

Abstract
A common challenge in the analysis of genomics data is trying to understand the underlying phenomenon in the context of all complex interactions taking place on various signaling pathways. A statistical approach using various models is universally used to identify the most relevant pathways in a given experiment. Here, we show that the existing pathway analysis methods fail to take into consideration important biological aspects and may provide incorrect results in certain situations. By using a systems biology approach, we developed an impact analysis that includes the classical statistics but also considers other crucial factors such as the magnitude of each gene’s expression change, their type and position in the given pathways, their interactions, etc. The impact analysis is an attempt to a deeper level of statistical analysis, informed by more pathway-specific biology than the existing techniques. On several illustrative data sets, the classical analysis produces both false positives and false negatives, while the impact analysis provides biologically meaningful results. This analysis method has been implemented as a Web-based tool, Pathway-Express, freely available as part of the Onto-Tools (http://vortex.cs.wayne.edu).
Together with the ability of generating a large amount of data per experiment, high-throughput technologies also brought the challenge of translating such data into a better understanding of the underlying biological phenomena. Independent of the platform and the analysis methods used, the result of a high-throughput experiment is, in many cases, a list of differentially expressed genes. The common challenge faced by all researchers is to translate such lists of differentially expressed genes into a better understanding of the underlying biological phenomena and, in particular, to put this in the context of the whole organism as a complex system. In 2002, a computerized analysis approach using the Gene Ontology (GO) was proposed to deal with this issue (Khatri et al. 2002; Draghici et al. 2003). This approach takes a list of differentially expressed genes and uses a statistical analysis to identify the GO categories (e.g. biological processes, etc.) that are over- or under-represented in the condition under study. Given a set of differentially expressed genes, this approach compares the number of differentially expressed genes found in each category of interest with the number of genes expected to be found in the given category just by chance. If the observed number is substantially different from the one expected just by chance, the category is reported as significant. A statistical model (e.g. hypergeometric) can be used to calculate the probability of observing the actual number of genes just by chance, i.e., a P-value. Currently, there are over 20 tools using this over-representation approach (ORA) (Khatri and Draghici 2005). In spite of its wide adoption, this approach has a number of limitations related to the type, quality, and structure of the annotations available. An alternative approach considers the distribution of the pathway genes in the entire list of genes and performs a functional class scoring (FCS), which also allows adjustments for gene correlations (Goeman et al. 2004; Pavlidis et al. 2004). Arguably the state of the art in the FCS category, the Gene Set Enrichment Analysis (GSEA) (Mootha et al. 2003; Subramanian et al. 2005; Tian et al. 2005), ranks all genes based on the correlation between their expression and the given phenotypes, and calculates a score that reflects the degree to which a given pathway P is represented at the extremes of the entire ranked list. The score is calculated by walking down the list of genes ordered by expression change. The score is increased for every gene that belongs to P and decreased for every gene that does not. Statistical significance is established with respect to a null distribution constructed by permutations.
Both ORA and FCS techniques currently used are limited by the fact that each functional category is analyzed independently without a unifying analysis at a pathway or system level (Tian et al. 2005). This approach is not well suited for a systems biology approach that aims to account for system level dependencies and interactions as well as to identify perturbations and modifications at the pathway or organism level (Stelling 2004). Several pathway databases such as KEGG (Ogata et al. 1999), BioCarta (http://www.biocarta.com), and Reactome (Joshi-Tope et al. 2005) currently describe metabolic pathway and gene signaling networks offering the potential for a more complex and useful analysis. A recent technique, ScorePage, has been developed in an attempt to take advantage of these types of data for the analysis of metabolic pathways (Rahnenfuhrer et al. 2004). Unfortunately, no such technique currently exists for the analysis of gene signaling networks. All pathway analysis tools currently available use one of the ORA approaches above and fail to take advantage of the much richer data contained in these resources. GenMAPP/MAPPfinder (Doniger et al. 2003; Dahlquist et al. 2002) and Gene-Sifter use a standardized Z-score. PathwayProcessor (Grosu et al. 2002), PathMAPA (Pan et al. 2003), Cytoscape (Shannon et al. 2003), and PathwayMiner (Pandey et al. 2004) use Fisher’s exact test. MetaCore uses a hypergeometric model, while ArrayXPath (Chung et al. 2004) offers both Fisher’s exact test and a false discovery rate (FDR). Finally, VitaPad (Holford et al. 2004) and Pathway Studio (Nikitin et al. 2003) focus on visualization alone and do not offer any analysis.
The approaches currently available for the analysis of gene signaling networks share a number of important limitations. First, these approaches consider only the set of genes on any given pathway and ignore their position in those pathways. This may be unsatisfactory from a biological point of view. If a pathway is triggered by a single gene product or activated through a single receptor and if that particular protein is not produced, the pathway will be greatly impacted, probably completely shut off. A good example is the insulin pathway (http://www.genome.ac.jp/KEGG/pathway/hsa/hsa04910.html). If the insulin receptor (INSR) is not present, the entire pathway is shut off. Conversely, if several genes are involved in a pathway but they only appear somewhere downstream, changes in their expression levels may not affect the given pathway as much.
Second, some genes have multiple functions and are involved in several pathways but with different roles. For instance, the above INSR is also involved in the adherens junction pathway as one of the many receptor protein tyrosine kinases. However, if the expression of INSR changes, this pathway is not likely to be heavily perturbed because INSR is just one of many receptors on this pathway. Once again, all these aspects are not considered by any of the existing approaches.
Probably the most important challenge today is that the knowledge embedded in these pathways about how various genes interact with each other is not currently exploited. The very purpose of these pathway diagrams is to capture some of our knowledge about how genes interact and regulate each other. However, the existing analysis approaches consider only the sets of genes involved on these pathways, without taking into consideration their topology. In fact, our understanding of various pathways is expected to improve as more data are gathered. Pathways will be modified by adding, removing or redirecting links on the pathway diagrams. Most existing techniques are completely unable to even sense such changes. Thus, these techniques will provide identical results as long as the pathway diagram involves the same genes, even if the interactions between them are completely redefined over time.
Finally, up to now the expression changes measured in these high-throughput experiments have been used only to identify differentially expressed genes (ORA approaches) or to rank the genes (FCS methods), but not to estimate the impact of such changes on specific pathways. Thus, ORA techniques will see no difference between a situation in which a subset of genes is differentially expressed just above the detection threshold (e.g., twofold) and the situation in which the same genes are changing by many orders of magnitude (e.g., 100-fold). Similarly, FCS techniques can provide the same rankings for entire ranges of expression values, if the correlations between the genes and the phenotypes remain similar. Even though analyzing this type of information in a pathway and system context would be extremely meaningful from a biological perspective, currently there is no technique or tool able to do this.
We propose a radically different approach for pathway analysis that attempts to capture all aspects above. An impact factor (IF) is calculated for each pathway incorporating parameters such as the normalized fold change of the differentially expressed genes, the statistical significance of the set of pathway genes, and the topology of the signaling pathway. We show on a number of real data sets that the intrinsic limitations of the classical analysis produce both false positives and false negatives while the impact analysis provides biologically meaningful results.
Impact analysis
Our goal is to develop an analysis model that would require both a statistically significant number of differentially expressed genes and biologically meaningful changes on a given pathway. In this model, the IF of a pathway Pi is calculated as the sum of two terms:
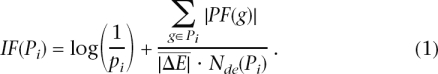 |
The first term is a probabilistic term that captures the significance of the given pathway Pi from the perspective of the set of genes contained in it. This term captures the information provided by the currently used classical statistical approaches and can be calculated using either an ORA (e.g., z-test [Doniger et al. 2003], contingency tables [Pan et al. 2003; Pandey et al. 2004], etc.), a FCS approach (e.g., GSEA; Mootha et al. 2003; Subramanian et al. 2005) or other more recent approaches (Robinson et al. 2004; Breslin et al. 2005; Tian et al. 2005). The pi value corresponds to the probability of obtaining a value of the statistic used at least as extreme as the one observed, when the null hypothesis is true. The results presented here were obtained using the hypergeometric model (Tavazoie et al. 1999; Draghici et al. 2003) in which pi is the probability of obtaining at least the observed number of differentially expressed gene, Nde, just by chance.
The second term in Equation 1 is a functional term that depends on the identity of the specific genes that are differentially expressed as well as on the interactions described by the pathway (i.e., its topology). In essence, this term sums up the absolute values of the perturbation factors (PFs) for all genes g on the given pathway Pi. The PF of a gene g is calculated as follows:
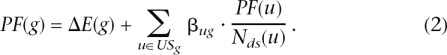 |
In this equation, the first term captures the quantitative information measured in the gene expression experiment. The factor ΔE (g) represents the signed normalized measured expression change of the gene g determined using one of the available methods (Quackenbush 2001; Churchill 2002; Draghici 2002; Yang and Speed 2002). The second term is a sum of all PFs of the genes u directly upstream of the target gene g, normalized by the number of downstream genes of each such gene Nds(u), and weighted by a factor βug, which reflects the type of interaction: βug = 1 for induction, βug = −1 for repression. (In KEGG, which is the source of the pathways used here, this information about the type of interaction is available for every link between two genes in the description of the pathway topology.) USg is the set of all such genes upstream of g. The second term here is similar to the PageRank index used by Google (Page et al. 1998), only we weight the downstream instead of the upstream connections (a Web page is important if other pages point to it, whereas a gene is important if it influences other genes).
Under the null hypothesis, which assumes that the list of differentially expressed genes only contains random genes, the likelihood that a pathway has a large IF is proportional to the number of such “differentially expressed” genes that fall on the pathway, which in turn is proportional to the size of the pathway. Thus, we need to normalize with respect to the size of the pathway by dividing the total perturbation by the number of differentially expressed genes on the given pathway, Nde(Pi). Furthermore, various technologies can yield systematically different estimates of the fold changes. For instance, the fold changes reported by microarrays tend to be compressed with respect to those reported by RT-PCR (Canales et al. 2006; Draghici et al. 2006). In order to make the IFs as independent as possible from the technology, and also comparable between problems, we also divide the second term in Equation 1 by the mean absolute fold change ΔE, calculated across all differentially expressed genes. Assuming that there are at least some differentially expressed genes anywhere in the data set, both ΔE and Nde(Pi) are different from zero so the second term is properly defined. (If there are no differentially expressed genes anywhere, the problem of finding the impact on various pathways is meaningless.)
It can be shown that the IFs correspond to the negative log of the global probability of having both a statistically significant number of differentially expressed genes and a large perturbation in the given pathway. IF values, if, will follow a Γ(2,1) distribution from which P-values can be calculated as P = (if + 1) ⋅ e−if (for details, see Supplemental materials).
The impact analysis proposed here extends and enhances the existing statistical approaches by incorporating the novel aspects discussed above. For instance, the second term of the gene perturbation (in Equation 2) increases the PF scores of those genes that are connected through a direct signaling link to other differentially expressed genes (e.g., the PFs of F5 and F11 in Fig. 1 are both increased because of the differentially expressed SERPINC1 and SERPINA1). This will yield a higher overall score for those pathways in which the differentially expressed genes are localized in a connected subgraph, as in this example. Interestingly, when the limitations of the existing approaches are forcefully imposed (e.g., ignoring the magnitude of the measured expression changes or ignoring the regulatory interactions between genes), the impact analysis reduces to the classical statistics and yields the same results. For instance, if there are no perturbations directly upstream of a given gene, the second term in Equation 2 is zero and the PF reduces to the measured expression change ΔE, which is the classical way of assessing the impact of a condition upon a given gene. A more detailed discussion of various particular cases is included in the Supplemental materials.
Figure 1.

The complement and coagulation cascade as affected by treatment with palmitate in a hepatic cell line. There are seven differentially expressed genes (red, up-regulated; blue, down-regulated) out of 69 total genes. All classical ORA models would give any other pathway with the same proportion of genes a similar P-value, disregarding the fact that six out of these seven genes are involved in the same region of the pathway, closely interacting with each other. Both ORA and GSEA would yield exactly the same significance value to this pathway even if the diagram were to be completely redesigned by future discoveries. In contrast, the impact factor can distinguish between this pathway and any other pathway with the same proportion of differentially expressed gene, as well as take into account any future changes to the topology of the pathway.
Results
We have used this pathway analysis approach to analyze several data sets. A first such set includes genes associated with better survival in lung adenocarcinoma (Beer et al. 2002). These genes have the potential to represent an important tool for the therapeutic decision, and if the correct regulatory mechanisms are identified, they could also be potential drug targets. The expression values of the 97 genes associated with better survival identified by Beer and colleagues were compared between the cancer and healthy groups. These data were then analyzed using a classical ORA approach (hypergeometric model), a classical FCS approach (GSEA), and our impact analysis. Figure 2 shows a comparison between the results obtained with the three approaches.
Figure 2.

A comparison between the results of the classical probabilistic approaches (A, hypergeometric; B, GSEA) and the results of the pathway impact analysis (C) for a set of genes differentially expressed in lung adenocarcinoma. The pathways marked with green are considered most likely to be linked to this condition in this experiment. The ones in red are unlikely to be related. The ranking of the pathways produced by the classical approaches is very misleading. According to the hypergeometric model, the most significant pathways in this condition are: prion disease, focal adhesion, and Parkinson’s disease. Two out of these three are likely to be incorrect. GSEA yields cell cycle as the most enriched pathway in cancer, but three out of the four subsequent pathways are clearly incorrect. In contrast, all three top pathways identified by the impact analysis are relevant to the given condition. The impact analysis is also superior from a statistical perspective. According to both hypergeometric and GSEA, no pathway is significant at the usual 1% or 5% levels on corrected P-values. In contrast, according to the impact analysis, the cell cycle is significant at 1%, and focal adhesion and Wnt signaling are significant at 5% and 10%, respectively.
From a statistical perspective, the power of both classical techniques appears to be very limited. The corrected P-values do not yield any pathways at the usual 0.01 or 0.05 significance levels, independently of the type of correction. If the significance levels were to be ignored and the techniques used only to rank the pathways, the results would continue to be unsatisfactory. According to the classical ORA analysis, the most significantly affected pathways in this data set are prion disease, focal adhesion, and Parkinson’s disease. In reality, both prion and Parkinson’s diseases are pathways specifically associated to diseases of the central nervous system and are unlikely to be related to lung adenocarcinomas. In this particular case, prion disease ranks at the top only due to the differential expression of LAMB1. Since this pathway is rather small (14 genes), every time any one gene is differentially expressed, the hypergeometric analysis will rank it highly. A similar phenomenon happens with Parkinson’s disease, indicating that this is a problem associated with the method rather than with a specific pathway. At the same time, pathways highly relevant to cancer such as cell cycle and Wnt signaling are ranked in the lower half of the pathway list. The most significant pathways reported as enriched in cancer by GSEA (Subramanian et al. 2005) are cell cycle, Huntington’s disease, DRPLA, Alzheimer’s disease, and Parkinson’s disease (see Fig. 2). Among these, only cell cycle is relevant, while Huntington’s, Alzheimer’s and Parkinson’s diseases are clearly incorrect. However, although ranked first, cell cycle is not significant in GSEA, even at the most lenient 10% significance and with the least conservative correction.
In contrast, the impact analysis reports cell cycle as the most perturbed pathway in this condition and also as highly significant from a statistical perspective (P = 1.6 × 10−6). Since early articles on the molecular mechanisms perturbed in lung cancers (Slebos and Rodenhuis 1989; Nau et al. 1985) until the most recent articles on this topic (Panani and Roussos 2006; Coe et al. 2006), there is a consensus that the cell cycle is highly deranged in lung cancers. Moreover, cell cycle genes have started to be considered both as potential prognostic factors and therapeutic targets (Vincenzi et al. 2006). The second most significant pathway as reported by the impact analysis is focal adhesion. An inspection of this pathway (shown in Fig. 3) shows that in these data, both ITG and RTK receptors are perturbed, as well as the VEGF ligand. Because these three genes appear at the very beginning and affect both entry points controlling this pathway, their perturbations are widely propagated throughout the pathway. Furthermore, the CRK oncogene was also found to be up-regulated. Increased levels of CRK proteins have been observed in several human cancers, and over-expression of CRK in epithelial cell cultures promotes enhanced cell dispersal and invasion (Rodrigues et al. 2005). For this pathway, the impact analysis yields a raw P-value of 0.005, which remains significant even after the FDR correction (P = 0.048), at the 5% level. In contrast, the ORA analysis using the hypergeometric model yields a raw P-value of 0.155 (FDR corrected to 0.627), while the GSEA analysis yields a raw P-value of 0.16 (FDR corrected to 0.384). For both techniques, not even the raw P-values are significant at the usual levels of 5% or 10%. This is not a mere accident but an illustration of the intrinsic limitations of the classical approaches. These approaches completely ignore the position of the genes on the given pathways, and therefore, they are not able to identify this pathway as being highly impacted in this condition. Note that any ORA approach will yield the same results for this pathway for any set of four differentially expressed genes from the set of genes on this pathway. Similarly, GSEA will yield the same results for any other set of four genes with similar expression values (yielding similar correlations with the phenotype). Both techniques are unable to distinguish between a situation in which these genes are upstream, potentially commandeering the entire pathway as in this example, or randomly distributed throughout the pathway.
Figure 3.

The focal adhesion pathway as impacted in lung adenocarcinoma vs. normal. In this condition, both ITG and RTK receptors are perturbed, as well as the VEGF ligand. Because these three genes appear at the very beginning and affect both entry points controlling this pathway, their perturbations are widely propagated throughout the pathway and this pathway appears as highly impacted. All classical approaches completely ignore the positions of the genes on the given pathways and fail to identify this pathway as significant.
The third pathway as ranked by the impact analysis is Wnt signaling (FDR corrected P = 0.055, significant at 10%). The importance of this pathway is well supported by independent research. At least three mechanisms for the activation of Wnt signaling pathway in lung cancers have been recently identified: (1) over-expression of Wnt effectors such as Dvl, (2) activation of a non-canonical pathway involving MAPK (previously known as JNK), and (3) repression of Wnt antagonists such as WIF (Mazieres et al. 2005). Mazieres and colleagues also argue that the blockade of Wnt pathway may lead to new treatment strategies in lung cancer.
In the same data set, Huntington’s disease, Parkinson’s disease, prion disease, and Alzheimer’s disease have low IFs (corrected P-values of >0.20), correctly indicating that they are unlikely to be relevant in lung adenocarcinomas.
A second data set includes genes identified as being associated with poor prognosis in breast cancer (van’t Veer et al. 2002). Figure 4 shows a comparison among the classical hypergeometric approach, GSEA, and the pathway impact analysis. Based on these data, GSEA finds no significantly impacted pathways at any of the usual 5% or 10% levels. In fact, the only FDR-corrected value below 0.25, in the entire data set is 0.11, corresponding to the ubiquitin mediated proteolysis pathway. Furthermore, GSEA’s ranking does not appear to be useful for these data, with none of the cancer-related pathways being ranked toward the top. The most significant signaling pathway according to the hypergeometric analysis, cell cycle, is also the most significant in the impact analysis. However, the agreement between the two approaches stops here. In terms of statistical power, according to the classical hypergeometric model, there are no other significant pathways at either 5% or 10% significance on the corrected P-values. If we were to ignore the usual significance thresholds and only consider the ranking, the third highest pathway according to the hypergeometric model is Parkinson’s disease. In fact, based on current knowledge, Parkinson’s disease is unlikely to be related to rapid metastasis in breast cancer. At the same time, the impact analysis finds several other pathways as significant. For instance, focal adhesion is significant with an FDR-corrected P-value of 0.03. In fact, a link between focal adhesion and breast cancer has been previously established (Golubovskaya et al. 2002; van Nimwegen and van de Water 2006). In particular, PTK2 (previously known as FAK), a central gene on the focal adhesion pathway, has been found to contribute to cellular adhesion and survival pathways in breast cancer cells, which are not required for survival in nonmalignant breast epithelial cell (Beviglia et al. 2003). Recently, it has also been shown that Doxorubicin, an anti-cancer drug, caused the formation of well-defined focal adhesions and stress fibers in mammary adenocarcinoma MTLn3 cells early after treatment (van Nimwegen et al. 2006). Consequently, the PTK2/PI-3 kinase/PKB signaling route within the focal adhesion pathway has been recently proposed as the mechanism through which Doxorubicin triggers the onset of apoptosis (van Nimwegen et al. 2006).
Figure 4.

A comparison between the results of the classical (ORA) probabilistic approach (A), GSEA (B), and the results of the pathway impact analysis (C) for a set of genes associated with poor prognosis in breast cancer. The pathways marked with green are well supported by the existing literature. The ones in red are unlikely to be related. After correcting for multiple comparisons, GSEA fails to identify any pathway as significantly impacted in this condition at any of the usual significance levels (1%, 5%, or 10%). The hypergeometric model pinpoints cell cycle as the only significant pathway. Relevant pathways such as focal adhesion, TGF-beta signaling, and MAPK do not appear as significant from a hypergeometric point of view. While agreeing on the cell cycle, the impact analysis also identifies the three other relevant pathways as significant at the 5% level.
TGF-beta signaling (P = 0.032) and MAPK (P = 0.064) are also significant. Both fit well with previous research results. TGF-beta1, the main ligand for the TGF-beta signaling pathway, is known as a marker of invasiveness and metastatic capacity of breast cancer cells (Todorovic-Rakovic 2005). In fact, it has been suggested as the missing link in the interplay between estrogen receptors and ERBB2 (previously known as HER-2; human epidermal growth factor receptor 2) (Todorovic-Rakovic 2005). Furthermore, plasma levels of TGF-beta1 have been used to identify low-risk postmenopausal metastatic breast cancer patients (Nikolic-Vukosavljevic et al. 2004). Finally, MAPK has been shown to be connected not only to cancer in general but to this particular type of cancer. For instance the proliferative response to progestin and estrogen was shown to be inhibited in mammary cells microinjected with inhibitors of MAP kinase pathway (Chen et al. 2001). Also, it is worth noting the gap between the P-values for regulation of actin cytoskeleton (P = 0.111), which may be relevant in cancer, and the next pathway, Parkinson’s disease (P = 0.239), which is irrelevant in this condition.
A third data set involves a set of differentially expressed genes obtained by studying the response of a hepatic cell line when treated with palmitate (Swagell et al. 2005). Figure 5 shows the comparison between the classical statistical analysis (ORA) and the pathway impact analysis. (The GSEA analysis requires expression values for all genes; since this experiment was performed with a custom array and not all values are publicly available, GSEA could not be applied here.) The classical statistical analysis yields three pathways significant at the 5% level: complement and coagulation cascades, focal adhesion, and MAPK. The impact analysis agrees on all these but also identifies several additional pathways. The top four pathways identified by the impact analysis are well supported by the existing literature. There are several studies that support the existence of a relationship between different coagulation factors, present in the complement and coagulation cascades pathway, and palmitate. Sanders et al. (1999), for instance, demonstrated that a high palmitate intake affects factor VII coagulant (FVIIc) activity. Interestingly, Figure 1 shows not only that this pathway has a higher than expected proportion of differentially expressed genes but also that six out of seven such genes are involved in the same region of the pathway, suggesting a coherently propagated perturbation. The focal adhesion and tight junction pathways involve cytoskeletal genes. Swagell et al. (2005) considered the presence of the cytoskeletal genes among the differentially expressed genes as very interesting and hypothesized that the down-regulation of these cytoskeletal genes indicates that palmitate decreases cell growth. Finally, the link between MAPK and the palmitate was established by Susztak et al. (2005), who showed that p38 MAP kinase is a key player in the palmitate-induced apoptosis.
Figure 5.

A comparison between the results of the classical probabilistic approach (A) and the results of the impact analysis (B) for a set of genes found to be differentially expressed in a hepatic cell line treated with palmitate. Green pathways are well supported by literature evidence, while red pathways are unlikely to be relevant. The classical statistical analysis yields three pathways significant at the 5% level: complement and coagulation cascades, focal adhesion, and MAPK. The impact analysis agrees on these three pathways but also identifies several additional pathways. Among these, tight junction is well supported by the literature.
Conclusions
A statistical approach using various models is commonly used in order to identify the most relevant pathways in a given experiment. This approach is based on the set of genes involved in each pathway. We identified a number of additional factors that may be important in the description and analysis of a given biological pathway. Based on these, we developed a novel impact analysis method that uses a systems biology approach in order to identify pathways that are significantly impacted in any condition monitored through a high-throughput gene expression technique. The impact analysis incorporates the classical probabilistic component but also includes important biological factors that are not captured by the existing techniques: the magnitude of the expression changes of each gene, the position of the differentially expressed genes on the given pathways, the topology of the pathway that describes how these genes interact, and the type of signaling interactions between them. The results obtained on several independent data sets show that the proposed approach is very promising. This analysis method has been implemented as a Web-based tool, Pathway-Express, freely available as part of the Onto-Tools (http://vortex.cs.wayne.edu).
Acknowledgments
This material is based upon work supported by the following grants: NSF DBI-0234806, CCF-0438970, 1R01HG003491-01A1, 1U01CA117478-01, 1R21CA100740-01, 1R01NS045207-01, 5R21EB000990-03, 2P30 CA022453-24. Onto-Tools currently runs on equipment provided by Sun Microsystems EDU 7824-02344-U and by NIH(NCRR) 1S10 RR017857-01. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the NSF, NIH, DOD, or any other of the funding agencies.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6202607
References
- Beer D.G., Kardia S.L., Huang C.-C., Giordano T.J., Levin A.M., Misek D.E., Lin L., Chen G., Gharib T.G., Thomas D.G., Kardia S.L., Huang C.-C., Giordano T.J., Levin A.M., Misek D.E., Lin L., Chen G., Gharib T.G., Thomas D.G., Huang C.-C., Giordano T.J., Levin A.M., Misek D.E., Lin L., Chen G., Gharib T.G., Thomas D.G., Giordano T.J., Levin A.M., Misek D.E., Lin L., Chen G., Gharib T.G., Thomas D.G., Levin A.M., Misek D.E., Lin L., Chen G., Gharib T.G., Thomas D.G., Misek D.E., Lin L., Chen G., Gharib T.G., Thomas D.G., Lin L., Chen G., Gharib T.G., Thomas D.G., Chen G., Gharib T.G., Thomas D.G., Gharib T.G., Thomas D.G., Thomas D.G., et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- Beviglia L., Golubovskaya V., Xu L., Yang X., Craven R.J., Cance W.G., Golubovskaya V., Xu L., Yang X., Craven R.J., Cance W.G., Xu L., Yang X., Craven R.J., Cance W.G., Yang X., Craven R.J., Cance W.G., Craven R.J., Cance W.G., Cance W.G. Focal adhesion kinase N-terminus in breast carcinoma cells induces rounding, detachment and apoptosis. Biochem. J. 2003;373:201–210. doi: 10.1042/BJ20021846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin T., Krogh M., Peterson C., Troein C., Krogh M., Peterson C., Troein C., Peterson C., Troein C., Troein C. Signal transduction pathway profiling of individual tumor samples. BMC Bioinformatics. 2005;6:163. doi: 10.1186/1471-2105-6-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canales R.D., Luo Y., Willey J.C., Austermiller B., Barbacioru C.C., Boysen C., Hunkapiller K., Jensen R.V., Knight C.R., Lee K.Y., Luo Y., Willey J.C., Austermiller B., Barbacioru C.C., Boysen C., Hunkapiller K., Jensen R.V., Knight C.R., Lee K.Y., Willey J.C., Austermiller B., Barbacioru C.C., Boysen C., Hunkapiller K., Jensen R.V., Knight C.R., Lee K.Y., Austermiller B., Barbacioru C.C., Boysen C., Hunkapiller K., Jensen R.V., Knight C.R., Lee K.Y., Barbacioru C.C., Boysen C., Hunkapiller K., Jensen R.V., Knight C.R., Lee K.Y., Boysen C., Hunkapiller K., Jensen R.V., Knight C.R., Lee K.Y., Hunkapiller K., Jensen R.V., Knight C.R., Lee K.Y., Jensen R.V., Knight C.R., Lee K.Y., Knight C.R., Lee K.Y., Lee K.Y., et al. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat. Biotechnol. 2006;24:1115–1122. doi: 10.1038/nbt1236. [DOI] [PubMed] [Google Scholar]
- Chen Z., Gibson T.B., Robinson F., Silvestro L., Pearson G., Xu B., Wright A., Vanderbilt C., Cobb M.H., Gibson T.B., Robinson F., Silvestro L., Pearson G., Xu B., Wright A., Vanderbilt C., Cobb M.H., Robinson F., Silvestro L., Pearson G., Xu B., Wright A., Vanderbilt C., Cobb M.H., Silvestro L., Pearson G., Xu B., Wright A., Vanderbilt C., Cobb M.H., Pearson G., Xu B., Wright A., Vanderbilt C., Cobb M.H., Xu B., Wright A., Vanderbilt C., Cobb M.H., Wright A., Vanderbilt C., Cobb M.H., Vanderbilt C., Cobb M.H., Cobb M.H. MAP kinases. Chem. Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- Chung H.-J., Kim M., Park C.H., Kim J., Kim J.H., Kim M., Park C.H., Kim J., Kim J.H., Park C.H., Kim J., Kim J.H., Kim J., Kim J.H., Kim J.H.2004ArrayXPath: Mapping and visualizing microarray gene-expression data with integrated biological pathway resources using scalable vector graphics Nucleic Acids Res. 32 :W460–W464. 10.1093/nar/gkh476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill G.A. Fundamentals of experimental design for cDNA microarrays. Nat. Genet. 2002;32:490–495. doi: 10.1038/ng1031. (Suppl. S) [DOI] [PubMed] [Google Scholar]
- Coe B.P., Lockwood W.W., Girard L., Chari R., Macaulay C., Lam S., Gazdar A.F., Minna J.D., Lam W.L., Lockwood W.W., Girard L., Chari R., Macaulay C., Lam S., Gazdar A.F., Minna J.D., Lam W.L., Girard L., Chari R., Macaulay C., Lam S., Gazdar A.F., Minna J.D., Lam W.L., Chari R., Macaulay C., Lam S., Gazdar A.F., Minna J.D., Lam W.L., Macaulay C., Lam S., Gazdar A.F., Minna J.D., Lam W.L., Lam S., Gazdar A.F., Minna J.D., Lam W.L., Gazdar A.F., Minna J.D., Lam W.L., Minna J.D., Lam W.L., Lam W.L. Differential disruption of cell cycle pathways in small cell and non-small cell lung cancer. Br. J. Cancer. 2006;94:1927–1935. doi: 10.1038/sj.bjc.6603167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlquist K., Salomonis N., Vranizan K., Lawlor S., Conklin B., Salomonis N., Vranizan K., Lawlor S., Conklin B., Vranizan K., Lawlor S., Conklin B., Lawlor S., Conklin B., Conklin B. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat. Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- Doniger S.W., Salomonis N., Dahlquist K.D., Vranizan K., Lawlor S.C., Conklin B.R., Salomonis N., Dahlquist K.D., Vranizan K., Lawlor S.C., Conklin B.R., Dahlquist K.D., Vranizan K., Lawlor S.C., Conklin B.R., Vranizan K., Lawlor S.C., Conklin B.R., Lawlor S.C., Conklin B.R., Conklin B.R. MAPPfinder: using Gene Ontology and GenMAPP to create a global gene expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S. Statistical intelligence: Effective analysis of high-density microarray data. Drug Discov. Today. 2002;7:S55–S63. doi: 10.1016/s1359-6446(02)02292-4. [DOI] [PubMed] [Google Scholar]
- Draghici S., Khatri P., Martins R.P., Ostermeier G.C., Krawetz S.A., Khatri P., Martins R.P., Ostermeier G.C., Krawetz S.A., Martins R.P., Ostermeier G.C., Krawetz S.A., Ostermeier G.C., Krawetz S.A., Krawetz S.A. Global functional profiling of gene expression. Genomics. 2003;81:98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- Draghici S., Khatri P., Eklund A.C., Szallasi Z., Khatri P., Eklund A.C., Szallasi Z., Eklund A.C., Szallasi Z., Szallasi Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006;22:101–109. doi: 10.1016/j.tig.2005.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman J.J., de van Geer S.A., de Kort F., van Houwelingen H.C., de van Geer S.A., de Kort F., van Houwelingen H.C., de Kort F., van Houwelingen H.C., van Houwelingen H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics. 2004;20:93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- Golubovskaya V., Beviglia L., Xu L.H., Earp H.S., Craven R., Cance W., Beviglia L., Xu L.H., Earp H.S., Craven R., Cance W., Xu L.H., Earp H.S., Craven R., Cance W., Earp H.S., Craven R., Cance W., Craven R., Cance W., Cance W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J. Biol. Chem. 2002;277:38978–38987. doi: 10.1074/jbc.M205002200. [DOI] [PubMed] [Google Scholar]
- Grosu P., Townsend J.P., Hartl D.L., Cavalieri D., Townsend J.P., Hartl D.L., Cavalieri D., Hartl D.L., Cavalieri D., Cavalieri D. Pathway processor: A tool for integrating whole-genome expression results into metabolic networks. Genome Res. 2002;12:1121–1126. doi: 10.1101/gr.226602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holford M., Li N., Nadkarni P., Zhao H., Li N., Nadkarni P., Zhao H., Nadkarni P., Zhao H., Zhao H. VitaPad: Visualization tools for the analysis of pathway data. Bioinformatics. 2004;21:1596–1602. doi: 10.1093/bioinformatics/bti153. [DOI] [PubMed] [Google Scholar]
- Joshi-Tope G., Gillespie M., Vasrik I., D’Eustachio P., Schmidt E., de Bone B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., Gillespie M., Vasrik I., D’Eustachio P., Schmidt E., de Bone B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., Vasrik I., D’Eustachio P., Schmidt E., de Bone B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., D’Eustachio P., Schmidt E., de Bone B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., Schmidt E., de Bone B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., de Bone B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., Jassal B., Gopinath G.R., Wu G.R., Matthews L., Gopinath G.R., Wu G.R., Matthews L., Wu G.R., Matthews L., Matthews L., et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P., Draghici S., Draghici S. Ontological analysis of gene expression data: Current tools, limitations, and open problems. Bioinformatics. 2005;21:3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P., Draghici S., Ostermeier G.C., Krawetz S.A., Draghici S., Ostermeier G.C., Krawetz S.A., Ostermeier G.C., Krawetz S.A., Krawetz S.A. Profiling gene expression using Onto-Express. Genomics. 2002;79:266–270. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- Mazieres J., He B., You L., Xu Z., Jablons D.M., He B., You L., Xu Z., Jablons D.M., You L., Xu Z., Jablons D.M., Xu Z., Jablons D.M., Jablons D.M. Wnt signaling in lung cancer. Cancer Lett. 2005;222:1–10. doi: 10.1016/j.canlet.2004.08.040. [DOI] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Carlsson E., Ridderstrale M., Laurila E., Ridderstrale M., Laurila E., Laurila E., et al. Pgc-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nau M.M., Brooks B.J., Battey J., Sausville E., Gazdar A.F., Kirsch I.R., McBride O.W., Bertness V., Hollis G.F., Minna J.D., Brooks B.J., Battey J., Sausville E., Gazdar A.F., Kirsch I.R., McBride O.W., Bertness V., Hollis G.F., Minna J.D., Battey J., Sausville E., Gazdar A.F., Kirsch I.R., McBride O.W., Bertness V., Hollis G.F., Minna J.D., Sausville E., Gazdar A.F., Kirsch I.R., McBride O.W., Bertness V., Hollis G.F., Minna J.D., Gazdar A.F., Kirsch I.R., McBride O.W., Bertness V., Hollis G.F., Minna J.D., Kirsch I.R., McBride O.W., Bertness V., Hollis G.F., Minna J.D., McBride O.W., Bertness V., Hollis G.F., Minna J.D., Bertness V., Hollis G.F., Minna J.D., Hollis G.F., Minna J.D., Minna J.D., et al. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature. 1985;318:69–73. doi: 10.1038/318069a0. [DOI] [PubMed] [Google Scholar]
- Nikitin A., Egorov S., Daraselia N., Mazo I., Egorov S., Daraselia N., Mazo I., Daraselia N., Mazo I., Mazo I. Pathway studio—The analysis and navigation of molecular networks. Bioinformatics. 2003;19:2155–2157. doi: 10.1093/bioinformatics/btg290. [DOI] [PubMed] [Google Scholar]
- Nikolic-Vukosavljevic D., Todorovic-Rakovic N., Demajo M., Ivanovic V., Neskovic B., Markicevic M., Neskovic-Konstantinovic Z., Todorovic-Rakovic N., Demajo M., Ivanovic V., Neskovic B., Markicevic M., Neskovic-Konstantinovic Z., Demajo M., Ivanovic V., Neskovic B., Markicevic M., Neskovic-Konstantinovic Z., Ivanovic V., Neskovic B., Markicevic M., Neskovic-Konstantinovic Z., Neskovic B., Markicevic M., Neskovic-Konstantinovic Z., Markicevic M., Neskovic-Konstantinovic Z., Neskovic-Konstantinovic Z. Plasma TGF-beta1-related survival of postmenopausal metastatic breast cancer patients. Clin. Exp. Metastasis. 2004;21:581–585. doi: 10.1007/s10585-004-4978-1. [DOI] [PubMed] [Google Scholar]
- Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M., Sato K., Fujibuchi W., Bono H., Kanehisa M., Fujibuchi W., Bono H., Kanehisa M., Bono H., Kanehisa M., Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page L., Brin S., Motwani R., Winograd T., Brin S., Motwani R., Winograd T., Motwani R., Winograd T., Winograd T. The pagerank citation ranking: Bringing order to the Web. Technical report. Stanford University; Palo Alto, CA.: 1998. [Google Scholar]
- Pan D., Sun N., Cheung K.-H., Guan Z., Ma L., Holford M., Deng X., Zhao H., Sun N., Cheung K.-H., Guan Z., Ma L., Holford M., Deng X., Zhao H., Cheung K.-H., Guan Z., Ma L., Holford M., Deng X., Zhao H., Guan Z., Ma L., Holford M., Deng X., Zhao H., Ma L., Holford M., Deng X., Zhao H., Holford M., Deng X., Zhao H., Deng X., Zhao H., Zhao H. PathMAPA: A tool for displaying gene expression and performing statistical tests on metabolic pathways at multiple levels for Arbidopsis. BMC Bioinformatics. 2003;4:56. doi: 10.1186/1471-2105-4-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panani A.D., Roussos C., Roussos C. Cytogenetic and molecular aspects of lung cancer. Cancer Lett. 2006;239:1–9. doi: 10.1016/j.canlet.2005.06.030. [DOI] [PubMed] [Google Scholar]
- Pandey R., Guru R.K., Mount D.W., Guru R.K., Mount D.W., Mount D.W. Pathway Miner: Extracting gene association networks from molecular pathways for predicting the biological significance of gene expression microarray data. Bioinformatics. 2004;20:2156–2158. doi: 10.1093/bioinformatics/bth215. [DOI] [PubMed] [Google Scholar]
- Pavlidis P., Qin J., Arango V., Mann J.J., Sibille E., Qin J., Arango V., Mann J.J., Sibille E., Arango V., Mann J.J., Sibille E., Mann J.J., Sibille E., Sibille E. Using the gene ontology for microarray data mining: A comparison of methods and application to age effects in human prefrontal cortex. Neurochem. Res. 2004;29:1213–1222. doi: 10.1023/b:nere.0000023608.29741.45. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Computational analysis of microarray data. Nat. Rev. Genet. 2001;2:418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- Rahnenfuhrer J., Domingues F. S., Maydt J., Lengauer T., Domingues F. S., Maydt J., Lengauer T., Maydt J., Lengauer T., Lengauer T. Calculating the statistical significance of changes in pathway activity from gene expression data. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1055. article16. http://www.bepress.com/sagmb/vol3/iss1/art16/ [DOI] [PubMed] [Google Scholar]
- Robinson P.N., Wollstein A., Bohme U., Beattie B., Wollstein A., Bohme U., Beattie B., Bohme U., Beattie B., Beattie B. Ontologizing gene-expression microarray data: Characterizing clusters with gene ontology. Bioinformatics. 2004;20:979–981. doi: 10.1093/bioinformatics/bth040. [DOI] [PubMed] [Google Scholar]
- Rodrigues S., Fathers K., Chan G., Zuo D., Halwani F., Meterissian S., Park M., Fathers K., Chan G., Zuo D., Halwani F., Meterissian S., Park M., Chan G., Zuo D., Halwani F., Meterissian S., Park M., Zuo D., Halwani F., Meterissian S., Park M., Halwani F., Meterissian S., Park M., Meterissian S., Park M., Park M. CrkI and CrkII function as key signaling integrators for migration and invasion of cancer cells. Mol. Cancer Res. 2005;3:183–194. doi: 10.1158/1541-7786.MCR-04-0211. [DOI] [PubMed] [Google Scholar]
- Sanders T.A., de Grassi T., Miller G.J., Humphries S.E., de Grassi T., Miller G.J., Humphries S.E., Miller G.J., Humphries S.E., Humphries S.E. Dietary oleic and palmitic acids and postprandial factor VII in middle-aged men heterozygous and homozygous for factor VII R353Q polymorphism. Am. J. Clin. Nutr. 1999;69:220–225. doi: 10.1093/ajcn/69.2.220. [DOI] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowskis B., Ideker T., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowskis B., Ideker T., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowskis B., Ideker T., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowskis B., Ideker T., Wang J.T., Ramage D., Amin N., Schwikowskis B., Ideker T., Ramage D., Amin N., Schwikowskis B., Ideker T., Amin N., Schwikowskis B., Ideker T., Schwikowskis B., Ideker T., Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slebos R.J., Rodenhuis S., Rodenhuis S. The molecular genetics of human lung cancer. Eur. Respir. J. 1989;2:461–469. [PubMed] [Google Scholar]
- Stelling J. Mathematical models in microbial systems biology. Curr. Opin. Microbiol. 2004;7:513–518. doi: 10.1016/j.mib.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Pomeroy S.L., Golub T.R., Lander E.S., Golub T.R., Lander E.S., Lander E.S., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susztak K., Ciccone E., McCue P., Sharma K., Bttinger E.P., Ciccone E., McCue P., Sharma K., Bttinger E.P., McCue P., Sharma K., Bttinger E.P., Sharma K., Bttinger E.P., Bttinger E.P. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005;2:e45. doi: 10.1371/journal.pmed.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swagell C., Henly D., Morris C.P., Henly D., Morris C.P., Morris C.P. Expression analysis of a human hepatic cell line in response to palmitate. Biochem. Biophys. Res. Commun. 2005;328:432–441. doi: 10.1016/j.bbrc.2004.12.188. [DOI] [PubMed] [Google Scholar]
- Tavazoie S., Hughes J.D., Campbell M.J., Cho R.J., Church G.M., Hughes J.D., Campbell M.J., Cho R.J., Church G.M., Campbell M.J., Cho R.J., Church G.M., Cho R.J., Church G.M., Church G.M. Systematic determination of genetic network architecture. Nat. Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Tian L., Greenberg S.A., Kong S.W., Altschuler J., Kohane I.S., Park P.J., Greenberg S.A., Kong S.W., Altschuler J., Kohane I.S., Park P.J., Kong S.W., Altschuler J., Kohane I.S., Park P.J., Altschuler J., Kohane I.S., Park P.J., Kohane I.S., Park P.J., Park P.J.2005Discovering statistically significant pathways in expression profiling studies Proc. Natl. Acad. Sci. 10213544 – 13549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic-Rakovic N. Tgf-beta 1 could be a missing link in the interplay between er and her-2 in breast cancer. Med. Hypotheses. 2005;65:546–551. doi: 10.1016/j.mehy.2005.03.019. [DOI] [PubMed] [Google Scholar]
- van Nimwegen M.J., de van Water B., de van Water B. Focal adhesion kinase: A potential target in cancer therapy. Biochem. Pharmacol. 2006;73:597–609. doi: 10.1016/j.bcp.2006.08.011. [DOI] [PubMed] [Google Scholar]
- van Nimwegen M.J., Huigsloot M., Camier A., Tijdens I.B., de van Water B., Huigsloot M., Camier A., Tijdens I.B., de van Water B., Camier A., Tijdens I.B., de van Water B., Tijdens I.B., de van Water B., de van Water B. Focal adhesion kinase and protein kinase b cooperate to suppress doxorubicin-induced apoptosis of breast tumor cells. Mol. Pharmacol. 2006;70:1330–1339. doi: 10.1124/mol.106.026195. [DOI] [PubMed] [Google Scholar]
- van’t Veer L.J., Dai H., de van Vijver M.J., He Y.D., Hart A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveenothers A.T., Dai H., de van Vijver M.J., He Y.D., Hart A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveenothers A.T., de van Vijver M.J., He Y.D., Hart A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveenothers A.T., He Y.D., Hart A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveenothers A.T., Hart A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveenothers A.T., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveenothers A.T., Peterse H.L., van der Kooy K., Marton M.J., Witteveenothers A.T., van der Kooy K., Marton M.J., Witteveenothers A.T., Marton M.J., Witteveenothers A.T., Witteveenothers A.T., et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Vincenzi B., Schiavon G., Silletta M., Santini D., Perrone G., Di Marino M., Angeletti S., Baldi A., Tonini G., Schiavon G., Silletta M., Santini D., Perrone G., Di Marino M., Angeletti S., Baldi A., Tonini G., Silletta M., Santini D., Perrone G., Di Marino M., Angeletti S., Baldi A., Tonini G., Santini D., Perrone G., Di Marino M., Angeletti S., Baldi A., Tonini G., Perrone G., Di Marino M., Angeletti S., Baldi A., Tonini G., Di Marino M., Angeletti S., Baldi A., Tonini G., Angeletti S., Baldi A., Tonini G., Baldi A., Tonini G., Tonini G. Cell cycle alterations and lung cancer. Histol. Histopathol. 2006;21:423–435. doi: 10.14670/HH-21.423. [DOI] [PubMed] [Google Scholar]
- Yang Y.H., Speed T., Speed T. Design issues for cDNA microarray experiments. Nat. Rev. Genet. 2002;3:579–588. doi: 10.1038/nrg863. [DOI] [PubMed] [Google Scholar]