering process removed all alignment problems, we manually evaluated 17 random pairs of identical SNPs from periodic DNA in the UCSC Genome Browser (Kent et al. 2002). None of the SNP pairs could be explained by ambiguity in the alignment. If alignment problems should explain the observed excess of pairs of identical SNPs (56.8% observed vs. 37.0% expected; see Results), the probability of observing no ambiguity alignments in the 17 SNP pairs is <0.001.
By applying the above filtering criteria, we ended up with 4,576,203 SNPs out of a total of 10,430,753 SNPs in dbSNP125 (Sherry et al. 1999). The majority of these SNPs (57.2%) are validated in the HapMap project (The International HapMap Consortium 2003; The International HapMap Consortium, in prep.).
The data set containing all genotyped HapMap SNPs were downloaded from the HapMap site (http://www.hapmap.org/genotypes/; build 21a, NCB1 35), including all redundant, unfiltered SNPs and all individuals from all populations (The International HapMap Consortium 2003; The International HapMap Consortium, in prep.).
HapMap-ENCODE regions
The HapMap-ENCODE regions used are the seven random ENCODE regions that have been resequenced by HapMap; i.e., Enr. 112, 113, 123, 131, 213, 232, and 321 (The ENCODE Project Consortium 2004, 2007). The regions were found using the “ENCODE Regions” tracks from the UCSC genome browser (Karolchik et al. 2004). To avoid biases due to selection of regions, we used only the seven randomly picked HapMap-ENCODE regions and not the three nonrandomly picked regions. The filtering criteria described above were also applied to the SNPs in these seven regions.
Periodic DNA
To identify periodic DNA, we first marked all SNPs from dbSNP125 in the reference sequence (hg17) to get a marked reference sequence. When looking for a periodic pattern in a piece of marked sequence, we allowed that both alleles of a SNP could be used to form the periodic pattern. A sequence is then defined as periodic DNA, with period p, if it fulfils the following three criteria: (1) The minimum length is 9 bp. This criterion is used because it has been shown that sequences with at length of at least 9 bp are more likely to create rearrangements (Gore et al. 2006). (2) The pattern (e.g., AT in ATATATATAT) is repeated at least three times. (3) There are at most p/4 bp that do not match a periodic pattern of period p in the sequence.
This is implemented by looking at one period (p) at a time. For each p, a window of 3p bp (or 9 bp if p = 1, 2) is moved over the entire marked reference sequence (criteria a and b), and the window is marked as periodic DNA if the pattern meets criterion c.
Finally, all marked windows are collapsed into regions of periodic DNA, and the smallest possible period is assigned to each region.
The criterion of at most p/4 mismatches ensures that short segments of periodic DNA (9–12 bp) have a perfect periodic pattern, whereas the longer segments are allowed to have a few mismatches.
Estimation of expected frequencies
To estimate the expected frequencies of pairs of identical SNPs, we estimated the expected ratio of identical versus different pairs of SNPs for each distance (d), and multiplied the result with the observed frequency of pairs of different SNPs. The expected ratio for each d is found using the following steps:
The frequencies of all 16 combinations of base pairs in distance d in the genome are counted.
The frequencies of the six types of SNPs (A/C, A/G, A/T, C/G, C/T, G/T) are counted.
- The probability of obtaining a specific SNP pair (g,h) (where g and h are any of the six types) by random mutation in distance d is calculated as
Here i and j run over A, G, T, C, and pd(i,j) is the frequency of base pair(i,j) in distance d as found in Step 1. The term pd(g,h|i,j) is found from Step 2 by restricting the possible mutations to those that can be obtained from(i,j), that is, if i is A, then only the three SNP types involving A are possible.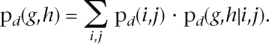
- The expected ratio is calculated as
where g, h run over A/C, A/G, A/T, C/G, C/T, G/T, and g ≠ h.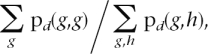
Test for over-representation of pairs of identical SNPs
To test for an over-representation of pairs of identical SNPs, we used a coin tossing test to compare the expected frequency of pairs of identical SNPs to the observed frequency. The overall expected frequency of pairs of identical SNPs is calculated as
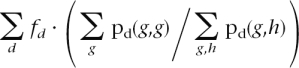 |
for g, h running over A/C, A/G, A/T, C/G, C/T, and G/T. Here fd is the observed frequency of SNP pairs in distance d. Note that ∑g,hpd(g,h) is the probability of obtaining any SNP pair in distance d.
Software
All data were analyzed using Python (http://www.python.org), and R (http://www.R-project.org) (R Development Core Team 2006). All scripts are available upon request.
Acknowledgments
We thank Frank Grønlund Jørgensen and Mikkel Heide Schierup for helpful discussions, and Enette Berndt Knudsen for excellent technical assistance. C.W. is supported by the Danish Cancer Society. P.V. is supported by the Lundbeck Foundation, Denmark.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6223207
References
- Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E., Massa H.F., Trask B.J., Eichler E.E., Trask B.J., Eichler E.E., Eichler E.E. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E., Adams M.D., Myers E.W., Li P.W., Eichler E.E., Myers E.W., Li P.W., Eichler E.E., Li P.W., Eichler E.E., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A.G., Nielsen R., Signorovitch J., Matise T.C., Glanowski S., Heil J., Winn-Deen E.S., Holden A.L., Lai E., Nielsen R., Signorovitch J., Matise T.C., Glanowski S., Heil J., Winn-Deen E.S., Holden A.L., Lai E., Signorovitch J., Matise T.C., Glanowski S., Heil J., Winn-Deen E.S., Holden A.L., Lai E., Matise T.C., Glanowski S., Heil J., Winn-Deen E.S., Holden A.L., Lai E., Glanowski S., Heil J., Winn-Deen E.S., Holden A.L., Lai E., Heil J., Winn-Deen E.S., Holden A.L., Lai E., Winn-Deen E.S., Holden A.L., Lai E., Holden A.L., Lai E., Lai E. Linkage disequilibrium and inference of ancestral recombination in 538 single-nucleotide polymorphism clusters across the human genome. Am. J. Hum. Genet. 2003;73:285–300. doi: 10.1086/377138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A.G., Hubisz M.J., Bustamante C.D., Williamson S.H., Nielsen R., Hubisz M.J., Bustamante C.D., Williamson S.H., Nielsen R., Bustamante C.D., Williamson S.H., Nielsen R., Williamson S.H., Nielsen R., Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad D.F., Andrews T.D., Carter N.P., Hurles M.E., Pritchard J.K., Andrews T.D., Carter N.P., Hurles M.E., Pritchard J.K., Carter N.P., Hurles M.E., Pritchard J.K., Hurles M.E., Pritchard J.K., Pritchard J.K. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman D., White S.J., Potter S., Eichler E.E., Dunnen J.T.D., Brookes A.J., White S.J., Potter S., Eichler E.E., Dunnen J.T.D., Brookes A.J., Potter S., Eichler E.E., Dunnen J.T.D., Brookes A.J., Eichler E.E., Dunnen J.T.D., Brookes A.J., Dunnen J.T.D., Brookes A.J., Brookes A.J. Complex SNP-related sequence variation in segmental genome duplications. Nat. Genet. 2004;36:861–866. doi: 10.1038/ng1401. [DOI] [PubMed] [Google Scholar]
- Freeman J.L., Perry G.H., Feuk L., Redon R., McCarroll S.A., Altshuler D.M., Aburatani H., Jones K.W., Tyler-Smith C., Hurles M.E., Perry G.H., Feuk L., Redon R., McCarroll S.A., Altshuler D.M., Aburatani H., Jones K.W., Tyler-Smith C., Hurles M.E., Feuk L., Redon R., McCarroll S.A., Altshuler D.M., Aburatani H., Jones K.W., Tyler-Smith C., Hurles M.E., Redon R., McCarroll S.A., Altshuler D.M., Aburatani H., Jones K.W., Tyler-Smith C., Hurles M.E., McCarroll S.A., Altshuler D.M., Aburatani H., Jones K.W., Tyler-Smith C., Hurles M.E., Altshuler D.M., Aburatani H., Jones K.W., Tyler-Smith C., Hurles M.E., Aburatani H., Jones K.W., Tyler-Smith C., Hurles M.E., Jones K.W., Tyler-Smith C., Hurles M.E., Tyler-Smith C., Hurles M.E., Hurles M.E., et al. Copy number variation: New insights in genome diversity. Genome Res. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- Gore J.M., Ran F.A., Ornston L.N., Ran F.A., Ornston L.N., Ornston L.N. Deletion mutations caused by DNA strand slippage in Acinetobacter baylyi. Appl. Environ. Microbiol. 2006;72:5239–5245. doi: 10.1128/AEM.00283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. A mechanism for gene conversion in fungi. Genet. Res. 1964;5:282–304. doi: 10.1017/S0016672308009476. [DOI] [PubMed] [Google Scholar]
- Hwang D.G., Green P., Green P. Inaugural article: Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl. Acad. Sci. 2004;101:13994–14001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Jeffreys A.J., Barber R., Bois P., Buard J., Dubrova Y.E., Grant G., Hollies C.R.H., May C.A., Neumann R., Panayi M., Barber R., Bois P., Buard J., Dubrova Y.E., Grant G., Hollies C.R.H., May C.A., Neumann R., Panayi M., Bois P., Buard J., Dubrova Y.E., Grant G., Hollies C.R.H., May C.A., Neumann R., Panayi M., Buard J., Dubrova Y.E., Grant G., Hollies C.R.H., May C.A., Neumann R., Panayi M., Dubrova Y.E., Grant G., Hollies C.R.H., May C.A., Neumann R., Panayi M., Grant G., Hollies C.R.H., May C.A., Neumann R., Panayi M., Hollies C.R.H., May C.A., Neumann R., Panayi M., May C.A., Neumann R., Panayi M., Neumann R., Panayi M., Panayi M., et al. Human minisatellites, repeat DNA instability and meiotic recombination. Electrophoresis. 1999;20:1665–1675. doi: 10.1002/(SICI)1522-2683(19990101)20:8<1665::AID-ELPS1665>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Jurka J. Repbase Update: A database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J., Sugnet C.W., Haussler D., Kent W.J., Haussler D., Kent W.J., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler A.D., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler A.D., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler A.D., Roskin K.M., Pringle T.H., Zahler A.M., Haussler A.D., Pringle T.H., Zahler A.M., Haussler A.D., Zahler A.M., Haussler A.D., Haussler A.D. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C., Raymond M.D., Kwok P.-Y., Raymond M.D., Kwok P.-Y., Kwok P.-Y. Distribution of human SNPs and its effect on high-throughput genotyping. Hum. Mutat. 2006;27:249–254. doi: 10.1002/humu.20286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin B. Genes VIII. Prentice-Hall; Upper Saddle River, NJ: 2004. [Google Scholar]
- Pe’er I., Chretien Y.R., de Bakker P.I.W., Barrett J.C., Daly M.J., Altshuler D.M., Chretien Y.R., de Bakker P.I.W., Barrett J.C., Daly M.J., Altshuler D.M., de Bakker P.I.W., Barrett J.C., Daly M.J., Altshuler D.M., Barrett J.C., Daly M.J., Altshuler D.M., Daly M.J., Altshuler D.M., Altshuler D.M. Biases and reconciliation in estimates of linkage disequilibrium in the human genome. Am. J. Hum. Genet. 2006;78:588–603. doi: 10.1086/502803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team . R: A language and environment for statistical computing. Foundation for Statistical Computing; Vienna, Austria: 2006. [Google Scholar]
- Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., Fiegler H., Shapero M.H., Carson A.R., Chen W., Shapero M.H., Carson A.R., Chen W., Carson A.R., Chen W., Chen W., et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S.T., Ward M., Sirotkin K., Ward M., Sirotkin K., Sirotkin K. dbSNP—Database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999;9:677–679. [PubMed] [Google Scholar]
- Stoneking M. Single nucleotide polymorphisms. From the evolutionary past. Nature. 2001;409:821–822. doi: 10.1038/35057279. [DOI] [PubMed] [Google Scholar]
- Tuzun E., Sharp A.J., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D., Sharp A.J., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D., Haugen E., Hayden H., Albertson D., Pinkel D., Hayden H., Albertson D., Pinkel D., Albertson D., Pinkel D., Pinkel D., et al. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Walsh P.S., Fildes N.J., Reynolds R., Fildes N.J., Reynolds R., Reynolds R. Sequence analysis and characterization of stutter products at the tetranucleotide repeat locus vWA. Nucleic Acids Res. 1996;24:2807–2812. doi: 10.1093/nar/24.14.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J.L., Wong C., Wong C. Mutation of human short tandem repeats. Hum. Mol. Genet. 1993;2:1123–1128. doi: 10.1093/hmg/2.8.1123. [DOI] [PubMed] [Google Scholar]