

Abstract
Alzheimer’s disease is a major illness of dementia characterized by the presence of amyloid plaques, neurofibrillary tangles, and extensive neuronal apoptosis. However, the mechanism behind neuronal apoptosis in the Alzheimer’s-diseased brain is poorly understood. This study underlines the importance of neutral sphingomyelinase in fibrillar Aβ peptide-induced apoptosis and cell death in human primary neurons. Aβ1–42 peptides induced the activation of sphingomyelinases and the production of ceramide in neurons. Interestingly, neutral (N-SMase), but not acidic (A-SMase), sphingomyelinase was involved in Aβ1–42-mediated neuronal apoptosis and cell death. Aβ1–42-induced production of ceramide was redox-sensitive, as reactive oxygen species were involved in the activation of N-SMase but not A-SMase. Aβ1–42 peptides induced the NADPH oxidase-mediated production of superoxide radicals in neurons that was involved in the activation of N-SMase, but not A-SMase, via hydrogen peroxide. Consistently, superoxide radicals generated by hypoxanthine and xanthine oxidase also induced the activation of N-SMase, but not A-SMase, through a catalase-sensitive pathway. Furthermore, antisense knockdown of p22phox, a subunit of NADPH oxidase, inhibited Aβ1–42-induced neuronal apoptosis and cell death. These studies suggest that fibrillar Aβ1–42 peptides induce neuronal apoptosis through the NADPH oxidase-superoxide-hydrogen peroxide-NS-Mase-ceramide pathway.
Alzheimer’s disease (AD)1 is a neurodegenerative disorder resulting in progressive neuronal death and memory loss. Neuropathologically, the disease is characterized by neurofibrillary tangles and neuritic plaques composed of aggregates of amyloid-β (Aβ) protein, a 40–43 amino acid proteolytic fragment derived from the amyloid precursor protein (1). The importance of Aβ in AD has been shown by means of several transgenic animal studies. The overexpression of mutant amyloid precursor protein results in neuritic plaque formation and synapse loss and correlative memory deficits, as well as behavioral and pathological abnormalities similar to those found in AD (2). Although deposition of Aβ peptides is one of the primary causes of neuronal loss in AD (1, 2), the mechanism by which Aβ causes neuronal loss is largely unknown. Increased TUNEL staining in postmortem AD brains suggests that neurons in the brains of AD patients die through apoptosis (3, 4). Consistently, overexpression of the Aβ peptides intracellularly in transgenic mice causes chromatin segmentation, condensation, and increased TUNEL staining (5-7). Cell culture studies have also shown that Aβ peptides are apoptotic and cytotoxic to neuronal cells (8, 9). However, the mechanism by which Aβ peptides lead to neuronal loss is poorly understood.
Although sphingomyelin was initially considered only a structural component of plasma membrane, several investigations established the involvement of sphingolipids and its metabolites in the key events of signal transduction associated with cell regulation, cell differentiation, and apoptosis (10, 11). Because ceramide, the lipid second messenger molecule, produced from the degradation of sphingomyelin by sphingomyelinases (neutral and acidic) induces apoptosis and cell death in various cell types, including glial and neuronal cells (10-15), we decided to investigate the effect of Aβ peptides on the induction of ceramide production in neuronal cells. Here we report that fibrillar Aβ1–42 peptides induce the activation of sphingomyelinases and the production of ceramide in human primary neurons. We also show that the activation of neutral, but not acidic, sphingomyelinase plays the vital role in neuronal apoptosis and that NADPH oxidase-mediated superoxide production in neurons is responsible for Aβ-induced activation of neutral sphingomyelinase.
MATERIALS AND METHODS
Reagents
Neurobasal medium and B27 supplement were purchased from Invitrogen. Human Aβ peptides 1–42 and 42–1 were obtained from Bachem Bioscience. Antibodies against p22phox were purchased from Santa Cruz Biotechnology. Phosphorothioate-labeled antisense and scrambled oligodeoxynucleotides were synthesized in the DNA-synthesizing facility of Invitrogen.
Isolation of Human Primary Neurons
Human primary neurons were prepared as described by Zhang et al.(16) with some modifications. All of the experimental protocols were reviewed and approved by the Institutional Review Board (IRB number 224–01-FB) of the University of Nebraska Medical Center. Briefly, 11–17-week-old fetal brains obtained from the Human Embryology Laboratory (University of Washington, Seattle, WA) were dissociated by trituration and trypsinization (0.25% trypsin in PBS at 37 °C for 15 min). The trypsin was inactivated with 10% heat-inactivated fetal bovine serum (Mediatech). The dissociated cells were filtered through 380- and 140-μm meshes (Sigma) and pelleted by centrifugation. The cell pellet was washed once with PBS and once with Neurobasal medium containing 2% B27 and 1% antibiotic-antimycotic mixture (Sigma). In the first step, neurons were enriched by allowing the cells (3 × 106/ml) to adhere to poly-d-lysine-coated plates or coverslips for 5 min. Nonadherent cells were removed, and adherent cells (mostly neurons) were further treated with 10 μm Ara-C to prevent the proliferation of dividing cells. After 10 days of Ara-C treatment, the cells were used for this study. More than 98% of this preparation was positive for microtubule-associated protein-2 (MAP-2), a marker for neurons.
Preparation of Fibrillar Aβ
Fibrillar Aβ1–42 and control reverse peptide Aβ42–1 (Bachem Bioscience) were prepared by incubating freshly solubilized peptides at 50 μm in sterile distilled water at 37 °C for 5 days (17).
Treatment of Primary Neurons
During treatment with fibrillar Aβ peptides, the cells were incubated in Neurobasal medium containing 2% B27 supplement without antioxidant (B27-AO) (Invitrogen).
Assay of Neutral and Acidic Sphingomyelinases (N-SMase and A-SMase)
Activities of SMase(s) were assayed as described by Liu et al. (18). Briefly, after stimulation with fibrillar Aβ peptides, the cells were washed with PBS, harvested in PBS, divided into two halves, and centrifuged. The fraction for N-SMase was resuspended in buffer A (100 mm Tris-HCl, pH 7.4, 0.1% Triton X-100, 1 mm EDTA, and protease inhibitors), and the cell suspension was sonicated and centrifuged at 500 × g at 4 °C for 5 min. The supernatant was used as the enzyme source for N-SMase. The reaction mixture contained enzyme preparation in buffer A containing 5 nmol of [14C]sphingomyelin, 5 nmol of phosphatidylserine, 5 mm dithiothreitol, and 5 mm MgCl2 in a final volume of 100 μl. Similarly, the fraction for A-SMase was resuspended in buffer B (100 mm sodium acetate, pH 5.0, 0.1% Triton X-100, and protease inhibitors). The cell suspension was sonicated and centrifuged. The supernatant was used as the source of A-SMase. The activity of A-SMase was measured in a 100-μl reaction mixture consisting of the enzyme preparation in buffer B and 5 nmol of [14C]sphingomyelin. The enzyme reaction was initiated by the addition of 50 μl of substrate and stopped by the addition of 1.5 ml of chloroform:methanol (2:1, v/v) and 0.2 ml of water. After vortexing and phase separation, the aqueous phase was removed for counting.
Lipid Extraction
Approximately 5 × 105 cells were exposed to fibrillar Aβ peptides for different periods of time, and the lipids were extracted as described previously (19, 20).
Quantification of Ceramide Levels by Diacylglycerol Kinase Assay
Ceramide content was quantified using diacylglycerol (DAG) kinase and [γ-32P]ATP as described earlier (19, 20). Briefly, dried lipids were solubilized in 20 μl of an octyl β-d-glucoside/cardiolipin solution (7.5% octyl β-d-glucoside, 5 mm cardiolipin in 1 mm diethylenetriaminepentaacetic acid) by sonication in a sonicator bath. The reaction was then carried out in a final volume of 100 μl containing the 20-μl sample solution, 50 mm imidazole HCl, pH 6.6, 50 mm NaCl, 12.5 mm MgCl2, 1 mm EGTA, 2 mm dithiothreitol, 6.6 μg of DAG kinase, and 1 mm [γ-32P]ATP (specific activity of 1–5 × 105 cpm/nmol) for 30 min at room temperature. The labeled ceramide-1-phosphate was resolved with a solvent system consisting of methyl acetate:n-propyl alcohol:chloroform:methanol:0.25% KCl in water:acetic acid (100:100:100:40:36:2). A standard sample of ceramide was phosphorylated under identical conditions and developed in parallel. Both standard and experimental samples had an identical RF value (0.46). Quantification of ceramide1-phosphate was carried out by autoradiography and densitometric scanning using a Fluor Chem 8800 imaging system (Alpha Innotech Corporation). Values are expressed either as arbitrary units (absorbance) or as percent of control, considering control as 100%. Statistical comparisons were made using one-way analysis of variance followed by Student’s t test.
Assay of Superoxide Production
Superoxide production by human primary neurons was detected by LumiMax™ superoxide anion-detection kit (Stratagene) following the manufacturer’s protocol. Briefly, 5 × 105 cells suspended in 100 μl of superoxide anion assay medium were added to 100 μl of reagent mixture containing 0.2 mm luminol, 0.25 mm enhancer, and either fibrillar Aβ1–42 or Aβ42–1 at 1 μm in superoxide anion assay medium. Light emission was recorded at regular intervals in a TD-20/20 Luminometer (Turner Designs).
Assay of Hydrogen Peroxide (H2O2) Production
The production of H2O2 by human primary neurons was quantified by a colorimetric H2O2 assay kit (Assay Designs, Inc.) following the manufacturer’s protocol. Briefly, different amounts of supernatants were allowed to react with 100 μl of a color reagent containing xylenol orange in an acidic solution with sorbitol and ammonium iron sulfate followed by reading of the optical density at 550 nm.
Immunostaining
Coverslips containing 200–300 cells/mm2 were fixed with 4% paraformaldehyde for 20 min followed by treatment with cold ethanol (−20 °C) for 5 min and 2 rinses in PBS. The samples were blocked with 3% bovine serum albumin in PBS containing Tween 20 (PBST) for 30 min and incubated in PBST containing 1% bovine serum albumin and goat anti-p22phox (1:50), rabbit anti-GFAP (1:50), or goat anti-MAP-2 (1:50), as described previously (21, 22). After three washes in PBST (15 min each), the slides were further incubated with Cy5 (Jackson ImmunoResearch Laboratories, Inc.). For negative controls, a set of culture slides was incubated under similar conditions without the primary antibodies. The samples were mounted and observed under a Bio-Rad MRC1024ES confocal laser scanning microscope.
Fragment End Labeling of DNA
Fragmented DNA was detected in situ by the terminal deoxynucleotidyltransferase-mediated binding of 3′-OH ends of DNA fragments generated in response to fibrillar Aβ1–42, using a commercially available kit (TdT FragEL™) from Calbiochem. Briefly, cover slips were treated with 20 μg/ml proteinase K for 15 min at room temperature and washed prior to terminal deoxynucleotidyltransferase staining.
Cell Viability Measurement
Mitochondrial activity was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma). The cells were grown on 24-well culture plates with 500 μl of medium and treated with various reagents according to the experimental design. At the end of the treatment period, 300 μl of culture medium were removed from each well, and 20 μl of MTT solution (5 mg/ml) were added and incubated for 1 h.
Lactate Dehydrogenase Measurement
The activity of lactate dehydrogenase (LDH) was measured using the direct spectrophotometric assay using an assay kit from Sigma.
RESULTS
Fibrillar Aβ1–42 Peptides Induce the Production of Ceramide in Human Primary Neurons
Because the fibrillar form of Aβ is commonly found in the senile plaques in AD brains (1), we examined whether fibrillar Aβ1–42 is capable of inducing apoptosis in human primary neurons. As demonstrated in Fig. 1, fibrillar Aβ1–42, but not reverse (Aβ42–1), peptides markedly induced the formation of apoptotic bodies. Because ceramide is an important inducer of apoptosis in different cells (10-15), and cell-permeable C2-ceramide induced apoptosis in human fetal neurons (Fig. 1), we examined the effect of fibrillar Aβ1–42 and Aβ42–1 on the induction of ceramide production. We have found that fibrillar Aβ1–42, but not Aβ42–1, induced the production of ceramide in human primary neurons at different times of incubation. Within 15 min of treatment, fibrillar Aβ1–42 peptides were able to induce the level of ceramide by more than 2-fold, and with further increase in duration of treatment, the level of ceramide increased markedly (Fig. 2A). After 10 h of treatment of Aβ1–42, a ~12-fold increase in ceramide production was observed (Fig. 2A). In contrast to the fibrillar form, soluble Aβ1–42 peptides were weakly efficient in inducing the level of ceramide (Fig. 2A). For example, soluble Aβ1–42 peptides were unable to induce the production of ceramide at 15 or 30 min of stimulation. However, after 6 h of stimulation, soluble Aβ1–42 peptides induced a 4-fold increase in ceramide production compared with an 8-fold increase by fibrillar Aβ1–42 (Fig. 2A). Although Aβ1–42-induced production of ceramide was dose-dependent, Aβ1–42 effectively induced the production of ceramide even at 0.25 μm (Fig. 2B). Because Aβ1–42 induced neuronal apoptosis effectively at 1 μm, we have chosen the particular dose for further studies. On the other hand, under similar treatment conditions, fibrillar reverse (Aβ42–1) peptides were unable to induce the production of ceramide (Fig. 2A), suggesting the specificity of the effect. Because DAG kinase phosphorylates both DAG and ceramide using [γ-32P]ATP as substrate, both lipids can be quantified in the same assay. In contrast to a time-dependent increase in ceramide production, the level of DAG was unchanged at different time points of stimulation (Fig. 2A).
Fig. 1. Fibrillar Aβ1–42, but not Aβ42–1, peptides induce apoptosis in human primary neurons.

A, cells were incubated with either 1 μm fibrillar Aβ peptides or different concentrations of C2-ceramide (C2-Cer) for 6 h in Neurobasal medium containing 2% B27-AO followed by TUNEL. B, digital images were collected under bright-field setting using a 40× objective. TUNEL-positive cells were counted manually in four different images of each of three coverslips by three individuals blinded to the experiment. Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percent of control accordingly. Results are mean ± S.D. of three different experiments.
Fig. 2. Fibrillar Aβ1–42, but not Aβ42–1, peptides induce the production of ceramide and the activation of neutral (N-SMase) and acidic (A-SMase) sphingomyelinases in human primary neurons.
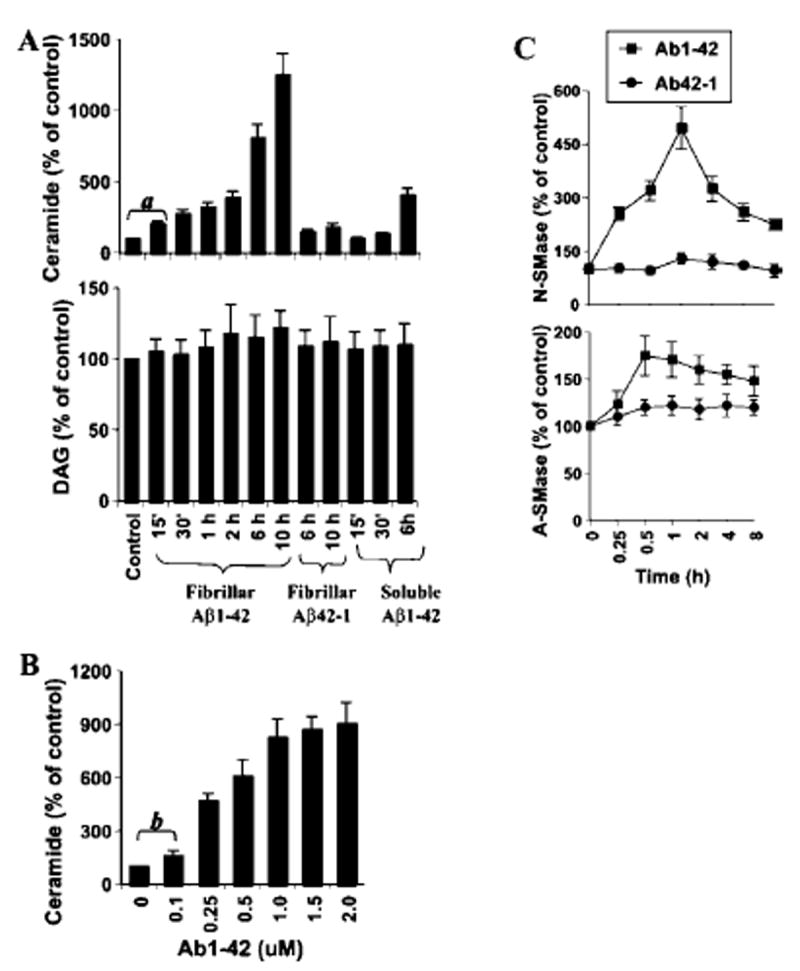
Cells were incubated with 1 μm of fibrillar Aβ1–42, fibrillar Aβ42–1, or soluble Aβ1–42 in Neurobasal medium containing 2% B27-AO for different time intervals. Lipids were extracted, and ceramide and DAG contents (A) were determined. a, p < 0.001 versus control. B, after 6 h of incubation with different concentrations of Aβ1–42, ceramide contents were determined. b, p < 0.05 versus control. C, after incubation with either fibrillar Aβ1–42 or Aβ42–1 at 1 μm for different time intervals, activities of N-SMase and A-SMase were assayed in total cell extract.
Fibrillar Aβ1–42 Peptides Induce the Activation of Sphingomyelinases in Human Primary Neurons
Five distinct sphingomyelinases have been identified based upon their pH optima, cellular localization, and cation dependence (10, 11). However, the neutral membrane-bound Mg2+-independent sphingomyelinase (N-SMase) and the lysosomal acid pH optima sphingomyelinase (A-SMase) have been the best studied for their roles in ceramide generation (10, 11). Therefore, we investigated whether fibrillar Aβ1–42 peptides are capable of inducing the activation of N-SMase and A-SMase. Marked induction of N-SMase activity was observed even at 15 min of treatment with Aβ1–42, with the maximum induction (~5-fold) observed at 1 h (Fig. 2C). However, the induction of N-SMase activation decreased afterward. On the other hand, Aβ42–1 peptides were unable to induce the activation of N-SMase at different time points of stimulation. Although Aβ1–42, but not Aβ42–1, peptides also induced the activation A-SMase showing the peak at 30 min, the level of its induction was much lower than that of N-SMase (Fig. 2C). Because TNF-α is a prototype inducer of N-SMase (10, 18), we were prompted to investigate whether TNF-α had any role in fibrillar Aβ1–42-induced neuronal activation of N-SMase. However, neutralizing antibodies against human TNF-α capable of blocking TNF-α-induced activation N-SMase had no effect on Aβ1–42-induced activation of N-SMase in human primary neurons (data not shown).
Role of N-SMase and A-SMase in Aβ1–42-induced Apoptosis and Cell Death in Human Primary Neurons
Antisense oligonucleotides provide the most effective tool with which to investigate the in vivo functions of different proteins in human primary neurons. Therefore, we adopted the antisense-knockdown technique to investigate the role of N-SMase and A-SMase in Aβ1–42-induced neuronal apoptosis and cell death. Using the method of Bloomfield and Giles (23) and based on the translational initiation site, we selected different antisense (ASO) and scrambled (ScO) oligonucleotide sequences against human N-SMase and A-SMase and screened their efficacy to inhibit Aβ1–42-induced activation of respective SMase in primary neurons. After screening of several antisenses, we found the following specific ASO and ScO against N-SMase and A-S-Mase: N-SMase, ASO, 5′-CAGCGAGCCCGTCCACCAGCC-3′; ScO, 5′-CACGCGTCCGACGCCGCACGA-3′ and A-SMase, ASO, 5′-GACATCTCGGAGCCGGGGCA-3′; ScO, 5′-GGAAAC-CCGGTTAGGCCCGG-3′.
As observed in Fig. 3, ASO against N-SMase dose-dependently inhibited Aβ1–42-induced activation of N-SMase but not A-SMase. Similarly, ASO against A-SMase inhibited Aβ1–42-induced activation of A-SMase but not N-SMase. On the other hand, respective ScO did not influence the activation of either N-SMase or A-SMase (Fig. 3).
Fig. 3. Specificities of antisense oligonucleotides against N-SMase and A-SMase.
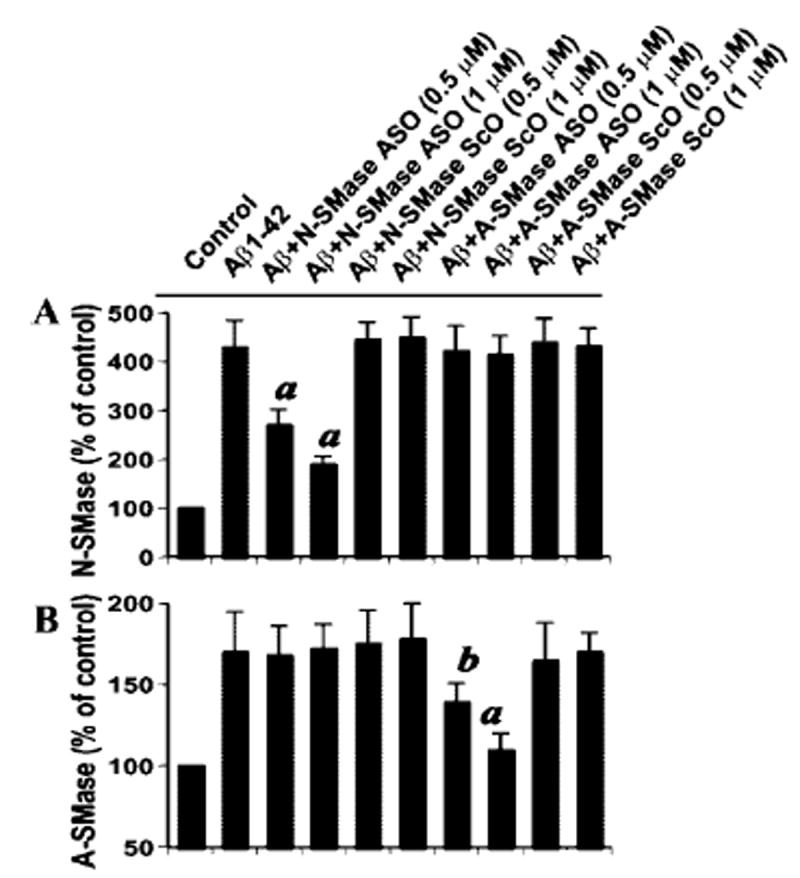
Human primary neurons received different concentrations of either antisense oligonucleotides (ASO) or scrambled oligonucleotides (ScO) against N-SMase and A-SMase. After 40 h of incubation, the cells were treated with 1 μm fibrillar Aβ1–42, and after 1 h of treatment, activities of N-SMase (A) and A-SMase (B) were measured. Results are expressed as mean ± S.D. of three separate experiments. a, p < 0.001; b, p < 0.05 versus Aβ1–42.
Next, we investigated the effect of antisense knockdown of N-SMase and/or A-SMase on Aβ1–42-induced neuronal apoptosis and cell death. Under the experimental condition of ASO/ScO treatment, the control neurons showed a few apoptotic bodies, but Aβ1–42 treatment resulted in marked increase in apoptosis (Fig. 4, A and B). However, ASO, but not ScO, against N-SMase markedly blocked Aβ1–42-induced neuronal apoptosis (Fig. 4, A and B). On the other hand, antisense knockdown of A-SMase had no effect on Aβ1–42-induced apoptosis (Fig. 4B). After 18 h of treatment, Aβ1–42 reduced cell viability, as evidenced by a decrease in MTT metabolism (Fig. 4C). Interestingly, ASO against N-SMase, but not A-SMase, effectively prevented Aβ1–42-induced loss of MTT metabolism. Similarly, Aβ1–42 also induced an increase in LDH release, and treatment of cells with ASO against N-SMase, but not A-SMase, resulted in significant reduction in LDH release (Fig. 4C). On the other hand, ScO against N-SMase had no effect on Aβ1–42-induced loss of MTT metabolism and increase in LDH release (Fig. 4C). These studies suggest that activation of N-SMase, but not A-SMase, plays the key role in fibrillar Aβ1–42-mediated neuronal apoptosis and cell death.
Fig. 4. Antisense knockdown of N-SMase, but not A-SMase, protects human primary neurons against Aβ1–42-induced apoptosis and cell death.

Cells received either ASO or ScO at 1 μm against N-SMase and A-SMase. After 40 h of incubation, the cells were treated with 1 μm fibrillar Aβ1–42, and after 6 h of stimulation, apoptotic events were detected by TUNEL (A). TUNEL-positive cells were counted manually in four different images of each of three coverslips by three individuals blinded to the experiment (B). After 18 h of stimulation, cell viability (C) was examined by the metabolism of MTT and the release of LDH. Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percent of control accordingly. Results are mean ± S.D. of three different experiments. a, p < 0.001 versus control. c, p < 0.001 versus Aβ1–42. n.s., not significant.
Involvement of Reactive Oxygen Species in Aβ1–42-induced Production of Ceramide and Activation of N-SMase in Human Primary Neurons
Next we investigated the mechanisms by which Aβ1–42 induced the activation of N-SMase in neurons. Earlier observations (12, 18) that reactive oxygen species are involved in cytokine-induced production of ceramide and activation of N-SMase prompted us to investigate whether reactive oxygen species play a role in Aβ1–42-induced production of ceramide and activation of N-SMase in neurons. We examined the effect of antioxidants, N-acetylcysteine (NAC) and diphenyliodonium (DPI), on Aβ1–42-induced production of ceramide and activation of SMases. Interestingly, both NAC and DPI dose-dependently prevented Aβ1–42-induced generation of ceramide (Fig. 5A), suggesting that Aβ1–42-induced ceramide generation in neurons is redox-sensitive. Next, we investigated whether Aβ1–42-induced activation of N-SMase and/or A-SMase is/are redox-sensitive. Although both NAC and DPI dose-dependently inhibited Aβ1–42-induced activation of N-SMase (Fig. 5B), these two antioxidants had no effect on the activation of A-SMase (Fig. 5C), suggesting that Aβ1–42-induced activation of N-SMase, but not A-SMase, is redox-sensitive.
Fig. 5. Effects of NAC and DPI on fibrillar Aβ1–42-induced production of ceramide and activation of N-SMase and A-SMase in human primary neurons.

Cells preincubated with either different concentrations of NAC or DPI for 1 h received 1 μm fibrillar Aβ1–42. A, after 6 h of incubation, the cells were washed with Hanks’ balanced salt solution and scraped off. Lipids were extracted, and the level of ceramide was measured. After 1 h of incubation, activities of N-SMase (B) and A-SMase (C) were assayed in total cell extracts. Control group served as 100%, and data from other groups were expressed as percent of control. Results are mean ± S.D. of three different experiments. a, p < 0.001; b, p < 0.05 versus Aβ1–42.
Involvement of NADPH Oxidase in Aβ1–42-induced Activation of N-SMase in Human Primary Neurons
In an attempt to identify the reactive oxygen species-producing molecule that couples fibrillar Aβ1–42 to N-SMase in neurons, we examined the production of superoxide in response to fibrillar Aβ peptides using the LumiMax™ superoxide anion-detection kit (Stratagene). Interestingly, treatment of primary neurons with fibrillar Aβ1–42, but not Aβ42–1, peptides resulted in time-dependent release of superoxide (Fig. 6). The production of superoxide was observed as early as 30 s of stimulation, which peaked at 50 s of stimulation and decreased afterward. On the other hand, the production of superoxide by Aβ1–42 was close to the control value, even after 120 s of stimulation (Fig. 6). This Aβ1–42-induced production of superoxide was completely inhibited when the cells were pretreated with 25 units/ml superoxide dismutase for 15 min (data not shown). On the other hand, catalase (25 units/ml) had no effect on fibrillar Aβ1–42-induced superoxide production (data not shown), suggesting that the LumiMax™ superoxide kit detects superoxide but not H2O2.
Fig. 6. Fibrillar Aβ1–42, but not Aβ42–1, peptides induce the production of superoxide in human primary neurons.
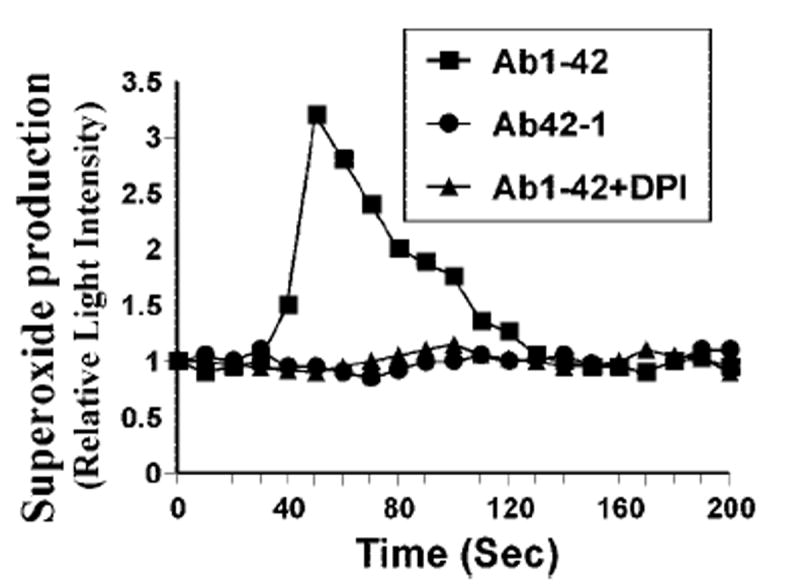
Cells were incubated with either fibrillar Aβ1–42 or Aβ42–1 at1 μm, and at different intervals (measured in seconds), superoxide production was assayed in whole cells. In the other set, the cells were preincubated with 2 μm DPI for 1 h, received 1 μm fibrillar Aβ1–42, and the production of superoxide was assayed. Results are the mean of two separate experiments.
Because DPI inhibited Aβ1–42-induced activation of N-S-Mase (Fig. 5B), we examined whether DPI was capable of inhibiting Aβ1–42-induced production of superoxide. Fig. 6 shows that Aβ1–42 failed to induce the production of superoxide in neurons pretreated with DPI, suggesting that Aβ1–42-induced production of superoxide is DPI-sensitive. Because DPI is an inhibitor of NADPH oxidase, the enzyme that catalyzes the production of superoxide, we investigated whether primary neurons expressed NADPH oxidase. Immunofluorescence analysis in Fig. 7A showed that control neurons abundantly expressed p22phox, a subunit of NADPH oxidase. To confirm that p22phox is responsible for Aβ1–42-induced production of superoxide in neurons, we used ASO against human p22phox. We selected several antisenses and screened for their efficacy to inhibit the protein expression of p22phox by Western blot. In the following, ASO, but not scrambled oligonucleotides (ScO), against p22phox effectively inhibited the expression of p22phox in neurons (data not shown): p22phox, ASO, 5′-CGC-CAGCGCCTGCTCGTTGGC-3′; ScO, 5′-GGCCTTCCCGGTTA-GCCCCGG-3′.
Fig. 7. Role of NADPH oxidase-mediated production of superoxide in fibrillar Aβ1–42-induced activation of N-SMase and cell death in human primary neurons.
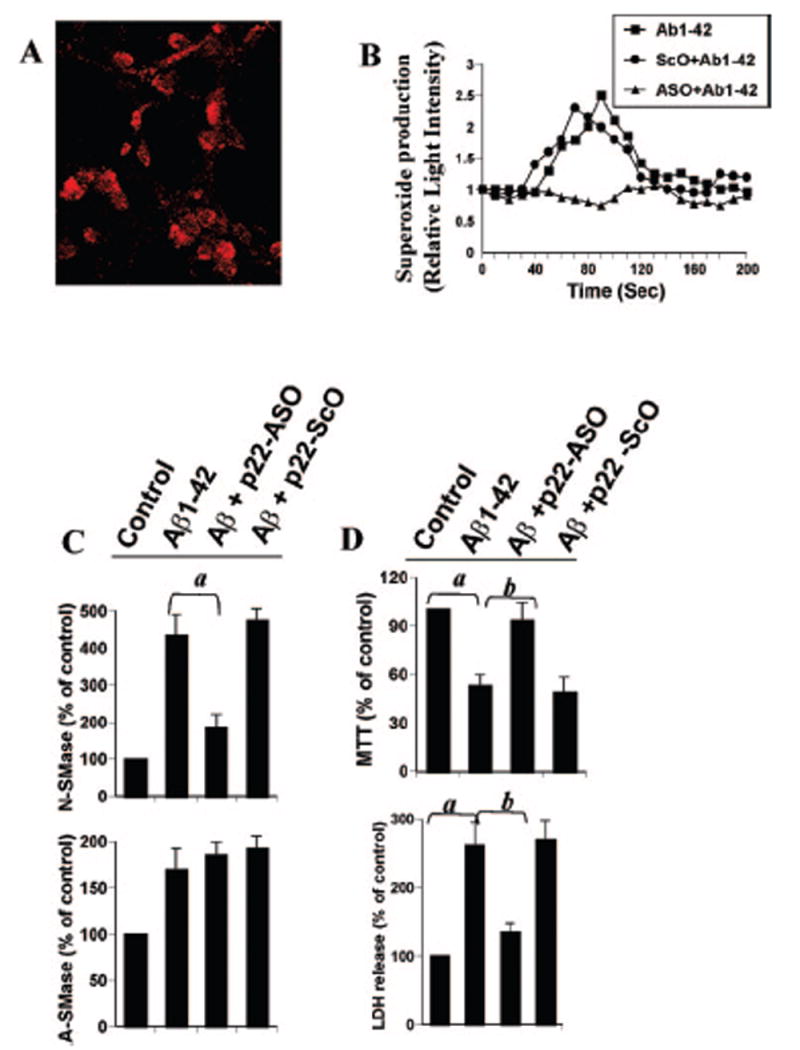
A, cells were immunostained with antibodies against p22phox. B, cells received either ASO or ScO at 1 μm against p22phox, and after 40 h of incubation, the cells were treated with 1 μm fibrillar Aβ1–42. At different intervals (measured in seconds), superoxide production was assayed in whole cells using the LumiMax™ superoxide anion detection kit (Stratagene). Results are the mean of two separate experiments. C, after 30 min of treatment with Aβ1–42, activities of N-SMase and A-SMase were assayed. D, after 18 h of stimulation with Aβ1–42, cell viability was examined by the metabolism of MTT and the release of LDH. Control group served as 100%, and data from other groups were expressed as percent of control. Results are mean ± S.D. of three different experiments. a, p < 0.001 versus control. b, p < 0.001 versus Aβ1–42.
It is apparent from Fig. 7B that ASO, but not ScO, against human p22phox markedly inhibited Aβ1–42-induced production of superoxide, suggesting that Aβ1–42 peptides induce the production of superoxide through NADPH oxidase. Because Aβ1–42-induced activation of N-SMase is redox-sensitive, we next examined whether p22phox is coupled to Aβ1–42-induced activation of N-SMase. Consistent with the inhibition of Aβ1–42-induced activation of N-SMase, but not A-SMase, by DPI (Fig. 5), ASO against p22phox inhibited Aβ1–42-induced activation of N-SMase but not A-SMase (Fig. 7C), suggesting that Aβ1–42 peptides induce the activation of N-SMase through NADPH oxidase-mediated generation of superoxide. Therefore, we were prompted to investigate whether antisense knockdown of p22phox could save the neurons against Aβ1–42 challenge. Consistent with the involvement of N-SMase in Aβ-induced cell death and the coupling of p22phox to the activation of N-SMase, ASO, but not ScO, against p22phox effectively prevented Aβ1–42-induced loss of MTT metabolism and release of LDH (Fig. 7D). These results suggest that Aβ1–42 peptides induce neuronal cell death via NADPH oxidase-mediated activation of N-SMase.
Do Superoxide Radicals Alone Induce the Activation of N-SMase in Human Primary Neurons?
Because superoxide radicals released from NADPH oxidase are involved in Aβ-induced activation of N-SMase, we investigated whether superoxide radicals alone were capable of activating N-SMase. Superoxide radicals were generated by the exogenous addition of hypoxanthine and xanthine oxidase. Although the addition of either hypoxanthine or xanthine oxidase alone to neurons was unable to induce the activation of N-SMase (data not shown), the addition of both effectively induced the activation of N-SMase (Fig. 8A). As expected, marked stimulation of Aβ-induced activation of N-SMase was also observed after treatment with hypoxanthine and xanthine oxidase (Fig. 8A). These studies suggest that superoxide radicals alone are sufficient to induce the activation of N-SMase and that these radicals are also capable of stimulating Aβ-induced activation of N-SMase. On the other hand, exogenous production of superoxide radicals had no effect on the activation of A-SMase (data not shown).
Fig. 8. Involvement of H2O2 in Aβ1–42- and superoxide-induced activation of N-SMase in human primary neurons.

A, cells were treated with 1 μm Aβ1–42 in the presence or absence of 5 μm hypoxanthine (H) and 0.5 milliunits/ml xanthine oxidase (XO). At different intervals (measured in minutes), activity of N-SMase was assayed. B, in one set, cells were treated with 1 μm Aβ1–42 and in the other, 1 μm Aβ1–42 was also added to media only. At different intervals (measured in minutes), the level of H2O2 was measured in supernatant, as described under “Materials and Methods.” Control group served as 100%, and data from other groups were expressed as percent of control. C, cells received either ASO or ScO at 1 μm against p22phox, and after 40 h of incubation, the cells were treated with 1 μm fibrillar Aβ1–42. After 15 min of stimulation, H2O2 production was assayed in supernatants. Results are mean ± S.D. of three different experiments. a, p < 0.001 versus Aβ1–42 + vehicle. Cells pretreated with different amounts of either superoxide dismutase (SOD) or catalase (Cat) for 15 min were stimulated with either 1 μm fibrillar Aβ1–42 (D) or the combination of H (5 μm) and XO (0.5 milliunits/ml) (E). After 30 min of incubation, activity of N-SMase was assayed in total cell extracts. Control group served as 100%, and data from other groups were expressed as percent of control. Results are mean ± S.D. of three different experiments.
Because superoxide radicals are not stable and could be converted to H2O2 by enzymatic and/or nonenzymatic fashion and H2O2 alone also induces the activation of N-SMase (18), we investigated whether superoxide itself or H2O2 derived from superoxide was involved in the activation of N-SMase. Consistent with the production of superoxide, Aβ1–42 also induced the production of H2O2 (Fig. 8B). Although the production of superoxide in response to Aβ1–42 was observed as early as 30 s of stimulation peaking at 50 s (Fig. 6), Aβ1–42 peptides were unable to induce the production of H2O2 at intervals of seconds (Fig. 8B). However, marked increase in H2O2 was observed after 5 or 15 min of stimulation with fibrillar Aβ1–42 (Fig. 8B). As different metal ions interact directly with Aβ1–42 to produce H2O2 in a cell-free manner in vitro (24), we examined the possibility that media was providing H2O2 response to Aβ1–42. Consistently, Aβ caused an increase in H2O2 production in media alone but at a lower level than in neurons (Fig. 8B). Next, we were prompted to investigate whether this increase in H2O2 was coupled to NADPH oxidase-mediated superoxide production. Therefore, we examined whether antisense knockdown of p22phox could inhibit Aβ1–42-induced production of H2O2 in primary neurons. Inhibition of Aβ1–42-induced production of H2O2 by ASO, but not ScO, against p22phox (Fig. 8C) suggests that at least a part of H2O2 is generated from superoxide released from NADPH oxidase. Next, we investigated whether Aβ1–42 was actually inducing the activation of N-SMase via H2O2. Although both superoxide dismutase and catalase inhibited Aβ1–42-induced activation of N-SMase, the extent of inhibition by catalase was much more than superoxide dismutase (Fig. 8D). Almost complete inhibition of Aβ1–42-induced activation of N-SMase was observed with 25 or 50 units/ml catalase (Fig. 8D), suggesting that H2O2, but not superoxide radicals, may be involved in Aβ1–42-induced activation of N-SMase. Thereafter, we examined whether superoxide radicals generated from hypoxanthine and xanthine oxidase also induced the activation of N-SMase via H2O2. Similar to the inhibition of Aβ1–42-induced activation of N-SMase, catalase also exerted a much greater inhibitory effect than superoxide dismutase on superoxide-induced activation of N-SMase (Fig. 8E), suggesting that superoxide radicals are also induced the activation of N-SMase in neurons via H2O2.
DISCUSSION
Several studies support a role for hydrolysis of sphingomyelin as a stress-activated signaling mechanism in which ceramide plays an important role in growth suppression and apoptosis in various cell types, including glial and neuronal cells (10-15). Ceramide activates proteases and caspases, the molecules responsible for apoptosis, and the activation of proteases and caspases and induction of apoptosis by ceramide are inhibited by overexpression of bcl-2. Again ceramide has been shown to induce PP2A-mediated dephosphorylation of bcl-2 and thereby loss of antiapoptotic function (reviewed in Ref. 10). The addition of exogenous ceramides or sphingomyelinase to cells induces the stress-activated protein kinase (SAPK) pathway, and a dominant negative mutant of SEK1, the protein kinase responsible for phosphorylation and activation of SAPK, attenuates ceramide-induced apoptosis (reviewed in Ref. 10). Recently, it has been shown that ceramide also inhibits stimulus-mediated activation of protein kinase B, a key determinant of cell fate (25). Previously, ceramide was shown to activate a kinase called ceramide-activated protein kinase (or CAPK), which has been recently identified as the kinase suppressor of Ras (11). This kinase suppressor of Ras also plays a role in the ceramide-induced apoptotic pathway (11). These observations suggest that various molecules, such as Bcl-2, SAPK, protein kinase B, and kinase suppressor of Ras, function downstream of ceramide in the apoptotic pathway. Although the importance of apoptosis in AD pathogenesis has been supported by evidence describing increased TUNEL staining and activated caspases in postmortem analysis of AD brain (3-7) and Aβ has been identified as one of the potential candidates for neuronal apoptosis in AD brain, the mechanism by which Aβ peptides induce neuronal apoptosis is poorly understood. Several lines of evidence presented in this work clearly suggest that fibrillar Aβ peptides induce neuronal apoptosis through the activation of neutral sphingomyelinase (N-SMase). First, fibrillar Aβ1–42, but not Aβ42–1, peptides induced the production of ceramide in human primary neurons. Second, fibrillar Aβ1–42, but not Aβ42–1, peptides markedly induced the activation of N-SMase. On the other hand, the activity of A-SMase increased marginally in response to these peptides. Third, antisense knockdown of N-SMase, but not A-SMase, protected neurons from Aβ-induced apoptosis and cell death. These results emphasize an important role of N-SMase, but not A-SMase, in Aβ-induced neuronal damage. Furthermore, our studies also suggest that the sphingomyelin cycle may play an important role during neurodegeneration in AD brain. Consistently, a recent finding from Mattson and colleagues (26) has demonstrated accumulation of long chain ceramides during normal brain aging and in the brains of AD patients.
Since the discovery of the sphingomyelin cycle, several inducers have been shown to be coupled to N-SMase, including 1α,25-dihydroxyvitamin D3, radiation, antibody cross-linking, TNF-α, IL-1β, Fas, nerve growth factor, brefeldin A, and serum deprivation (10, 11). The mechanisms involved in the activation of N-SMase in response to TNF-α are becoming clear. For example, TNF-α initiates the pathway through TNFR1 (55-kDa receptor) leading to phospholipase A2 activation, generation of arachidonic acid, and subsequent activation of N-SMase (10). Although binding of Fan to the N-SMase-activating domain of TNFR1 is required for the activation of N-SMase, binding of TNF receptor-associated death domain to the same receptor induces the activation of A-SMase (27). In addition, proteases have also been implicated in the pathway leading from TNF-α to the activation of N-SMase (10, 11). However, mechanisms involved in fibrillar Aβ-mediated activation of N-SMase in neurons are not known.
Previously it was found that the activation of N-SMase, but not A-SMase, in TNF-α-stimulated cells is redox-sensitive (18). When this manuscript was in preparation, it was shown by Lee et al. (28) that Aβ induces the death of oligodendrocytes by activating the N-SMase-ceramide cascade via an oxidative mechanism. Here we delineate that fibrillar Aβ peptides induce the activation of N-SMase in human primary neurons through NADPH-oxidase-sensitive superoxide production. First, antioxidants inhibited Aβ-induced neuronal activation of N-SMase but not A-SMase. Second, Aβ1–42, but not Aβ42–1, peptides induced the production of superoxide in neurons. Third, DPI, a specific inhibitor of NADPH oxidase, inhibited the Aβ1–42-induced production of superoxide. Fourth, primary neurons expressed p22phox, a subunit of NADPH oxidase, and antisense knockdown of p22phox strongly inhibited Aβ-induced production of superoxide, suggesting that Aβ induces the production of superoxide via NADPH oxidase. Fifth, Aβ1–42 peptides also induced the production of H2O2 in neurons, and antisense knockdown of p22phox reduced Aβ-induced production of H2O2, suggesting that superoxide generated from NADPH oxidase was actually converted to H2O2. Because H2O2 is responsible for the activation of N-SMase, but not A-SMase (18), antisense knockdown of p22phox inhibited Aβ1–42-induced activation of N-SMase but not A-SMase. Sixth, the generation of superoxide radicals by hypoxanthine and xanthine oxidase was sufficient to induce the activation of N-SMase, but not A-SMase, in neurons via H2O2. Furthermore, fibrillar Aβ peptides were unable to induce apoptosis and cell death in p22phox knockout human primary neurons. Taken together, these studies support the model in which Aβ peptides induce neuronal apoptosis and cell death via the NADPH oxidase-superoxide-H2O2-NSMase pathway.
Although the local concentration of fibrillar Aβ peptides present in the brain microenvironment of AD patients may differ from the concentration we used in primary neurons and the in vitro situation of human fetal neurons in culture may not truly resemble the in vivo situation of neurons in the brain of AD patients, our results clearly point out N-SMase as a possible therapeutic target to halt neuronal damage in AD and other neurodegenerative disorders.
Acknowledgments
We thank Dr. Xiaojuan Liu for her help in cell culture and Dr. You Zhou of the University of Nebraska at Lincoln for his help in microscopy.
Footnotes
This study was supported by Grants NS39940 and AG19487 from National Institutes of Health.
The abbreviations used are: AD, Alzheimer’s disease; Aβ, amyloid-β; N-SMase, neutral sphingomyelinase; A-SMase, acidic sphingomyelinase; PBS, phosphate-buffered saline; DAG, diacylglycerol; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; LDH, lactate dehydrogenase; TNF-α, tumor necrosis factor α; NAC, N-acetylcysteine; DPI, diphenyliodonium; SAPK stress-activated protein kinase; TUNEL, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick-end labeling.
References
- 1.Martin JB. N Engl J Med. 1999;340:1970–1980. doi: 10.1056/NEJM199906243402507. [DOI] [PubMed] [Google Scholar]
- 2.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 3.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LHT, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 4.Smale G, Nichols NR, Brady DR, Finch CE, Horton WE., Jr Exp Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 5.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 6.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. J Neurosci. 1996;16:5795–5811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 8.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bastianetto S, Ramassamy C, Dore S, Christen Y, Poirier J, Quirion R. Eur J Neurosci. 2000;12:1882–1890. doi: 10.1046/j.1460-9568.2000.00069.x. [DOI] [PubMed] [Google Scholar]
- 10.Hannun YA. Science. 1996;274:1855–1858. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 11.Pettus BJ, Chalfant CE, Hannun YA. Biochim Biophys Acta. 2002;1585:114–125. doi: 10.1016/s1388-1981(02)00331-1. [DOI] [PubMed] [Google Scholar]
- 12.Singh I, Pahan K, Khan M, Singh AK. J Biol Chem. 1998;273:20354–20362. doi: 10.1074/jbc.273.32.20354. [DOI] [PubMed] [Google Scholar]
- 13.Brugg B, Mitchel PP, Agid Y, Ruberg M. J Neurochem. 1996;66:733–739. doi: 10.1046/j.1471-4159.1996.66020733.x. [DOI] [PubMed] [Google Scholar]
- 14.Wiesner DA, Dawson G. J Neurochem. 1996;66:1418–1425. doi: 10.1046/j.1471-4159.1996.66041418.x. [DOI] [PubMed] [Google Scholar]
- 15.Keane RW, Srinivasan A, Foster LM, Testa MP, Ord T, Nonner D, Wang HG, Reed JC, Bredesen DE, Kayalar C. J Neurosci Res. 1997;48:168–180. doi: 10.1002/(sici)1097-4547(19970415)48:2<168::aid-jnr9>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, McLaughlin R, Goodyer C, LeBlanc A. J Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu B, Andrieu-Abadie N, Levade T, Zhang P, Obeid LM, Hannun YA. J Biol Chem. 1998;273:11313–11320. doi: 10.1074/jbc.273.18.11313. [DOI] [PubMed] [Google Scholar]
- 19.Pahan K, Sheikh FG, Khan M, Namboodiri AM, Singh I. J Biol Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- 20.Pahan K, Khan M, Singh I. J Neurochem. 2000;75:576–582. doi: 10.1046/j.1471-4159.2000.0750576.x. [DOI] [PubMed] [Google Scholar]
- 21.Dasgupta S, Zhou Y, Jana M, Banik NL, Pahan K. J Immunol. 2003;170:3874–3882. doi: 10.4049/jimmunol.170.7.3874. [DOI] [PubMed] [Google Scholar]
- 22.Dasgupta S, Jana M, Zhou Y, Fung YK, Ghosh S, Pahan K. J Immunol. 2004;173:1344–1354. doi: 10.4049/jimmunol.173.2.1344. [DOI] [PubMed] [Google Scholar]
- 23.Bloomfeild MR, Giles IG. Biochem Soc Trans. 1992;20(suppl):293S. doi: 10.1042/bst020293s. [DOI] [PubMed] [Google Scholar]
- 24.Opazo C, Huang X, Cherny RA, Moir RD, Roher AE, White AR, Cappai R, Masters CL, Tanzi RE, Inestrosa NC, Bush AI. J Biol Chem. 2002;277:40302–40308. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 25.Powell DJ, Hajduch E, Kular G, Hundal HS. Mol Cell Biol. 2003;23:7794–7808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, Kronke M. Cell. 1996;86:937–947. doi: 10.1016/s0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- 28.Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, Chen S, Hsu CY. J Cell Biol. 2004;164:123–131. doi: 10.1083/jcb.200307017. [DOI] [PMC free article] [PubMed] [Google Scholar]