

Abstract
There is a dire need for novel therapeutics to treat the dangerous malarial parasite, Plasmodium falciparum. Recently, the X-ray crystal structure of enoyl-acyl carrier protein reductase (ENR) in complex with triclosan has been determined and provides an opportunity for the rational design of novel inhibitors targeting the active site of ENR. Here we report the discovery of several compounds by virtual screening and their experimental validation as high potency PfENR inhibitors.
Introduction
Malaria is the second leading cause of death in the world, after tuberculosis. Of the four species in the Plasmodium genus that causes malaria, Plasmodium falciparum is the most widespread in humans and is the predominant cause of severe disease and death. Traditionally, the antimalarial drugs chloroquine and sulfadoxine have been used to treat malaria by targeting heme polymerase. Unfortunately, the chronic use of these antimalarials has slowly led to resistance in P. falciparum, rendering the drugs much less effective, and ultimately resulting in treatment failure; choloquine and sulfadoxine have shown failure rates of 10-60% [1].
To circumvent these issues, a key malarial enzyme, enoyl-acyl carrier protein reductase (ENR) has been investigated as an attractive target because it plays an important role in membrane construction and energy production in the parasite, and does not have any human homologues. ENR was originally identified as a target for Mycobacterium tuberculosis [4,5] and E. coli [6], and has recently been proposed as a target for P. falciparum [7]. A widely used antibiotic, triclosan is a potent inhibitor of PfENR enzyme activity and Plasmodium growth both in vitro and in vivo [8]. Triclosan has been approved for use as a topical antibacterial drug and can be found in many soaps and toothpastes. It has been shown to be effective in reducing and controlling bacterial contamination on the hands and on treated products. However, triclosan is not orally bioavailable. A number of nanotechnological approaches were recently evaluated for the development of an oral delivery method capable of systemic release of triclosan [9]. In an effort to optimize triclosan binding, two chemical derivatives were found to exhibit biochemical inhibition of PfENR [7]. Unfortunately, these derivatives showed activities no better than triclosan itself.
The lack of suitable alternatives to replace chloroquine and sulfadoxine, and the limited extent to which triclosan can be optimized, threatens efforts to control the rapid spread of drug-resistant strains of this parasite, and underscores the need for pharmaceutical discoveries to overcome existing barriers [10]. Recently, the X-ray crystal structure of PfENR was determined in complex with triclosan [7]. This gave a clear description of the active site and the mode of interaction of triclosan with the target enzyme. This information motivated a rational design approach to develop alternatives to triclosan that target the ENR enzyme of P. falciparum. Here we report a virtual screen against PfENR, which resulted in a series of compounds showing enzymatic inhibition of PfENR and growth inhibition of P. falciparum cell culture within an order of magnitude of triclosan. These novel molecular scaffolds present an opportunity for further optimization into more potent antimalarial compounds.
Materials and Methods
VLS method
The ICM program [16] was used to screen the ChemBridge Express database (San Diego, CA) for chemical compounds that were able to fit into the ENR pocket. The VLS method has been described previously [15]. Briefly, it uses Monte Carlo global energy optimization for a flexible ligand and a rigid receptor represented as energy maps. The procedure combines large-scale random moves with gradient local minimization at every step. The scoring function then discriminates a small number of binders from hundreds of thousands of non-binders.
The crystal structure of ENR (1VRW) was used as the target for VLS. The coordinates of the 2.43 Å resolution structure include the NAD+ cofactor. It is believed that any newly discovered small-molecule inhibitor may interact with NAD+, and therefore this cofactor was retained in the active site during VLS. The choice of active site residues and calculated maps were obtained from a pocket modeling analysis of ENR (unpublished results). This putative inhibitor binding pocket includes the following residues from chain B: 106, 111, 131, 133, 134, 216-220, 222, 223, 237, 241, 266-269, 274, 277, 278, 281, 285, 312-323 as well as residues 368, 369, 372, and 373 from chain D.
VLS was run on 336,600 compounds from the ChemBridge Express Library (San Diego, CA). This library was used because it is enriched with drug-like compounds based on 3D pharmacophore analysis. During the course of VLS, the method produces 3-D coordinates of the best docking pose. These coordinates were displayed in the active site of ENR for analysis and comparison. As a benchmark for hit scoring, triclosan, a known inhibitor, was docked into the ENR active site pocket. This virtual dock produced a score of −40, and thus was used as a minimum score for novel binders. (Smaller numbers indicate a better score). Compounds scoring better than −50 were clustered by chemical similarity. Structurally similar compounds noted by visual inspection were then eliminated to reduce redundancy and increase diversity. This screen resulted in 750 compounds showing ICM scores better that −50.
Next, these compounds were subjected to a theoretical measure of toxicity by submission to PreADMET [17], a web-based application for predicting ADME data. This ADME prediction considers two indicators of cell permeability, Caco-2 and MDCK. Selecting for compounds with high Caco-2 scores (>7) yields a list of 260 compounds. Further screening this list for compounds with high MDCK scores (>50) narrowed the list to 169 compounds. These compounds were experimentally evaluated for their inhibitory effect on ENR activity.
PfENR Expression and Purification
The P. falciparum ENR was cloned as previously described [7]. BL21(DE3) Codon+-RIL cells (Novagen) harboring the expression plasmids were grown in Terrific broth. When the A600 reached 0.8, the cells were induced with 1 mM isopropyl-1-thio-β-D-galactopyranoside for 5 h at 37 °C. Cell pellets were resuspended in buffer A (20 mM Tris/HCl, pH 8.0, 500 mM NaCl, 50 mM imidazole) and disrupted using a French press. The filtered supernatant was applied to a metal chelate affinity column loaded with nickel. The column was washed with buffer B (20 mM Tris/HCl, pH 8.0, 500 mM NaCl, 150 mM imidazole) and eluted with buffer C (20 mM Tris/HCl, pH 8.0, 500 mM NaCl, 400 mM imidazole). The protein was concentrated using Centriprep 30 and applied to a Superdex 75 size-exclusion column equilibrated with buffer D (20 mM Tris/HCl, pH 7.5, 150 mM NaCl).
Enzyme Assay
All experiments were carried out on a Shimadzu UV-1201 UV-visible spectrophotometer at 25 °C in 20 mM Tris/HCl, pH 7.6, 150 mM NaCl. Kinetic parameters were determined spectrophotometrically by following the oxidation of NADH to NAD+ at 340 nm (ε = 6.3 mM−1cm−1). Km and Vmax values for crotonoyl-CoA were determined at a fixed and saturating concentration of NADH (200 μM) and by varying the substrate concentration (0–500 μM). Km and Vmax values for NADH were determined at variable concentrations of NADH and a fixed and saturating concentration of crotonoyl-CoA (500 μM). Kinetic parameters were obtained by fitting the initial velocity data to the Michaelis-Menten equation.
Inhibition constants were determined under conditions of saturating substrate (500 μM crotonoyl-CoA, 200 μM NADH) and variable inhibitor concentration. Values for Ki were determined from the x-intercept of a Dixon plot, assuming uncompetitive inhibition. Mean values of two independent experiments are reported for kinetic parameters and inhibition data.
Whole Cell Assay
Parasite susceptibilities to antimalarial drugs were measured in vitro using [3H]-hypoxanthine assays [10]. Briefly, sorbitol-synchronized predominantly ring-stage cultures were seeded in duplicate in 96-well plates at a parasitemia and hematocrit of 0.4% and 1.6% respectively. [3H]-hypoxanthine (0.5 μCi per well) was added after 48 hr, and cells were harvested after a further 24 hr. IC50 values were determined by extrapolation from the linear portion of the dose-response curve, as previously described [10]. All compounds tested in this study were obtained from ChemBridge (San Diego, CA).
Results and Discussion
Virtual Library Screening
Virtual Library Screening (VLS) is rapidly becoming a popular alternative to high throughput screening (HTS). VLS is much cheaper and less time consuming, and allows researchers to narrow the search to a relatively small set of compounds, which are further experimentally validated in enzymatic and cell-based assays. We used the versatile computational program, ICM, to identify novel chemical scaffolds from which potent inhibitors against PfENR can be developed. The VLS method in ICM has shown considerable success with protein kinases [11,12], and has been used to discover antagonists for the thyroid hormone receptor [13], and EGFR [14]. The procedure is based on the fast docking of a flexible ligand to a target receptor pocket represented by energy maps. The scoring method uses the ligand-receptor interaction energy, conformational strain energy, conformational entropy loss, and desolvation effects [15]. ICM's VLS module was run on the ChemBridge database according to the method described in the Experimental Section, and resulted in 750 compounds with scores better that −50. Theoretical ADME prediction of this list resulted in 169 compounds, which were then tested experimentally.
PfENR and whole-cell assay
The 169 compounds generated by virtual screening were examined with an enzyme activity assay for their ability to inhibit ENR. Of these, 16 compounds showed > 45% inhibition at 50μM. The chemical structure of these compounds as well as their inhibition rates are shown in Table I.
Table 1.
Name, chemical structure, enzyme inhibition rates, and cell growth inhibition rates for triclosan and top 16 compounds arising from enzyme inhibition studies. For each compound, two separate cell lines (Dd2 and 3D7) and two inhibitory measurements (half-maximal and 90% maximal) were used for the whole cell experiments.
| Compound | Chemical Structure | % PfENR Activity Left at 50μM Inhibitor |
Cell line |
IC50 (μM) |
IC90 (μM) |
|---|---|---|---|---|---|
| Triclosan | 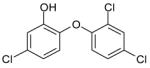 |
0.2 | Dd2 | 4.45 | 7.85 |
| 3D7 | 4.45 | 8.51 | |||
| CHBR7682275 |  |
3 | Dd2 | >100 | >100 |
| 3D7 | >100 | >100 | |||
| CHBR6842511 |  |
9 | Dd2 | >100 | >100 |
| 3D7 | >100 | >100 | |||
| CHBR6401523 | 17 | Dd2 | 74.33 | >100 | |
| 3D7 | >100 | >100 | |||
| CHBR5964423 |  |
20 | Dd2 | >100 | >100 |
| 3D7 | >100 | >100 | |||
| CHBR5217961 |  |
24 | Dd2 | 5.98 | 12.21 |
| 3D7 | 5.98 | 12.44 | |||
| CHBR5344752 | 25 | Dd2 | >100 | >100 | |
| 3D7 | >100 | >100 | |||
| CHBR6671761 | 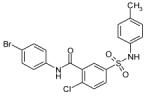 |
41 | Dd2 | 42.58 | 88.35 |
| 3D7 | 41.07 | 83.08 | |||
| CHBR5302316 | 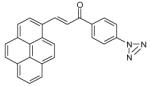 |
45 | Dd2 | >100 | >100 |
| 3D7 | >100 | >100 | |||
| CHBR5523126 |  |
46 | Dd2 | 73.65 | 107.79 |
| 3D7 | 78.78 | 110.31 | |||
| CHBR5927013 |  |
47 | Dd2 | >100 | >100 |
| 3D7 | >100 | >100 | |||
| CHBR5210857 | 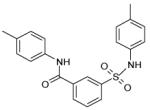 |
49 | Dd2 | 35.49 | 72.75 |
| 3D7 | 35.57 | 72.76 | |||
| CHBR5221057 | 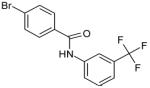 |
49 | Dd2 | 24.90 | 36.45 |
| 3D7 | 24.87 | 35.31 | |||
| CHBR5985919 | 49 | Dd2 | >100 | >100 | |
| 3D7 | >100 | >100 | |||
| CHBR5217954 |  |
49 | Dd2 | 9.14 | 13.48 |
| 3D7 | 11.27 | 23.61 | |||
| CHBR7671066 |  |
52 | Dd2 | 70.92 | 105.69 |
| 3D7 | 83.52 | >100 | |||
| CHBR6875566 |  |
53 | Dd2 | >100 | >100 |
| 3D7 | >100 | >100 |
The 16 promising compounds arising from the inhibition assay were tested in cell-based experiments (see Experimental Section). Three of these compounds, CHBR5217961, CHBR5217954, and CHBR5221057, showed IC50 values of 5.98μM, 9.14μM, and 24.90μM, respectively (Table I), which fall within an order of magnitude of triclosan (4.45μM).
Structural Modeling
The three top binders were modeled into the active site of ENR to examine interactions with protein residues. Structural analysis of the predicted binding poses of these compounds reveals important information. CHBR5217961 occupies a portion of the active site flanked on one side by the linker regions between β4-α5, as well as β5-α6, and on the other side by helix α7 (nomenclature from Perozzo, et al. 2002). Specifically, a hydrogen bonding interaction is observed between CHBR5217961 atom N2 and the phenolic hydroxyl atom of PfENR Tyr-277 (Figure I). Van der Waals contacts are also observed between the piperidinylsulfonyl of CHBR5217961 and Val-222. In addition, the hydroxybenzylidene ring of CHBR5217961 forms hydrophobic packing interactions with Tyr-267 of PfENR. Hydrophobic contacts also occur between the ligand benzohydrazide moiety and Asn-218.
Figure 1.
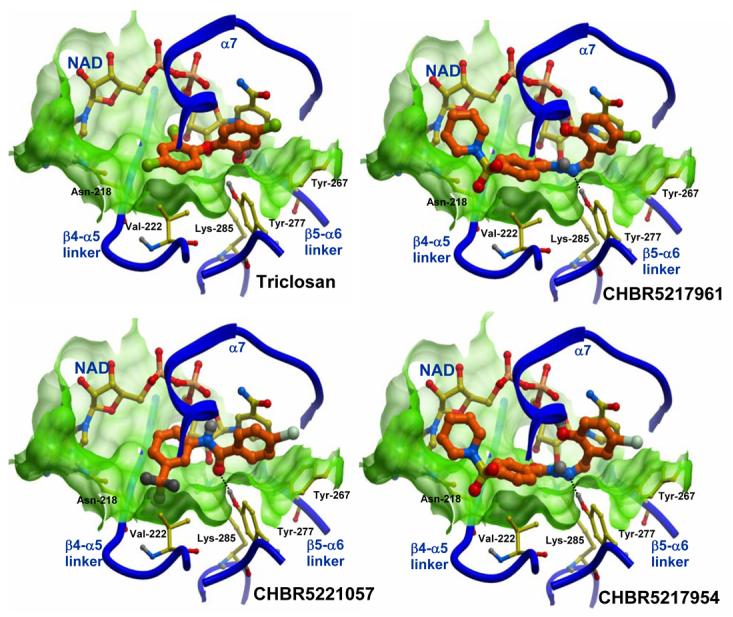
Zoom-in of the top three compounds from the whole cell experiments docked into the active site of ENR, and compared to the docked structure of triclosan. A skin-mesh representation of the active site pocket encompassing each ligand and the NAD+ cofactor is displayed in light-green. Regions of the active site not interacting with the inhibitors are omitted for clarity. Water molecules were not included in VLS experiments and therefore not shown. Hydrogen bonds are displayed with dotted lines. Carbon atoms of the enzyme and of NAD+ are colored yellow; each compound has its carbon atoms colored orange and is displayed with thicker bonds.
CHBR5221057 lies in the same active site groove and is positioned in a similar orientation as CHBR5217961. Atom O1 of the bromo-benzamide moiety in the inhibitor forms hydrogen bonding interaction with Tyr-277 of the protein. However, this compound does not extend as far towards the linker region between the β4 sheet and helix α5 and subsequently does not interact with Asn-218 as is the case with the other two inhibitors.
The CHBR5217954 inhibitor is similar to CHBR5217961, differing only at position 1 where a bromine atom is in place of a chlorine. Not surprisingly, the inhibitor overlays almost identically with that of CHBR5217961. As in CHBR5217961, atom N2 of the CHBR5217954 inhibitor forms a hydrogen bond with the hydroxyl atom of PfENR Tyr-277. Hydrophobic packing interactions occur between the inhibitor and residues Asn-218, Val-222, and Tyr-267 of the protein.
All three of the compounds overlap similarly to triclosan as viewed in a comparison (Figure I). Similar to triclosan, all three of the inhibitors selected for testing display a trio of edge face aryl-aryl interactions between an aromatic group in the compound, the nicotinamide ring in NAD+, and residues Tyr-267 and Tyr-277 of PfENR. These interactions are seemingly important to the affinity of the compound for the enzyme active site and may assist in stability of the complex. Tyr-277, along with Lys-285, have been proposed to be important to the catalytic mechanism of the enzyme [7]. It is likely that the hydrogen bond between the compounds and Tyr-277 of the enzyme prevents this residue from interacting with its natural substrate. It can therefore be proposed that a successful ENR inhibitor needs to interact with Tyr-277, and thus occupy it from interacting with its substrate, in order to block its role in catalysis.
It is also important to note that two of the inhibitors, CHBR5217961 and CHBR5217954, have bulky moieties protruding towards the ENR β4-α5 linker region, which is not present in the CHBR5221057 compound. Interestingly, compounds CHBR5217961 and CHBR5217954 have inhibitory rates almost double that of CHBR5221057. It is appealing to speculate that the extra bulky group may give those compounds a “handle” by which the enzyme may better position them within the active site groove. However, triclosan is considerably smaller than the tested compounds and does not contain a bulky group near the β4-α5 region.
Triclosan is unique among antimicrobials in that it forms a non-covalent complex with ENR, primarily via hydrogen bonds. Similarly, the inhibitors identified in the current study interact with ENR primarily through hydrogen bonds and hydrophobic interactions. These compounds have shown inhibitory potency similar to triclosan and may be better suited for oral bioavailability. To our knowledge, this is the first time virtual screening has been successfully applied to a malarial target. These promising inhibitors merit further investigation in vivo as potential therapeutic candidates.
Supplementary Material
Acknowledgments
This work was funded in part by NIH research grant 5R01GM071872-03 to RA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO World Malaria Report. World Health Organization; Geneva, Switzerland: 2005. [Google Scholar]
- 2.Freundlich JS, Anderson JW, Sarantakis D, Shieh HM, Yu M, et al. Synthesis, biological activity, and X-ray crystal structural analysis of diaryl ether inhibitors of malarial enoyl acyl carrier protein reductase. Part 1: 4′-substituted triclosan derivatives. Bioorg Med Chem Lett. 2005;15:5247–5252. doi: 10.1016/j.bmcl.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 3.Bray PG, Martin RE, Tilley L, Ward SA, Kirk K, et al. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol Microbiol. 2005;56:323–333. doi: 10.1111/j.1365-2958.2005.04556.x. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 5.Dessen A, Quemard A, Blanchard JS, Jacobs WR, Jr., Sacchettini JC. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science. 1995;267:1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- 6.Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, et al. Protein EnvM is the NADH-dependent enoyl-ACP reductase (FabI) of Escherichia coli. J Biol Chem. 1994;269:5493–5496. [PubMed] [Google Scholar]
- 7.Perozzo R, Kuo M, Sidhu AS, Valiyaveettil JT, Bittman R, et al. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J Biol Chem. 2002;277:13106–13114. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 8.Surolia N, Surolia A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat Med. 2001;7:167–173. doi: 10.1038/84612. [DOI] [PubMed] [Google Scholar]
- 9.Maestrelli F, Mura P, Alonso MJ. Formulation and characterization of triclosan sub-micron emulsions and nanocapsules. J Microencapsul. 2004;21:857–864. doi: 10.1080/02652040400015411. [DOI] [PubMed] [Google Scholar]
- 10.Fidock DA, Nomura T, Wellems TE. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol Pharmacol. 1998;54:1140–1147. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- 11.Kovacs JA, Chacon P, Abagyan R. Predictions of protein flexibility: first-order measures. Proteins. 2004;56:661–668. doi: 10.1002/prot.20151. [DOI] [PubMed] [Google Scholar]
- 12.Cavasotto CN, Kovacs JA, Abagyan RA. Representing receptor flexibility in ligand docking through relevant normal modes. J Am Chem Soc. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 13.Schapira M, Raaka BM, Das S, Fan L, Totrov M, et al. Discovery of diverse thyroid hormone receptor antagonists by high-throughput docking. Proc Natl Acad Sci U S A. 2003;100:7354–7359. doi: 10.1073/pnas.1131854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavasotto CN, Ortiz MA, Abagyan RA, Piedrafita FJ. In silico identification of novel EGFR inhibitors with antiproliferative activity against cancer cells. Bioorg Med Chem Lett. 2006 doi: 10.1016/j.bmcl.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 15.Abagyan R, Totrov M. High-throughput docking for lead generation. Curr Opin Chem Biol. 2001;5:375–382. doi: 10.1016/s1367-5931(00)00217-9. [DOI] [PubMed] [Google Scholar]
- 16.Abagyan R, Totrov M, Kuznetsov DA. ICM: A New Method for Protein Modeling and Design: Applications to Docking and Structure Prediction from the Distorted Native Conformation. J Comp Chem. 15:488–506. [Google Scholar]
- 17.Lee SK, Lee IH. The PreADME approach: Web-based program for rapid prediction of physiochemical, drug absorption and drug-like properties. Bournemouth, UK: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.