

Summary
The positive transcription elongation factor b (P-TEFb) is an essential regulator of viral gene expression during the life cycle of HIV-1. Its Cyclin T1 subunit forms a ternary complex with the viral Tat protein and the TAR RNA element thereby activating cyclin dependent kinase 9 (Cdk9), which stimulates transcription at the level of chain elongation. We report the structure of the cyclin box domain of human Cyclin T1 at a resolution of 2.67 Å. The structure was obtained by crystallographic analysis of a fusion protein composed of Cyclin T1 linked to the transactivator protein Tat from equine infectious anemia virus (EIAV), which is functionally and structurally related to HIV-1 Tat. The conserved cyclin box domain of Cyclin T1 exhibits structural features for interaction with physiological binding partners such as Cdk9. A recognition site for Cdk/Cyclin substrates is partly covered by a Cyclin T-specific insert, suggesting specific interactions with regulatory factors. The previously identified Tat/TAR recognition motif (TRM) forms a C-terminal helix that is partly occluded in the cyclin box repeat interface, while cysteine 261 is accessible to form an intermolecular zinc finger with Tat. Residues of the TRM contribute to a positively charged groove that may directly attract RNA molecules. The EIAV Tat protein instead appeared undefined from the electron density map suggesting that it is highly disordered. Functional experiments confirmed the TAR binding properties of the fusion protein and suggested residues on the second cyclin box repeat to contribute to Tat stimulated transcription.
Keywords: Cyclin T1, P-TEFb, Tat, transcription, crystal structure
Introduction
HIV-1 gene expression is the major determinant regulating the rate of virus replication and, consequently, progression to AIDS. The HIV genome encodes a transcriptional activator protein, Tat, which binds to the transactivator response (TAR) RNA element to enhance transcription at the level of RNA polymerase II (RNAPII) elongation.1 For efficient transcriptional activation in the infected cell the cellular cofactor Cyclin T1 (CycT1) is required. The conserved cyclin box domain of CycT1 is necessary and sufficient for its interaction with Tat/TAR.2,3 Cyclins are generally known as regulatory subunits of cyclin dependent kinases (Cdk’s), most of which are involved in cell cycle regulation.4 In contrast to cyclins A, B, D and E, that form complexes with Cdk’s involved in cell-cycle progression, T-type cyclins regulate the activity of Cdk9, a protein kinase important for transcriptional regulation.5–7 While detailed information is available about cyclins involved in cell-cycle regulation and progression, including molecular complexes with their cognate Cdk’s, substrates or inhibitory molecules,8–13 little is known about Cdk-Cyclin systems involved in transcriptional regulation.
Cyclin T was initially discovered as an 87 kDa Cyclin C-related protein that specifically interacts with HIV-1 Tat.2 At the same time, multiple Cyclin T subunits (T1, T2a, T2b) were identified as the obligate partner of the kinase Cdk9, which together constitute the positive transcription elongation factor P-TEFb.14 P-TEFb has been described to enable RNAPII to enter productive elongation by phosphorylating serine 2 within the heptad region of the C-terminal domain (CTD).6,15 This enables the polymerase to resist pausing caused by negative elongation factors, which limit polymerase processivity.16,17 Unlike other cyclins, CycT1 is constitutively expressed and protein levels of P-TEFb vary little throughout the cell cycle.18 Accordingly, P-TEFb was found to be regulated by the abundant small nuclear RNA 7SK and the regulatory protein Hexim1.19,20 Roughly half of nuclear P-TEFb in HeLa cells is sequestered in an inactive state; however, inactive and active P-TEFb complexes can rapidly interchange upon treatment with stress-inducing agents. Dysregulation of P-TEFb was found to cause cardiac muscle-cell hypertrophy and Hexim1 was recently described as a novel inhibitor of breast cell growth that is down-regulated by estrogens and decreased in breast tumours.21,22 Kinase activity of P-TEFb is thus tightly regulated to mediate cellular gene transcription.
The Cdk9/CycT1 pair serves not only as a general transcription factor but also as specific cellular cofactor of the HIV-1 Tat protein. Tat activates transcription of viral genes through direct binding to CycT1 and promotes cooperative binding to TAR, a stem-loop RNA structure formed at the 5′ end of the nascent viral transcript.23,24 This results in activation of viral gene expression at the level of transcript elongation. A critical cysteine (Cys261) of human CycT1 is thought to form an intermolecular zinc-finger with cysteines from Tat.3 Moreover, a Tat-TAR recognition motif (TRM) composed of basic residues at the C-terminal region of the cyclin box repeats that directly contacts the stem loop structure of TAR RNA is required for the interaction (Figure 1(a)).3,25 The interaction of Tat/TAR to CycT1 may finally be released by acetylation of a single lysine residue in Tat that serves to dissociate the complex and transfers Tat onto the elongating RNA polymerase II complex.26
Figure 1. Structure of the cyclin box domain of Cyclin T1.
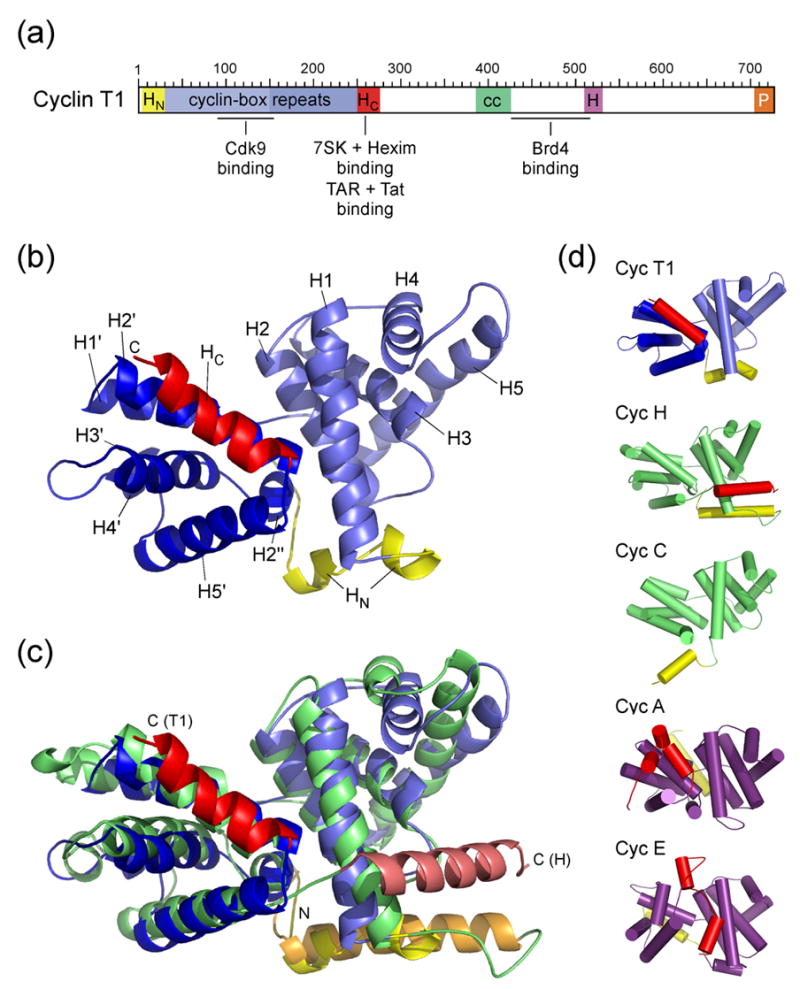
(a) Schematic diagram of the modular domain organization of human CycT1. The N-terminal cyclin domain contains the two canonical cyclin-box repeats and accessory N- and C-terminal helices. The central region is less conserved among different CycT’s but contains a proposed helical segment (cc, coiled coil) and a highly conserved histidine-rich segment (H). The C-terminal 20 residues are proline-rich (P). Proteins and RNA elements binding to CycT1 are indicated at their proposed interaction sites (Brd4, bromodomain-containing protein 4). (b) Overall structure of human CycT1 (8–263). Helices of the two repeats are denoted H1–H5 and H1′–H5′, coloured light and medium blue, respectively. N- and C-terminal helices (HN and HC) are shown in yellow and red, respectively. (c) Superimposition of CycT1 (blue) and human Cyclin H (green, AC: 1JKW). The two molecules were aligned on their cyclin box repeat structures (T1: 30–248; H: 49–262) resulting in an RMSD value of 2.2 Å. (d) Tube representation of A- and C-type cyclins. The orientation of all five molecules was aligned relative to helices H2 and H1′. N- and C-terminal helices of all cyclins are displayed in yellow and red, respectively. They are oriented differently in cyclins A8 (1FIN) and E12 (1W98) compared to cyclins C32 (1ZP2), H30 (1JKW) and T1 (2PK2, this study). Although CycT1 shares the closest structural similarity of the cyclin boxes to cyclin H, the orientation of its C-terminal helix aligns best to cyclin A.
Tat from equine infectious anemia virus (EIAV) shares about 60% similarity with HIV-1 Tat and is essential for efficient viral gene expression and replication.27,28 Here we report the crystallographic analysis of a fusion protein containing human CycT1 (1–281) at the N- and EIAV-Tat (eqTat, 22–78) at the C-terminus. Such fusion proteins have previously been shown to be biologically active in TAR-activated transcription elongation.29 The X-ray structure determined at a resolution of 2.67 Å reveals the detailed three-dimensional architecture of the cyclin box domain of CycT1 with the Tat component being largely disordered in our crystals. Regions important for the interactions with physiological binding partners such as the TRM give insights into the spatial arrangement of critical residues that build up the interface for its regulatory factors.
Results
Generation and structure determination of a CycT1-Tat fusion protein
The sequence of the CycT1 cyclin boxes shows highest similarity to Cyclin H with an identity of 19%, but due to low similarity of the N- and C-terminal segments flanking the two repeats, the presence of adjacent helices could not be predicted reliably. Thus, six different constructs of human CycT1 with different domain boundaries were generated, out of which (1–281) turned out to be most stable. Initial crystallization trials of this protein product failed potentially due to unspecific protein aggregation. Therefore, we fused the EIAV Tat protein C-terminal to CycT1 (1–281) using a 20 residue glycine-rich linker, which allows for widespread conformational flexibility in the formation of the complex. Equine Tat was chosen as stabilizing factor since it is supposed to contain only one zinc-binding site consisting of two cysteines and one histidine, with the fourth coordination site being possibly contributed by Cys261 of CycT1.23,3 In contrast, the HIV-1 Tat protein contains seven cysteines within 16 residues that complicate its recombinant expression and folding. Crystals of the CycT1-eqTat fusion protein that diffracted to 2.67 Å resolution were obtained using the hanging drop diffusion technique. But while electron density maps from diffraction experiments could be assigned for residues 8 to 263 of CycT1, the Tat protein remained largely disordered in our crystals and not fixed by intermolecular interactions. We therefore conclude that the presence of CycT1 is not sufficient to achieve a stable folding of the equine Tat protein.
Structure of the Cyclin T1 cyclin box repeat domain
The cyclin box structure of CycT1 adopts an elongated shape with two characteristic α-helical repeats, each consisting of five helices referred to as H1–H5 and H1′–H5′, respectively (Figure 1(b)). Both ends of the canonical repeats are flanked by N- and C-terminal helices, termed HN and HC, which fold in between the interface of the two repeats. Overall, the structure of CycT1 (30–248) excluding both terminal helices can be superimposed best to cyclin H30 (PDB accession number 1JKW) with a root mean-square deviation (RMSD) value of 2.24 Å for the Cα atom positions (Figure 1(c)). The individual cyclin box repeats can be aligned with those of cyclin H with RMSD-values of 1.96 Å and 2.45 Å, respectively. The higher structural and sequential similarity of the first repeat holds for cyclins in general and reflects the conserved Cdk-binding properties. A specific insert between helices H4 and H5 is on the kinase-binding reverse site and may have implications on interactions with effector molecules. Similarly, the second repeat exhibits a specific insert between helices H3′ and H4′ that is unique to T-type cyclins and leads to an extended bulged loop with highly conserved aromatic residues. A structure-based alignment of the cyclin box folds from cyclins A, E, C and H compared to CycT1 shows the differences in the length of individual helices and the similarity in sequence composition (Figure 2).
Figure 2.
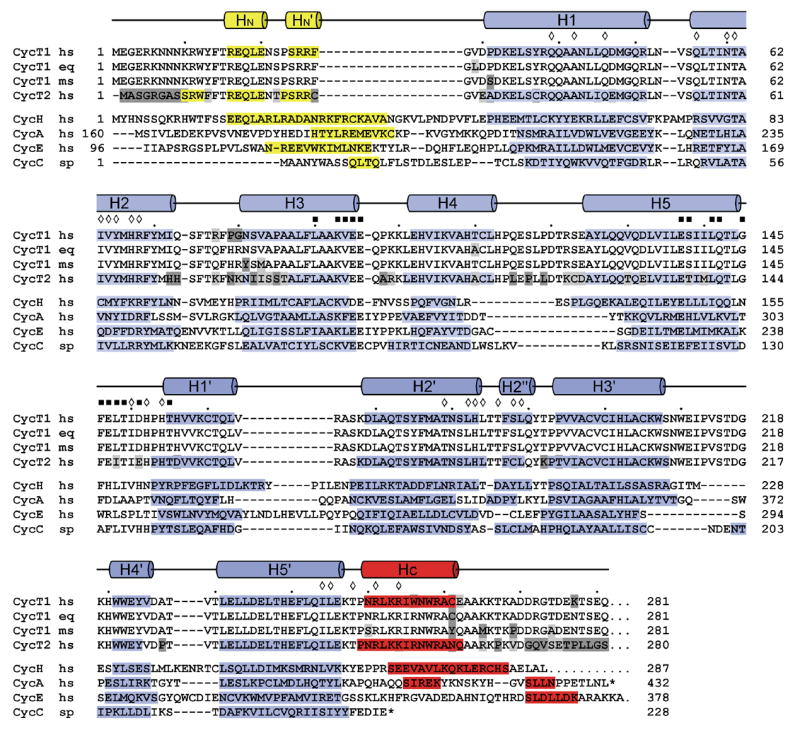
Structure-based sequence alignment of the cyclin box repeats from various cyclin proteins. The alignment is based on the structures of human Cyclin H (1JKW, ref. 30), human Cyclin A (1FIN, ref. 8), human Cyclin E (1W98, ref. 12), Cyclin C from S. pombe (1ZP2, ref. 32) and human CycT1 (2PK2, this study). Secondary structure elements of CycT1 are indicated on top, helices of cyclins H, A, E and C are displayed by bars with the N- and C-terminal helices coloured yellow and red, respectively. Residues within the interface of the two repeats in CycT1 are indicated by diamonds. Residues of CycT1 that are proposed to interact with Cdk9 are marked by black squares.
The interface of the two cyclin box repeats is composed of residues from helices H1 and H2 that interact primarily with residues of helix H2′ to form an anti-parallel three helix bundle. Interactions between the side chains from the second repeat (T179, L182 and H183) with residues from H1 (Q39, A42 and Q46) and H2 (N60, I63 and H67) compose the core of the repeat interface (see annotation in Figure 2). The repeat interface is sealed on the kinase-binding site by hydrogen bond interactions of residues H152 and H154 from the inter-repeat linker with the side chains of T61 and N60 of helix H2, respectively, as well as hydrophobic interactions between I150 and V64. On the opposite site, the C-terminal helix HC from residue (250–261) runs anti-parallel to H2′ and forms a characteristic surface that opens a groove into the cyclin box repeat interface. Both CycT1 cyclin box repeats finally match each other with an RMSD-value of 1.98 Å.
Interface and specificity to Cdk9
Cyclins activate Cdk’s through direct binding and induction of conformational changes in their kinase lobe. This initial binding step is followed by a phosphorylation reaction of a serine or threonine residue in the Cdk activation segment (T186 in human Cdk9). Subsequently, ionic interactions between three conserved arginine residues with the phosphothreonine induce conformational changes that lead to full activation of the kinase. Based on crystal structures of the complexes of Cdk2-Cyclin A (ref. 8–10) and Cdk2-Cyclin E (ref. 12) we identified residues in CycT1 that are supposed to interact with Cdk9. Residues in the cyclin that interact with the kinase locate on helices H3 and H5 of the first cyclin box repeat and the inter-repeat loop to helix H1′ (see annotation in Figure 2). Based on sequence alignments and mutual comparisons they can be divided in “general” and “specific” Cdk-interacting residues, in order to achieve specificity for the cognate kinase partner. Besides the conserved “general” amino acids, residues S138, Q142, G145, F146, E147, T149, D151 and T155 in CycT1 are likely to contribute to the specific recognition of Cdk9. However, these evaluations are based on homology comparisons that do not consider a specific insert of 5 residues in the kinase active centre of Cdk9. The precise identification of the kinase interface has thus to await structure determination of the P-TEFb complex.
Conformation of N- and C-terminal (TRM) helices
The presence and conformation of N- and C-terminal helices flanking the two cyclin box repeats is known to be critical for the activity and specificity of cyclins.31 These segments are less tightly associated to the canonical cyclin box structure and may change their conformation upon interaction with effectors or regulatory molecules. CycT1 contains a short N-terminal helix with the sequence motif REQLE that superimposes well with the first turn of the N-terminal helix in Cyclin H (EEQLA), although in Cyclin H the N-terminal region preceding H1 is 19 residues longer than in CycT1 (Figure 1(c) and Figure 2). The similar orientation relative to the cyclin boxes results from a conserved leucine residue (L19), that sticks into the repeat interface and forms hydrophobic interactions with M71, L189 and Q190 in CycT1. Residues preceding this helix form an elongated loop that runs towards the inter-repeat linker with N8 and N9 forming hydrogen bonds with D151 and E147, respectively. In addition, R68, which is highly conserved in all cyclins, forms an ionic interaction to the backbone carbonyl group of N8, which differs from observations in Cyclin H and Cyclin C, where the respective arginine coordinates the inter-repeat linker.30,32 Valine 29, which has been supposed to be involved in Tat binding,27 is directly preceding the first helix of the cyclin box repeat and partly accessible on the surface of CycT1 at a site close to the C-terminus of the two repeats.
The C-terminal helix (250–261) of CycT1 adopts a rather unexpected conformation running anti-parallel to helix H2′. Such conformation was so far only found in A-type cyclins (Figure 1(d)). In Cyclin H the C-terminal helix embraces the first cyclin box repeat,30 while Cyclin C from S. pombe, another cyclin involved in transcription regulation, does not contain a flanking helix in its coding frame.32 Similarly, the recently determined crystal structure of Cyclin K did not exhibit a C-terminal helix secluding the cyclin box repeat fold.33 In CycT1 this helix, which is also known as TRM segment,3 is coordinated to the second cyclin box repeat by residues R251, L252, I255, W256, W258 and R259, while N250, K253, R254, N257, A260 and C261 are exposed on the surface as is partly W256 (Figure 3(a)). Interestingly, N250, R259 and C261 were found to be required for Tat binding, whereas R251, L252, R254, I255 and W258 are needed for TAR RNA recognition.3 Since most of the residues required for TAR binding are hidden in the conformation of the TRM helix in our crystal structure and associate with the cyclin box, this observation suggests that the interaction might be indirect and demands the structural integrity of the TRM relative to the cyclin. Alternatively, the TRM helix may loose its coordination to the cyclin boxes and become accessible to interact directly with the RNA moiety as suggested before.34,35 The latter suggestion is supported by the observation that the TRM together with residues from the first cyclin box repeat (K33, R51) forms a positively charged surface that could attract negatively charged RNA (Figure 3(b)). The Tat protein which is itself highly basic may contribute to this binding site from a rather complementary surface.
Figure 3. The Tat-TAR recognition motif in Cyclin T1.
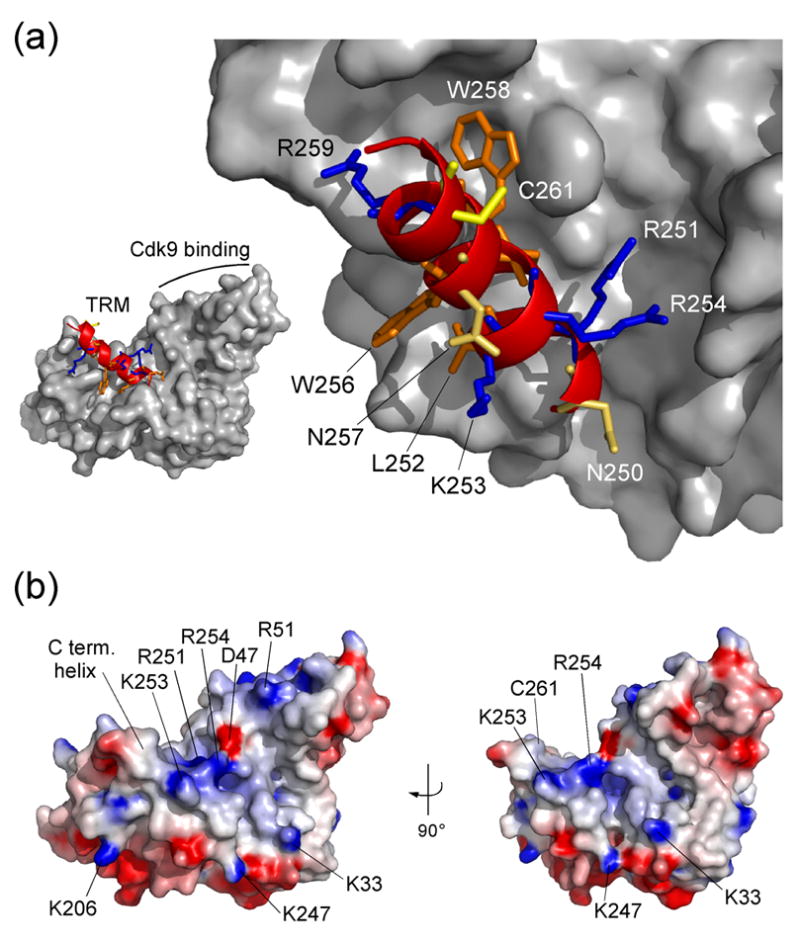
(a) Conformation of the C-terminal region from residue 247 to 263 as ribbon plot within the CycT1 structure. Positively charged residues K253 and R254, polar asparagines N250 and N257, and the aromatic W256 are accessible on the surface of the TRM structure. In contrast, side chains of R251, L252, I255 and W258 connect the C-terminal helix to the cyclin box repeats. Cystein 261 which might form an intermolecular zinc-finger with Tat is fully exposed on the surface. (b) Electrostatic surface potential of CycT1 (displayed from −8 kBT to +8 kBT). Basic residues of the TRM form a combinatorial surface with the first cyclin box repeat in between both cyclin boxes that could attract negatively charged RNA molecules of 7SK or TAR.
The CycT1-eqTat fusion protein is capable of TAR binding
The apparent flexibility of equine Tat in the crystals prompted us to analyse the functional activity of the fusion protein. We first performed electrophoretic mobility shift assays (EMSA) with a 30-mer EIAV TAR RNA fragment that was transcribed in vitro by the T7 promoter and polymerase system. Increasing concentrations of CycT1-eqTat led to a full shift of the RNA in the native gel at a slight molar excess of protein over RNA (Figure 4(a), lanes 1–6). These experiments were performed in the absence of EDTA and indicate the complex formation between CycT1-eqTat and EIAV TAR. Addition of EDTA to the CycT1-eqTat fusion protein prior to complex formation with TAR showed significantly less shifted RNA indicating the reduced ability to form a ternary complex (Figure 4(a), lanes 7, 8). These observations suggest that the presence of zinc in the CycT1-eqTat complex supports TAR binding, although the complex formation is not fully inhibited even at 20-fold higher concentration of EDTA over protein. We therefore conclude that the bacterially expressed CycT1-eqTat protein is capable of forming a specific complex with EIAV TAR which is sustained by the formation of an intermolecular zinc finger.
Figure 4. TAR-binding of the CycT1-Tat fusion protein and functional analysis of mutant CycT1.
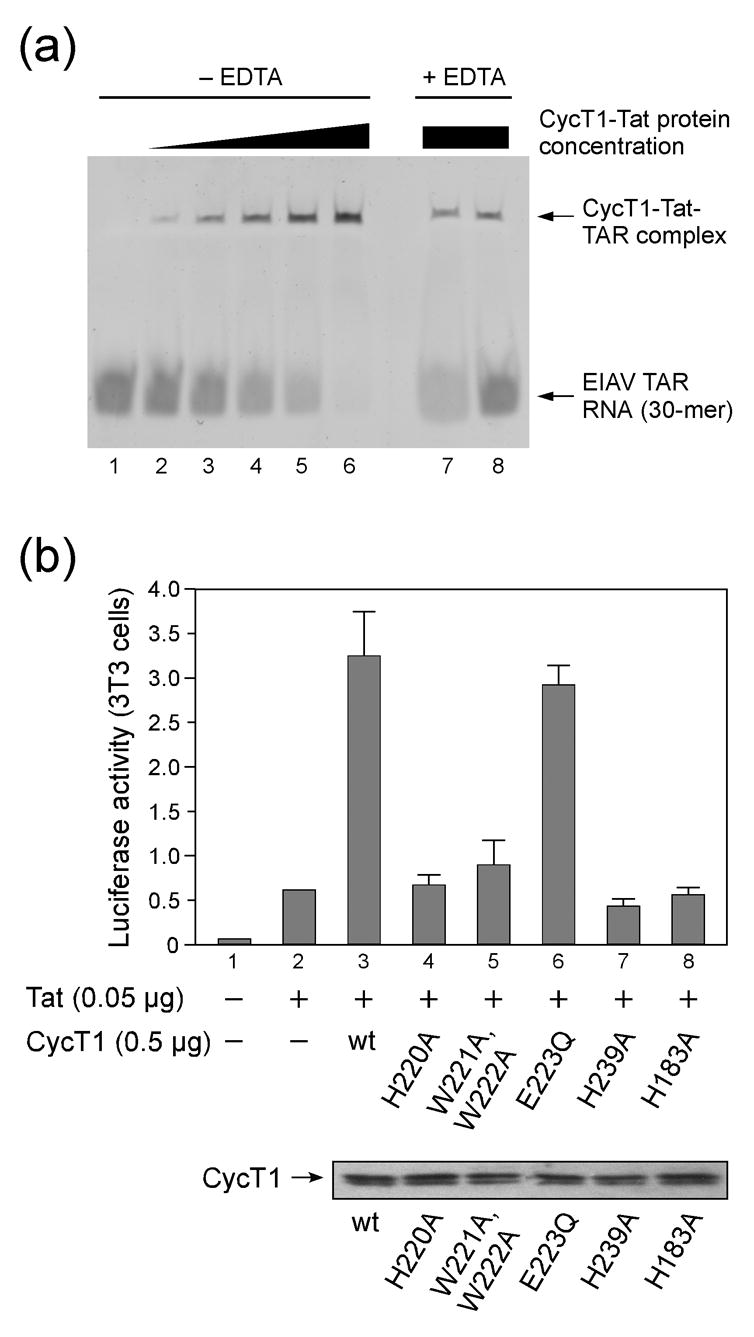
(a) Electrophoretic mobility shift assay of CycT1-eqTat with a 30-mer equine TAR RNA. Increasing concentrations from 1 to 5 μM of the fusion protein in a buffer solution containing equal concentrations of zinc led to a complete shift of the RNA, which was used at 2 μM concentration, indicating the complex formation with the protein (lanes 2 to 6). Addition of EDTA in four- and twenty-fold concentration over the zinc ion concentration diminished the complex formation (lanes 7 and 8). (b) Functional analysis of mutant CycT1 proteins in cells. Mutation of aromatic residues H220, W221 and W222 to alanine in the Cyclin T specific region around helices H3′ and H4′ of the second cyclin box repeat reduced transcriptional activity, as does H239 on helix H5′ adjacent to the TRM. Mutation of H183 which forms the core interface between the first and second cyclin box repeat similarly diminished the CycT1 activity. The lower panel presents expression levels of CycT1 proteins as indicated by the arrow.
We next performed functional experiments to analyse the effect of specific CycT1 mutants for transactivation. To this end, NIH 3T3 cells, in which the murine endogenous CycT1 is unable to support Tat-transactivation,3,36,37 were co-transfected with CycT1 and HIV-Luciferase in the presence or absence of Tat as described before.38 Transfection of both CycT1 and Tat led to a six-fold increase in Luciferase activity over Tat alone (Figure 4(b), lane 2, 3). Next, a series of mutants in helices H4′ and H5′ was analysed which are exposed on the second cyclin box repeat. In this CycT1-specific region bulky aromatic residues were chosen for mutation, since they often sustain RNA or DNA binding by stacking with the nucleotide bases and stabilize flexible regions. Histidine mutants H220A and H239A showed indeed impaired transcriptional activity as was similarly observed for the tryptophane double mutant W221A, W222A. In contrast, a glutamate mutation E223Q had no effect on transactivation. Finally, mutation of histidine 183 to alanine, which is in the core of the cyclin boxes interface, reduced the transcription activity to basal levels, suggesting that proper formation of the cyclin box fold is required for transcription activity. These data suggested, that residues in the second cyclin box repeat of CycT1 could sustain TAR binding in a cooperative mode with Tat and the TRM region.
Discussion
In this study, the crystallographic analysis of human CycT1 (1–281) fused to EIAV Tat (22–78) by a flexible linker of 20 residues revealed the structure of the cyclin box domain. The first canonical cyclin box repeat of CycT1 confirms the high structural homology to cyclin H, which is also shared by the more distantly related cyclins A and E (Figure 1). These similarities reflect the binding properties to cyclin-dependent kinases, whose interaction site is spanned by residues on helices H3, H5 and the interconnecting loop to the following cyclin box repeat. The second repeat instead showed a higher degree of divergence to other cyclins containing a unique insert between helices H3′ and H4′ that is less structured.
The viral transactivator protein, however, remained largely flexible in the complex and not defined in the electron density map. This observation is reminiscent of recent NMR studies on the HIV-1 Tat protein that found the Tat protein to be natively unfolded.39 Residues of the Tat-TAR recognition motif (TRM) in the C-terminal helix of CycT1 (250–262), however, were partly exposed on the surface and accessible for putative interactions. Particularly cysteine 261 which is thought to form an intermolecular zinc finger with cysteines of the Tat protein is accessible with its sulfur atom. Moreover, large gaps emerged at the site of the TRM motif in between the four molecules of the asymmetric unit that could accommodate the Tat component. The fusion protein was shown to be active in TAR binding and addition of EDTA diminished this interaction. While even folded species can in principle be disordered in crystals, the observation of the ill-defined Tat in our crystals is in line with the previous spectroscopic study.39 We therefore speculate that a stable tertiary structure of the coupling factor Tat, that goes beyond the coordination of the zinc ions, is only achieved upon simultaneous binding of both TAR RNA and the target protein CycT1.
The cyclins not only serve as Cdk-activating subunits but may also function in effector recognition through direct interaction with substrates or other regulating factors. In the crystal structures of the complex of Cdk2/CycA bound to its substrate peptides Cdc6, p107 or its inhibitor p27Kip1 a highly conserved RxL motif has been identified that coordinates the effectors to the cyclin molecule.13,10,9 This recruitment motif of the effector molecules is over 40 Å apart from the catalytic site of the kinase but acts in a cooperative mode with the kinase interacting motif to gain additional specificity for its recognition.13 The RxL motif was shown to interact with helices H1 and H3 of the first cyclin box repeat, bound in an extended conformation to the forthcoming helices. Although the key residue in RxL recognition, E220 of cyclin A, is the homologues residue D47 in CycT1, a specific structural feature of CycT1 may sterically inhibit such interaction. Compared to cyclins A and E, helix H4 of CycT1 is elongated by one additional turn and its succeeding loop. This segment is part of a T-type cyclin specific insert of 16 residues (compared to Cyclin A) in between helices H4 and H5 in the first cyclin box repeat (Figure 2). Hydrophobic interactions between helices H1 and H4 close the cleft between both helices and hide the subjacent helix H3, suggesting it unable to interact with RxL-based substrate motifs. This finding is in line with the observation that the CTD of RNAPII, which is the endogenous substrate of P-TEFb, contains no functional RxL motif close to its heptade region, while rather the histidine-rich segment around amino acid position 520 in CycT1 may coordinate the CTD.40 Thus, discrimination against different substrates and regulators of the various Cdk/cyclin pairs is achieved by individual conformational variability of the cyclin boxes.
But how might CycT1 recognize its regulating factors Hexim1 and 7SK snRNA? The conformation of the C-terminal TRM helix in CycT1 generates a parallel two-helical bundle together with helix H1 of the first cyclin box repeat that forms a positively charged groove (Figure 3). While the TRM seems rather loosely bound to the second cyclin box repeat, this groove could easily accommodate a helical structure as is the proposed coiled coil structure of the CycT-binding domain of Hexim1.41,42 Supporting evidence for such location of the Hexim1-binding site on CycT1 results from observations that both the P-TEFb regulating Hexim1 protein as well as the transcription stimulating Tat protein bind to CycT1 in a mutually exclusive manner.41,43 Besides the coupling proteins, the presence of the obligatory RNA partners 7SK for Hexim1 or TAR for Tat, respectively, were shown to sustain P-TEFb binding.19,20,2,25 These observations could be due to structural similarities in the stem loop folding of the two RNA molecules that may interact with similar binding sites on CycT1.44
But while Hexim1 necessarily requires the cyclin box repeats of CycT1 for P-TEFb regulation, it remains unclear how the kinase activity of Cdk9 is inhibited by Hexim1 and 7SK. It has been previously suggested that tyrosines at positions 203 or 271 of Hexim1 may block the ATP-binding site in Cdk9 similarly as a critical tyrosine in p27Kip1 was shown to inhibit the purine base coordination in the active centre of Cdk2.41,9 The spatial distance between these tyrosines and the putative CycT1 binding site of Hexim1 (284–313) should therefore be at least as large as for the substrate recognition assembly, in order for these tyrosines to locate at the kinase active centre. In the substrate Cdc6 there are about 20 residues in between the N-terminal (S/T)Px(K/R) kinase binding motif and the C-terminal RxL cyclin binding motif.13 In contrast, in p27Kip1 the amino acid chain follows a different path on the Cdk2/CycA structure with the inhibiting tyrosine being 58 residues apart downstream of the N-terminal RxL motif.9 The structure of the CycT1 cyclin box now defines the molecular basis for interacting factors that act catalytically on P-TEFb. Since the interaction between CycT1, Tat and TAR is an essential step in HIV-1 transcription, formation of this complex serves as an excellent target for anti-viral therapy.7,45 However, it is critical to identify and target viral specific pathways in order to develop safe and effective treatment, since P-TEFb regulates transcription of host cellular genes. Future structural studies on P-TEFb inhibiting and stimulating factors will be required to understand the molecular basis of transcription elongation regulation.
Materials and Methods
Plasmid cloning, protein expression and purification
An expression plasmid encoding human CycT1 (accession number AF048730) from aminoacid residues 1 to 281 was generated by PCR with primers containing NcoI (5′ sense primer) and SpeI (3′ antisense primer) restriction sites, respectively. A 20-residue linker GGT(GGGS)3GGGTS was encoded in the antisense primer at the 3′ end. EIAV Tat (acc. no. AF247394) from residue G22 to L78 was cloned with a SpeI restriction site at the 5′ end. Both fragments were cut with SpeI, ligated, again PCR amplified with primer containing NcoI and EcoRI restriction sites, and cloned into the pGEX-4T1 expression vector (Amersham Biosciences), modified with an additional TEV protease cleavage site, to construct a fusion protein with glutathione-S-transferase (GST). The clone was confirmed by DNA sequencing prior to expression.
For expression of the GST-CycT1-eqTat fusion protein the plasmid was transformed into BL21(DE3) E.coli cells (Novagen), expressed at 20°C and induced with 0.1 mM IPTG for overnight growth. Cells were lysed in buffer A (50 mM Tris/HCl pH 7.4, 500 mM NaCl, 5 mM β-mercaptoethanol) and cleared by spinning for 45 min at 30,000 g. The lysate was loaded onto a GSH-Sepharose column pre-equilibrated with buffer A containing 5% glycerol. After washing the column with buffer A containing 1 M NaCl and 5% glycerol, the GST-fusion protein was eluted with buffer A containing 10 mM glutathione, 5% glycerol. Peak fractions were dialysed in buffer A and the GST-tag was cleaved off at 4°C over 12 hrs with TEV protease, similarly as described.46 The CycT1-eqTat protein was further purified by size exclusion chromatography (S75 column) in 20 mM Tris/HCl (pH 7.4), 200 mM NaCl, 5 mM β-mercaptoethanol. Peak fractions containing CycT1-eqTat were analyzed by SDS-PAGE, concentrated to 20 mg/ml, aliquoted and stored at −80°C. Protein concentrations were determined by extinction coefficient measurements.
Crystallization
Initial crystals of CycT1-eqTat were obtained through a screening with the mosquito crystallization robot (TTP Labtech) using the sitting drop vapour diffusion technique in 96-well plate format with drop- and reservoir volumes of 0.2 and 100 μl, respectively. Crystals were routinely grown in 24-well-plates (Hampton Research) at 15°C by the hanging drop vapour diffusion technique. Equal volumes (1.5 μl/1.5 μl) of protein (20 mg/ml) and crystallization buffer (2.2 M ammonium sulphate. 5% of polyethylene glycol 400 and 50 mM MES pH 6.0) were mixed on a cover slip, which was subsequently suspended over a reservoir containing 500 μl of crystallization buffer. Clusters of small crystals appeared within 3 to 4 days; microseeding was used to obtain larger single crystals, with lowering the salt concentration in the reservoir solution to 1.9 M. The best crystals grew in about 10 days to a size of about 100 × 100 × 50 μm3. They were harvested by transfer to a cryoprotectant solution of 15% ethylene glycol in the mother-liquor, rapidly suspended in a nylon loop (Hampton Research) and flash-cooled in liquid nitrogen.
Data collection and crystal characterization
Data were collected at beamline ID29 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) using an ADSC Quantum Q105 CCD detector, at a wavelength of 0.9737 Å (Table 1). Crystals were kept at a temperature of 100 K in a stream of nitrogen gas for data collection. Data were processed and scaled using the XDS program package.47 The diffraction data initially suggested cubic crystals with space group I23, which implies the existence of 3-fold and 2-fold symmetry axes. However, analysis of the self rotation function showed that the 2-fold axes are not crystallographic but they are 2-fold twinning axes that run parallel to 2-fold non-crystallographic symmetry axes. Therefore, the system actually only has a crystallographic 3-fold axis with space group R3. The cell is represented by the hexagonal system with a = b = 203.83 Å, c = 124.79 Å, α = β = 90° and γ = 120°.
Table 1.
Data collection and refinement statistics.
| Beamline | ID29 ESRF Grenoble, France |
|---|---|
| Space group | R3 |
| Unit-cell parameter (Å) | a = 203.83, c = 124.79, γ = 120° |
| Wavelength (Å) | 0.9737 |
| Resolution range (Å)* | 100–2.67 (2.70–2.67) |
| Redundancy | 12.6 |
| Completeness (%) | 98.4 (95.2) |
| Rmerge (%) | 6.0 (32.3) |
| Rmeas. (%)a | 14.1 (70.0) |
| (I)/(Is) | 15.45 (3.21) |
| Rcryst (%); (no. of reflections) | 27.50 (53,216) |
| Rfree (%); (no. of reflections)b | 30.60 (1547) |
| Twin fraction | 0.187, 0.257, 0.178, 0.377 |
| R.m.s.d bond length (target) (Å) | 0.004 (0.005) |
| R.m.s.d bond angle distances (target) (Å) | 0.012 (0.012) |
| No. of protein atoms | 8,289 |
| No. of solvent atoms | 79 |
| Ramachandran plot (%)c | |
| Most favoured regions | 78.3 |
| Allowed regions | 20.4 |
| Generously allowed regions | 1.3 |
| Disallowed regions | 0.0 |
The measured R-factor was calculated as described.54
The data set for the calculation of Rfree included for each randomly selected reflection its twin-related mate and all symmetry equivalents. The target r.m.s. deviation from ideal values corresponds to an effective weighting of that restrained in least-squares calculations. R.m.s. deviation for bond angles expressed in terms of 1–3 separation.
As defined by PROCHECK.52
Values in brackets belong to the highest resolution shell.
Structure determination and refinement
Data analysis using the internet server by Yeates (1998) and by using the cumulative intensity distributions calculated with TRUNCATE (CCP4)48 indicated merohedral twinning in space group R3 as did reduced |E2−1| values from XPREP (Bruker AXS). The untreated twinned data were used for the structure determination. The unit cell dimensions, as well as the self-rotation function (CCP4), implied that several monomers were present in the asymmetric unit. A Matthews coefficient of 3.0 Å3/Da and a solvent content of 61% were obtained assuming four molecules in the asymmetric unit. The structure was solved by molecular replacement method using the program PHASER49 and the coordinates of cyclin T2 (2IVX, Debreczeni et al., Structural Genomics Consortium) as a search model. The model was refined using the program SHELXL97 (ref. 50), which uses the original twinned data for refinement, but de-twins the data for calculation of electron-density maps according to twin fraction (initial twinning ratios of 4 × 0.25), which is refined as well.
The test reflections (Rfree) were selected in thin resolution shells; to avoid possible correlations introduced by the twinning, all twin-related pairs belonged to either the reference or the working set49. Five conjugate-gradient cycles were performed using SHELXL97 (SHELXLPRO and SWAT, respectively). Except for rigid-body refinement, NCS restraints (NCSY option) for homologous regions of all four molecules were included in the refinement procedures. Several cycles of refinement of the twin fraction were performed. Equivalent torsion angles (absolute values) and atomic displacement parameters were restrained to similarity. The model was refined in alternating cycles of refinement and manual rebuilding using SHELXL97 and COOT (ref. 51), respectively. The quality of the structural model and its agreement with the structure factors were checked with programs PROCHECK (ref. 52). Data quality and refinement statistics are given in Table 1. The molecular diagrams were drawn using PYMOL (ref. 53).
Electrophoretic mobility shift assay
A 30-mer equine TAR RNA from nucleotide 217 to 246 of the LTR of EIAV (AC: AF247394) was generated with modified 5′ and 3′ ends (GGCCG and its reverse complement, respectively) for higher stability of the stem loop structure. The RNA was transcribed in vitro from an antisense single-stranded DNA oligonucleotide using the T7 promotor and polymerase system (Bioline). EMSAs were performed by incubating 2 μM of equine TAR RNA (192 ng TAR in 10 μl buffer solution) with increasing concentrations from 1 to 5 μM CycT1-eqTat fusion protein for 20 min at RT. Samples were subjected to 5% acrylamide gel electrophoresis and stained with ethidium bromide.
Transfection and luciferase assay
NIH 3T3 cells (0.1 million cells in 24 well plate) were co-transfected with 0.5 μg of pEF-CycT1 (1–280, wt or mutant constructs) and HIV-Luciferase, in the presence or absence of pTat (0.01 μg) using lipofectamine 2000 according to the manufacturer’s instruction (Invitrogen). Forty-eight hours after transfection, cells were harvested and lysed in passive lysis buffer (Promega). The protein concentration of the cell lysates were determined by Protein Assay kit (BioRad). Luciferase activities in the cell lysate were measured as described before.38
Coordinates
Atomic coordinates and the structure factors have been deposited in the Protein Data Bank (accession code 2PK2).
Acknowledgments
We thank Sabine Wohlgemuth for initial assistance on protein expression and purification, Karin Vogel-Bachmayr for RNA transcription and purification and Matija Peterlin and Ran Taube for stimulating discussions. We thank Dr. Carlos Frazao and Dr. Zbigniew Dauter for extensive discussion on crystal twinning. M.G. thanks Roger Goody for continuous support. K.F. is supported by NIH R21 AI62516 and the Center for AIDS Research (CFAR) at Case Western Reserve University. This work was supported by an EMBO long-term fellowship to K.A. and a DFG grant (GE-976/2) to M.G.
Abbreviations used
- CDK
cyclin-dependent kinases
- CTD
carboxy-terminal domain
- CycT1
Cyclin T1
- EIAV
equine infectious anemia virus
- eqTat
equine Tat
- HIV-1
human immunodeficiency virus type 1
- P-TEFb
positive transcription elongation factor b
- TAR
transactivation response element
- Tat
transcriptional transactivator protein
- TRM
Tat/TAR recognition motif
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jones KA, Peterlin BM. Control of RNA initiation and elongation at the HIV-1 promoter. Annu Rev Biochem. 1994;63:717–743. doi: 10.1146/annurev.bi.63.070194.003441. [DOI] [PubMed] [Google Scholar]
- 2.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 3.Garber ME, Wei P, KewalRamani VN, Mayall TP, Herrmann CH, Rice AP, Littman DR, Jones KA. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998;12:3512–3527. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 5.Shilatifard A, Conaway RC, Conaway JW. The RNA polymerase II elongation complex. Annu Rev Biochem. 2003;72:693–715. doi: 10.1146/annurev.biochem.72.121801.161551. [DOI] [PubMed] [Google Scholar]
- 6.Saunders A, Core LJ, Lis JT. Breaking barriers to transcription elongation. Nat Rev Mol Cell Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 7.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 9.Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 10.Brown NR, Noble ME, Endicott JA, Johnson LN. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat Cell Biol. 1999;1:438–443. doi: 10.1038/15674. [DOI] [PubMed] [Google Scholar]
- 11.Schulze-Gahmen U, Kim SH. Structural basis for CDK6 activation by a virus-encoded cyclin. Nat Struct Biol. 2002;9:177–181. doi: 10.1038/nsb756. [DOI] [PubMed] [Google Scholar]
- 12.Honda R, Lowe ED, Dubinina E, Skamnaki V, Cook A, Brown NR, Johnson LN. The structure of cyclin E1/CDK2: implications for CDK2 activation and CDK2-independent roles. EMBO J. 2005;24:452–463. doi: 10.1038/sj.emboj.7600554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng KY, Noble ME, Skamnaki V, Brown NR, Lowe ED, Kontogiannis L, Shen K, Cole PA, Siligardi G, Johnson LN. The role of the phospho-CDK2/cyclin A recruitment site in substrate recognition. J Biol Chem. 2006;281:23167–23179. doi: 10.1074/jbc.M600480200. [DOI] [PubMed] [Google Scholar]
- 14.Peng J, Zhu Y, Milton JT, Price DH. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998;12:755–762. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meinhart A, Kamenski T, Hoeppner S, Baumli S, Cramer P. A structural perspective of CTD function. Genes Dev. 2005;19:1401–1415. doi: 10.1101/gad.1318105. [DOI] [PubMed] [Google Scholar]
- 16.Marshall NF, Peng J, Xie Z, Price DH. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- 17.Wada T, Takagi T, Yamaguchi Y, Watanabe D, Handa H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J. 1998;17:7395–7403. doi: 10.1093/emboj/17.24.7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garriga J, Bhattacharya S, Calbo J, Marshall RM, Truongcao M, Haines DS, Grana X. CDK9 is constitutively expressed throughout the cell cycle, and its steady-state expression is independent of SKP2. Mol Cell Biol. 2003;23:5165–5173. doi: 10.1128/MCB.23.15.5165-5173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, Lania L, Bensaude O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol Cell Biol. 2003;23:4859–4869. doi: 10.1128/MCB.23.14.4859-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 21.Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, DeMayo FJ, Schneider MD. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med. 2002;8:1310–1317. doi: 10.1038/nm778. [DOI] [PubMed] [Google Scholar]
- 22.Wittmann BM, Fujinaga K, Deng H, Ogba N, Montano MM. The breast cell growth inhibitor, estrogen down regulated gene 1, modulates a novel functional interaction between estrogen receptor alpha and transcriptional elongation factor cyclin T1. Oncogene. 2005;24:5576–5588. doi: 10.1038/sj.onc.1208728. [DOI] [PubMed] [Google Scholar]
- 23.Karn J. Tackling Tat. J Mol Biol. 1999;293:235–254. doi: 10.1006/jmbi.1999.3060. [DOI] [PubMed] [Google Scholar]
- 24.Taube R, Fujinaga K, Wimmer J, Barboric M, Peterlin BM. Tat transactivation: a model for the regulation of eukaryotic transcriptional elongation. Virology. 1999;264:245–253. doi: 10.1006/viro.1999.9944. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Tamilarasu N, Hwang S, Garber ME, Huq I, Jones KA, Rana TM. HIV-1 TAR RNA enhances the interaction between Tat and cyclin T1. J Biol Chem. 2000;275:34314–34319. doi: 10.1074/jbc.M006804200. [DOI] [PubMed] [Google Scholar]
- 26.Kaehlcke K, Dorr A, Hetzer-Egger C, Kiermer V, Henklein P, Schnoelzer M, Loret E, Cole PA, Verdin E, Ott M. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol Cell. 2003;12:167–176. doi: 10.1016/s1097-2765(03)00245-4. [DOI] [PubMed] [Google Scholar]
- 27.Taube R, Fujinaga K, Irwin D, Wimmer J, Geyer M, Peterlin BM. Interactions between equine cyclin T1, Tat, and TAR are disrupted by a leucine-to-valine substitution found in human cyclin T1. J Virol. 2000;74:892–898. doi: 10.1128/jvi.74.2.892-898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albrecht TR, Lund LH, Garcia-Blanco MA. Canine cyclin T1 rescues equine infectious anemia virus tat trans-activation in human cells. Virology. 2000;268:7–11. doi: 10.1006/viro.1999.0141. [DOI] [PubMed] [Google Scholar]
- 29.Fujinaga K, Irwin D, Taube R, Zhang F, Geyer M, Peterlin BM. A minimal chimera of human cyclin T1 and Tat binds TAR and activates human immunodeficiency virus transcription in murine cells. J Virol. 2002;76:12934–12939. doi: 10.1128/JVI.76.24.12934-12939.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersen G, Busso D, Poterszman A, Hwang JR, Wurtz JM, Ripp R, Thierry JC, Egly JM, Moras D. The structure of cyclin H: common mode of kinase activation and specific features. EMBO J. 1997;16:958–967. doi: 10.1093/emboj/16.5.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noble ME, Endicott JA, Brown NR, Johnson LN. The cyclin box fold: protein recognition in cell-cycle and transcription control. Trends Biochem Sci. 1997;22:482–487. doi: 10.1016/s0968-0004(97)01144-4. [DOI] [PubMed] [Google Scholar]
- 32.Hoeppner S, Baumli S, Cramer P. Structure of the mediator subunit cyclin C and its implications for CDK8 function. J Mol Biol. 2005;350:833–842. doi: 10.1016/j.jmb.2005.05.041. [DOI] [PubMed] [Google Scholar]
- 33.Baek K, Brown RS, Birrane G, Ladias JA. Crystal structure of human Cyclin K, a positive regulator of cyclin-dependent kinase 9. J Mol Biol. 2007;366:563–573. doi: 10.1016/j.jmb.2006.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das C, Edgcomb SP, Peteranderl R, Chen L, Frankel AD. Evidence for conformational flexibility in the Tat-TAR recognition motif of cyclin T1. Virology. 2004;318:306–317. doi: 10.1016/j.virol.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Richter S, Ping YH, Rana TM. TAR RNA loop: a scaffold for the assembly of a regulatory switch in HIV replication. Proc Natl Acad Sci USA. 2005;99:7928–7933. doi: 10.1073/pnas.122119999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998;17:7056–7065. doi: 10.1093/emboj/17.23.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujinaga K, Taube R, Wimmer J, Cujec TP, Peterlin BM. Interactions between human cyclin T, Tat, and the transactivation response element (TAR) are disrupted by a cysteine to tyrosine substitution found in mouse cyclin T. Proc Natl Acad Sci USA. 1999;96:1285–1290. doi: 10.1073/pnas.96.4.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol. 2004;24:787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shojania S, O’Neil JD. HIV-1 Tat is a natively unfolded protein: The solution conformation and dynamics of reduced HIV-1 Tat (1–72) by NMR spectroscopy. J Biol Chem. 2006;281:8347–8356. doi: 10.1074/jbc.M510748200. [DOI] [PubMed] [Google Scholar]
- 40.Taube R, Lin X, Irwin D, Fujinaga K, Peterlin BM. Interaction between P-TEFb and the C-terminal domain of RNA polymerase II activates transcriptional elongation from sites upstream or downstream of target genes. Mol Cell Biol. 2002;22:321–331. doi: 10.1128/MCB.22.1.321-331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schulte A, Czudnochowski N, Barboric M, Schonichen A, Blazek D, Peterlin BM, Geyer M. Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J Biol Chem. 2005;280:24968–24977. doi: 10.1074/jbc.M501431200. [DOI] [PubMed] [Google Scholar]
- 42.Li Q, Price JP, Byers SA, Cheng D, Peng J, Price DH. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J Biol Chem. 2005;280:28819–28826. doi: 10.1074/jbc.M502712200. [DOI] [PubMed] [Google Scholar]
- 43.Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, Geyer M, Peterlin BM, Zhou Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007;35:2003–2012. doi: 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Egloff S, Van Herreweghe E, Kiss T. Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol Cell Biol. 2006;26:630–642. doi: 10.1128/MCB.26.2.630-642.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murchie AI, Davis B, Isel C, Afshar M, Drysdale MJ, Bower J, Potter AJ, Starkey ID, Swarbrick TM, Mirza S, Prescott CD, Vaglio P, Aboul-ela F, Karn J. Structure-based drug design targeting an inactive RNA conformation: exploiting the flexibility of HIV-1 TAR RNA. J Mol Biol. 2004;336:625–638. doi: 10.1016/j.jmb.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 46.Schonichen A, Alexander M, Gasteier JE, Cuesta FE, Fackler OT, Geyer M. Biochemical characterization of the diaphanous autoregulatory interaction in the formin homology protein FHOD1. J Biol Chem. 2006;281:5084–5093. doi: 10.1074/jbc.M509226200. [DOI] [PubMed] [Google Scholar]
- 47.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 48.Collaborative Computational Project number 4 (CCP4) Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 49.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Cryst. 2004;D60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 50.Sheldrick GM, Schneider TR. SHELXL: High-resolution refinement. Meth Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 51.Emsley P, Cowtan K. COOT: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 52.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 53.DeLano WL. The PYMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]
- 54.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]