

Abstract
Mycobacteria contain a large number of redundant genes whose functions are difficult to analyze in mutants because there are only two efficient antibiotic resistance genes available for allelic exchange experiments. Sequence-specific recombinbases such as the Flp recombinase can be used to excise resistance markers. Expression of the flpe gene from Saccharomyces cerevisiae is functional for this purpose in fast-growing Mycobacterium smegmatis but not in slow-growing mycobacteria such as M. bovis BCG or M. tuberculosis. We synthesized the flpm gene by adapting the codon usage to that prefered by M. tuberculosis. This increased the G+C content from 38% to 61%. Using the synthetic flpm gene, the frequency of removal of FRT-hyg-FRT cassette from the chromosome by the Flp recombinase was increased by more than 100-fold in M. smegmatis. In addition, 40% of all clones of M. bovis BCG had lost the hyg resistance cassette after transient expression of the flpm gene. Sequencing of the chromosomal DNA showed that excision of the FRT-hyg-FRT cassette by Flp was specific. These results show that the flpm encoded Flp recombinase is not only an improved genetic tool for M. smegmatis, but can also be used in slow growing mycobacteria such as M. tuberculosis for constructing unmarked mutations. Other more sophisticated applications in mycobacterial genetics would also profit from the improved Flp/FRT system.
Keywords: sequence-specific recombination, Flp recognition target, codon usage, unmarked mutation
1. INTRODUCTION
Mycobacterium tuberculosis is a major global health problem and causes about 2 million deaths per year. To understand mycobacterial pathogenesis at the molecular level, efficient and specific genetic systems for recombination, mutagenesis and complementation are required (Pelicic et al., 1998). In particular, the ability to construct mutants by allelic exchange is imperative to characterize the function of a particular gene. Considerable progress in constructing allelic exchange mutants in mycobacteria has been achieved using conditionally replicating temperature-sensitive plasmids (Pelicic et al., 1997) or specialized transducing mycobacteriophages (Lee et al., 1991). However, the main challenge in analyzing the functions of redundant genes is that only a few resistance genes are functional in mycobacteria. Due to their superior efficiency, the hyg gene from Streptomyces hygroscopicus and the aph genes are used for almost all knock-out experiments in mycobacteria (Kana and Mizrahi, 2004).
There are two strategies to construct unmarked mutations and to simultaneously solve the problem of limited resistance markers in mycobacteria. One is based on two consecutive allelic exchange reactions. This is tedious work for both construction and analysis of the mutants. Sequence-specific recombination provides a faster and more efficient strategy. Several site-specific recombination systems are used in E. coli. The most frequently used system is the Flp/FRT system from the 2 μm plasmid of Saccharomyces cerevisiae (Merlin et al., 2002). In addition, the Cre/loxP system of the bacteriophage P1 (Hasan et al., 1994), the TnpR/res system of the γδ transposon (Tsuda, 1998) and the ParA/res system of the broad-host-range plasmid RP4 (Denome et al., 1999) are known. In our previous work, we showed that the Flp-flanked DNA was removed by the Flp recombinase in M. smegmatis but not in M. bovis BCG or M. tuberculosis (Stephan et al., 2004). It was discussed that the low G+C content of 38% of the S. cerevisiae flpe gene may have impaired its expression in mycobacteria which have an average G+C content of >65 %. However, it was unknown why this affected expression more severely in slowly growing mycobacteria (Stephan et al., 2004). In this study, we describe the synthesis of a flpm gene whose codon-usage was adapted for efficient translation in mycobacteria. Using this mycobacterial flpm gene the efficiency of the Flp-mediated recombination process increased drastically both in M. smegmatis and in M. bovis BCG. The mycobacterial flpm gene is useful both for the construction of unmarked mutants and for the analysis of essential genes in mycobacteria. Thus, it adds another genetic tool to the growing toolbox used to dissect the pathogenesis of M. tuberculosis on a molecular level.
2. MATERIALS AND METHODS
2.1. Chemicals, enzymes and DNA
Hygromycin B was purchased from Calbiochem. All other chemicals were purchased from Merck, Roche or Sigma at the highest purity available. Enzymes for DNA restriction and modification were purchased from New England Biolabs. Isolation and modification of DNA was performed as described (Ausubel et al., 1987). Oligonucleotides were obtained from Integrated DNA Technologies.
2.2. Bacterial strains and growth conditions
Escherichia coli DH5α was used for cloning experiments and was routinely grown in Luria-Bertani broth at 37°C. M. smegmatis strains were grown at 37°C in Middlebrook 7H9 medium (Difco) supplemented with 0.2% glycerol and 0.05% Tween®80 or on Middlebrook 7H10 agar (Difco) supplemented with 0.2% glycerol. M. bovis BCG (strain Institut Pasteur) was grown in Middlebrook 7H9 broth (Difco) or on 7H10 agar plates supplemented with 0.2% glycerol and 10% OADC enrichment (BBL) at 37°C. Antibiotics were used when required at the following concentrations: hygromycin (200 μg ml−1 for E. coli; 50 μg ml−1 for mycobacteria) and kanamycin (50 μg ml−1 for E. coli; 30 μg ml−1 for mycobacteria).
2.3. Synthesis of the mycobacterial flpm gene
To increase the expression of S. cerevisiae flpe in mycobacteria, the codon usage of the flpe gene was altered to reflect the codon usage preferred by M. tuberculosis H37Rv (1,368,699 codons from 4,067 CDS, http://www.kazusa.or.jp/codon/). The codons which were chosen to replace rare codons of the flpe gene are shown in Table 1. This synthetic gene flpm was assembled from oligonucleotides. Briefly, the oligonucleotides (two 30-mers, forty 50-mers) were synthesized on a 25 nmol scale with no purification and dissolved in water to a final concentration of 100 μM each. To assemble the oligonucleotides, PCR reactions were performed as described (Withers-Martinez et al., 1999) with minor modifications. In order to obtain optimal amplification for G+C rich fragments, DMSO (Sigma) was added to all PCR reactions to a final concentration of 5% (v/v). The synthetic flpm gene was cloned into the E. coli pUC57 vector (Fermentas) by TA cloning and verified by DNA sequencing. This plasmid was named pUC57- flpm. The G+C content of the mycobacterial flp gene (flpm) and the flpe of S. cerevisiae is 61% and 38%, respectively (Fig. S1).
Table 1.
Frequency of excision of the FRT-hyg-FRT cassette by Flp recombinase encoded by flpe and flpm.
| Gene | Strain | Number of clones | Excision frequency | References |
|---|---|---|---|---|
| flpe | M. smegmatis | 40 | 5% | (Stephan et al., 2004) |
| M. smegmatis | 40 | 10% | (Stephan et al., 2004) | |
| M. smegmatis | 200 | 0.5% | This study | |
| M. bovis BCG | ND | 0% | (Stephan et al., 2004) | |
| M. bovis BCG | 200 | 0% | This study | |
| flpm | M. smegmatis | 200 | 64% | This study |
| M. smegmatis | 120 | 63% | This study | |
| M. bovis BCG | 200 | 40% | This study | |
| M. bovis BCG | 32 | 59% | This study |
ND: not determined
2.4. Construction of plasmids pML116 and pML597
The flpe expression vector pMN234 was constructed previously in our lab (Stephan et al., 2004). To obtain the flpm expression vector, pUC57-flpm was digested with HindIII/BamHI and the fragment was cloned into pMN234 using the same restriction sites. This plasmid was named pML597.
To integrate the FRT-flanked hyg cassette into the genomic attB site of mycobacteria, the plasmid pML116 (Fig. 1) was constructed by cloning the ClaI/PmeI flanked mycgfp2+ gene of pML113 (Wolschendorf et al., 2007) into the backbone of pMN403 using the same restriction sites (Kaps et al., 2001). pML116 contains the attP site for the L5 integrase, a FRT-flanked hyg cassette and an expression cassette for mycgfp2+ which encodes an enhanced green fluorescent protein (GFP) under psmyc promoter. The mycgfp2+ gene contains the same fluorescence enhancing mutations as gfp+ (Scholz et al., 2000) and was adapted to the mycobacterial codon usage (Niederweis et al., unpublished).
Fig. 1. Representation of the Flp-mediated excision of the hyg gene from a plasmid integrated into mycobacterial chromosomes.

The plasmid pML116 carries a hygromycin resistance gene (hyg) flanked by two FRT sites in direct orientation, a gfp reporter gene (mycgfp2+), the attP site of mycobacteriophage L5 used for site-specific integration into mycobacterial chromosomes, an ampicillin resistance gene (bla) and an origin of replication for E. coli (oriE). The localization of the oligonucleotides used for PCR and the size of amplified fragments in M. smegmatis and M. bovis BCG are indicated.
2.5. Transformation and site specific recombination
To integrate the FRT-flanked hyg cassette (pML116) into the genomic attB site of mycobacteria, a two-plasmid system derived from mycobacteriophage L5 was used. Briefly, the replicative vector pML102 (Stephan et al., 2005) carrying the L5 integrase gene (int) and the counterselection marker sacB was transformed into M. smegmatis and M. bovis BCG. These cells were transformed with the nonreplicative vector pML116 containing the phage attachment site attP and the FRT-flanked hyg cassette. Since the continued expression of L5 integrase can cause the excision of the integrated vector from the genome contributing to plasmid instability (Springer et al., 2001), cells were plated on 7H10 plates containing hygromycin and 10% sucrose to select for the insertion of pML116 and to counterselect against pML102. Single colonies were plated in parallel on 7H10 plates containing hygromycin and 7H10 plates containing kanamycin to confirm the loss of pML102. The integration of the plasmid resulted in clones resistant to hygromycin and sensitive to kanamycin. Vector integration into the chromosome was visualized by GFP fluorescence and confirmed by colony PCR. The M. smegmatis SMR5 and M. bovis BCG strains with integrated pML116 were named ML17 (attB::pML116) and ML37 (attB::pML116), respectively.
To compare the efficacies of the Flp recombinases encoded by flpe or by the synthetic flpm genes for removal of FRT-hyg-FRT cassette, the plasmids pMN234 (flpe) and pML597 (flpm) were transformed into competent cells of M. smegmatis ML17 and M. bovis BCG ML37 and plated on 7H10 plates containing kanamycin. One colony was picked from each plate and cultured in 7H9-kan medium for two days (M. smegmatis) or two weeks (M. bovis BCG) to saturation. Then serial dilutions in 7H9 medium ranging from 10−4–10−8 were plated on 7H10-kan plates. Two hundred colonies were picked from each plate and streaked in parallel on 7H10 and 7H10-hyg plates. The colonies which grew on 7H10 but not on 7H10-hyg plates were counted.
2.6. Analysis of chromosomal DNA by colony PCR
Colony PCR was used to analyze plasmid integration into the chromosome and removal of FRT-hyg-FRT cassette using PuReTaq™ Ready-To-Go™ PCR beads (GE Healthcare) according to the manufacturer’s protocol with the primers MGR19 (5′-CGACCAGGATCGGGACGACGCCGGTGAACAGCTCCTCGCC-3′) and downattP (5′-TGATCTGCGACGAACCACGACCTTGGTG-3′) for M. smegmatis, and primers MGR19 and attBbcg01 (5′-CAGGTTGACGACAAGATCCCCGTCGA-3′) for M. bovis BCG, respectively. The PCR products were gel-purified using GFX™ PCR DNA and gel band purification kit (GE Healthcare) and sequenced to verify the removal of the hyg gene using the specific primers downattP and attBbcg01.
3. RESULTS AND DISCUSSION
3.1. Expression system for a synthetic flp gene with a mycobacterial codon-usage
The goal of our work was to establish a system which can be used for consecutive genomic deletions in mycobacteria. Since resistance markers suitable for selection of allelic mutants are limited to hygromycin and kanamycin in these organisms, it is necessary to remove the marker from the chromosome for subsequent modifications. We chose the Flp/FRT system as it represents one of the best characterized recombination systems (Schweizer, 2003). In its resolvase reaction, the Flp recombinase binds to two recognition sites and excises the sequence between them, thereby leaving one FRT site in the chromosome without polar effect (Merlin et al., 2002). However, the native flpe gene of S. cerevisiae does not appear to be expressed in slowly growing mycobacteria (Stephan et al., 2004). The factors which could play a role for heterologous gene expression include codon bias (Kane, 1995), transcriptional promoters (Morris and Miller, 1992), the nucleotide sequences surrounding the N-terminus (Deana et al., 1998), mRNA stability (Stenstrom and Isaksson, 2002) and product toxicity (Cherepanov and Wackernagel, 1995). Analysis of the codon usage revealed that the flpe gene contains 57 codons (out of 424) used in mycobacteria with a frequency of less than 5 % (Table 1) and are therefore considered to be rare codons (Kane, 1995). Since rare codon gene expression can lead to misincorporation of amino acids, termination of translation, or frameshift errors (Calderone et al., 1996), we redesigned and synthesized the flpe gene using codons preferred by M. tuberculosis. To avoid an inbalanced tRNA pool due to the usage of only one codon for a particular amino acid (Kurland and Gallant, 1996), we used two codons preferred by mycobacteria for the amino acids Ala, Arg, Leu and Thr (Table S1). In addition, we also removed the singular restriction sites within the flpm gene, which increases the compatibility of the gene with various expression vectors. The flpe gene of S. cerevisiae is expressed from the constitutive promoter pimyc in the plasmid pMN234 (Stephan et al., 2004). We exchanged the flpe gene with the flpm gene to obtain pML597. Both plasmids contain the aph gene and are thereby selectable in hygromycin resistant strains. They also contain the rpsL gene as a counter-selectable marker for easy removal in rpsL mutants of mycobacteria which are streptomycin-resistant (Sander et al., 1995).
3.2. Integration of the marker plasmid into the chromosome of M. smegmatis and Flp-mediated removal of the hyg cassette
We constructed the integration vector pML116 as a carrier for the FRT-hyg-FRT cassette with two minimal FRT sites in direct orientation and a gfp reporter gene (Fig. 1). The integration was mediated by the L5 recombinase on the plasmid pML102 which was removed after integration by counterselection using 10% sucrose. The correct integration of the plasmid pML116 into the attB site was verified for 20 clones both by colony PCR and GFP fluorescence (Fig. 2A, B and C). One of the resulting strains was named M. smegmatis ML17 (attB::pML116). To compare the excision efficiencies mediated by the Flp recombinase encoded by the flpe and flpm genes, M. smegmatis ML17 containing the FRT-flanked hyg gene in the attB site was transformed with the plasmids pMN234 and pML597, respectively. The transformants were selected on plates containing kanamycin. One clone was chosen and grown for two days to saturation in liquid medium and then plated on plates containing kanamycin. To test whether FRT-hyg-FRT was excised, 200 clones were streaked in parallel on plates with and without hygromycin. Out of the 200 clones containing the flpe gene of S. cerevisiae only one clone did not grow on hygromycin plates indicating the loss of the hyg gene with a frequency of 0.5% (Table 1). These low excision frequencies upon expression of flpe were confirmed in multiple experiments using the FRT-hyg-FRT cassette in different chromosomal locations. The reason for the 10-fold higher excision frequencies as observed in previous experiments is unknown (Stephan et al., 2004). One explanation may be that the expansion of an early clone which has lost the hyg cassette would result in more hyg negative cells and in an apparently higher recombination frequency. If such an event contributed to the number hyg negative clones, the recombination frequency should be different for different excision experiments. Therefore, two clones were selected after transformation with the flpm expression plasmid pML597 and grown for two days to saturation in liquid medium before plating the culture on plates containing kanamycin. In experiments with both clones, the hyg gene was lost with a similar frequency of more than 60% (Table 1). The Flp-mediated removal of the hyg cassette was verified by colony PCR for eight of these clones (Fig. 2C). Clone #1 was named M. smegmatis ML27 (attB::pML116 hyg). Sequencing of the chromosomal DNA of the ML27 strain confirmed that the hyg gene of ML17 was specifically excised and replaced by one FRT site (Fig. 2D). These results demonstrate that the codon-usage adapted flpm gene confers a much higher recombination efficiency in M. smegmatis than the flpe gene of S. cerevisiae.
Fig. 2. Integration and removal of a FRT-hyg-FRT cassette from M. smegmatis.
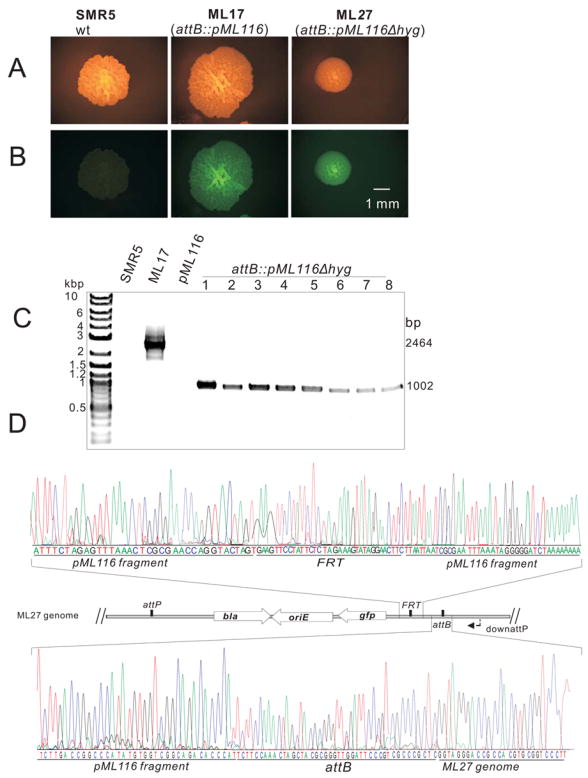
A and B: Microscopy of single colonies of M. smegmatis SMR5 (wt), ML17 (attB::pML116) and ML27 (attB::pML116Δhyg). Pictures of the same colonies were taken with an Olympus SZX12 (Model U-ULH) stereomicroscope equipped with a Zeiss AxioCam MRc camera and an Olympus fluorescence illuminator (100W mercury lamp) using a 20-fold magnification. A fluorescence filter cube (excitation: 470 nm, emission: 500 nm) was used to reveal fluorescence of the colonies. Note that the sizes of the colonies are different because they were incubated for different days. The scale bar is shown.
C: Agarose gel (1%) analysis of the PCR products amplified from chromosomal DNA. The primer pair MGR19 and downattP yielded no fragments for SMR5 and pML116 and fragments of 2,464 bp and 1,002 bp for ML17 and 8 different clones picked after flpm expression.
D: Partial sequencing results of the amplified 1,002 bp fragment from clone #1 which was named M. smegmatis ML27 (lane 1 of C) using primer downattP. The locations of the FRT and attB sites and parts of the pML116 fragment in the genome of ML27 are shown in the map.
3.3. Integration of the marker plasmid into the chromosome of M. bovis BCG and Flp-mediated removal of the hyg cassette
The integration vector pML116 was also used as a carrier for the FRT-hyg-FRT cassette and the gfp reporter gene in M. bovis BCG (Fig. 1). The integration was mediated by the L5 recombinase on the plasmid pML102 which was removed after integration by counterselection using 2% sucrose. The correct integration of the plasmid pML116 into the attB site was verified for 20 clones both by colony PCR and GFP fluorescence (Fig. 3A, B and C). One of the resulting strains was named M. bovis BCG ML37 (attB::pML116). To compare the efficacies of the Flp recombinases encoded by the flpe gene from S. cerevisiae and the flpm gene, M. bovis BCG ML37 containing the FRT-flanked hyg gene in the attB site was transformed with the plasmids pMN234 and pML597, respectively. The transformants were selected on plates containing kanamycin. One clone was chosen and grown for two weeks to saturation in liquid medium and then plated on plates containing kanamycin. To test whether FRT-hyg-FRT was excised, 200 clones were streaked in parallel on plates with and without hygromycin. Out of the 200 clones containing the flpe gene of S. cerevisiae, all of them grew on hygromycin plates indicating the loss of the hyg gene with a frequency of 0% (Table 1). Two clones were selected after transformation with the flpm expression plasmid pML597 and grown for two weeks to saturation in liquid medium before plating the culture on plates containing kanamycin. In both experiments, the hyg gene was lost with a frequency of at least 40% (Table 1). The Flp-mediated removal of the hyg cassette was verified by colony PCR for eight of these clones (Fig. 3C). Clone #1 was named M. bovis BCG ML47 (attB::pML116Δhyg). Sequencing of the chromosomal DNA of the ML47 strain confirmed that the hyg gene of ML37 was specifically excised and replaced by one FRT site (Fig. 3D). These results demonstrated that the codon-usage adapted flpm gene confers a much higher recombination efficiency in M. bovis BCG than the flpe gene of S. cerevisiae. These results further demonstrate that expression of the Flp recombinase is not toxic in mycobacteria as it was found for other bacteria including E. coli, Salmonella typhimurium and Pseudomonas aeruginosa (Cherepanov and Wackernagel, 1995).
Fig. 3. Integration and removal of FRT-hyg-FRT cassette from M. bovis BCG.
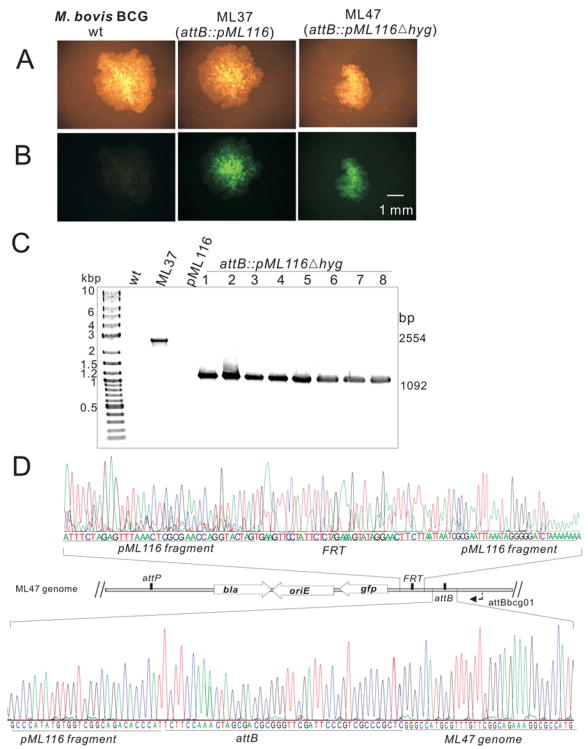
A and B: Microscopy of single colonies of M. bovis BCG (wt), ML37 (attB::pML116) and ML47 (attB::pML116Δhyg). Pictures of the same colonies were taken with an Olympus SZX12 (Model U-ULH) stereomicroscope equipped with a Zeiss AxioCam MRc camera and an Olympus fluorescence illuminator (100W mercury lamp) using a 20-fold magnification. A fluorescence filter cube (excitation: 470 nm, emission: 500 nm) was used to reveal fluorescence of the colonies. Note that the sizes of the colonies are different because they were incubated for different days. The scale bar is shown.
C: Agarose gel (1%) analysis of the PCR products amplified from chromosomal DNA. The primer pair MGR19 and attBbcg01 yielded no fragments for M. bovis BCG wt and pML116 and fragments of 2,554 bp and 1,092 bp for ML37 and 8 different clones picked after flpm expression.
D: Partial sequencing results of the amplified 1,092 bp fragment from clone #1 which was named M. bovis BCG ML47 (lane 1 of C) using primer attBbcg01. The locations of the FRT and attB sites and parts of the pML116 fragment in the genome of ML47 are shown in the map.
3.4. Role of codon usage for the expression of heterologous genes in mycobacteria
The codon distribution, genome G+C content, and the changes in codon usage are at least partly explained by a mutation-selection equilibrium between the different synonymous codons in each organism (Gustafsson et al., 2004). Although codon bias is not the only factor involved in protein expression, it has become increasingly clear that codon biases can have profound impacts on the expression of heterologous proteins (Kane, 1995). Indeed, the result that the codon-usage adapted flpm gene confers a much higher recombination efficiency both in M. smegmatis and M. bovis BCG than the flpe gene of S. cerevisiae, strongly indicates that mRNA transcribed from the flpe gene of S. cerevisiae was not efficiently translated in both organisms. This is consistent with previous observations that codon usage adaptation improved the expression of other heterologous genes with low G+C content in mycobacteria, such as Sm14 antigen of Schistosoma mansoni (Varaldo et al., 2006) and human immunodeficiency virus type 1 Gag (Kanekiyo et al., 2005). Taken together, these studies indicate that codon usage adaptation may be a generally useful approach to increase expression of heterologous genes in mycobacteria. It may also alleviate putative growth-inhibiting effects of depleting the tRNA pool of mycobacteria from rare tRNAs which may be needed for the synthesis of essential housekeeping genes or are involved in post-transcriptional regulatory processes.
3.5. Comparison of the sequence-specific recombination systems available for mycobacteria
In addition to the Flp/FRT system, the use of two other sequence-specific recombination systems in mycobacteria has been described: the Cre/loxP system of the bacteriophage P1 (Hasan et al., 1994) and the TnpR/res system of the γδ transposon (Tsuda, 1998). The minimal target sites of the Flp recombinase (423 amino acids) and the Cre recombinase (343 amino acids) have identical lengths of 34 bp ((Sternberg and Hamilton, 1981; McLeod et al., 1986), Table 2). Expression of these recombinases in a host which harbors two target sites in direct orientation results in excision of the flanked DNA fragment and leaves a single target site in the chromosome. We have shown that the excision frequencies for the Flp recombinase encoded by the flpm gene are similar to those for the Cre recombinase (unpublished data, Table 2). A notable difference between the target sites was revealed by sequence analysis: the FRT site has stop codons in reading frames 1 and 3, while the loxP site has stop codons in reading frames 2 and 3 with respect to the reading frame of the gene in which the site was inserted. This difference might become important if DNA within an operon should be deleted. In-frame deletions without interfering with translation was shown to be essential for efficient expression of downstream genes in operons (Liberati et al., 2006). The transposon γδ resolvase TnpR is also functional in mycobacteria (Malaga et al., 2003). However, the res site is 122 bp long (Heffron et al., 1979) and contains seven stop codons which are present in all three reading frames (Table 2). This may represent a disadvantage for the use of the γδ resolvase for small in-frame deletions or similar applications. The drastic increase of the excision frequency by the Flp recombinase expressed from the codon-usage adapted flpm gene indicates that the expression level is the major determinant of the activities of these enzymes in host cells.
Table 2.
Comparison of FRT, loxP and res recombination sites
| Recombinase (amino acids) | Target sites | Sequence (5′-3′) | Recombination frequency (%) | Length (bp) | |
|---|---|---|---|---|---|
| Msmeg | BCG/Mtb | ||||
| FLP (423) | FRT |
|
>60a | >40a | 34b |
| Cre (343) | loxP |
|
90c | 40c | 34d |
| γδ TnpR (186) | res |

|
100e | 3–5f | 122g |
The core regions of the FRT and loxP sites are underlined. The palindromic sequences of FRT and loxP are indicated with italic letters. There are three resolvase binding sites at the internal of res, which are indicated with box. The stop codons of translation in 5′-3′ orientation and 3′-5′ orientation are indicated with bold and shadow letters, respectively, for each recombination site.
this study
Song et al. (unpublished data using the plasmid pCreSacB1 and a hyg expression cassette flanked by loxP sites)
4. CONCLUSIONS
Future, more sophisticated expression and gene deletion constructs for mycobacteria require more tools. The flpm based Flp/FRT system provides an efficient site-specific recombination system which can be used for multiple applications including constructing unmarked mutants in fast- and slow-growing mycobacteria. Although the same FRT cassette can be repeatedly used for construction of consecutive mutations in the same chromosome, the presence of multiple chromosomal FRT sites can possibly lead to undesirable secondary effects, such as inversions or deletions between these sites. In P. aeruginosa, the combination of two mutations caused the Flp-mediated inversion of a 1.59-Mb chromosomal region at fairly high frequencies (Barekzi et al., 2000). Therefore, the combinatorial use of Cre/loxP, TnpR/res and Flp/FRT provides a new perspective for constructing consecutive deletions and other genetic modifications in mycobacteria.
Supplementary Material
Fig. S1: Alignment of the flpe and flpm genes. The sequence alterations in the flpm gene compared to the flpe gene are marked by boxes. The encoded amino acid sequence is shown under the nucleotide sequence in one letter code.
Table S1: Replacement of rare codons in the flpe gene The codon usage of 1,272 bpflpe gene and the occurrence of encoded 424 amino acids are indicated in this table. The synthesizedflpm gene was designed according to the codon usage ofMtb (bolded codon), which was obtained from 4,067 CDS (1,368,699 codons) of H37Rv. The codon usages offlpe and the genome ofM. tuberculosis H37Rv were analyzed using the Codon Usage Database at http://www.kazusa.or.jp/codon/. Codons used byflpe with a frequency of 5% or less inMtb are marked with a star.
Acknowledgments
We thank Jason Huff for critically reading the manuscript and Dr. Adrie Steyn for providing the Cre recombinase expression plasmid pCreSacB1. This work was supported by the grant AI063432 of the National Institutes of Health to MN.
Abbreviations
- bp
base pair(s)
- kbp
1000 bp
- Flp
S. cerevisiae recombinase
- FRT
Flp recognition target
- CDS
coding sequence
- hyg
hygromycin phosphotransferase encoding gene
- ori
origin of DNA replication
- PCR
polymerase chain reaction
- wt
wild-type
- kan
kanamycin
- hyg
hygromycin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidmann JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. John Wiley & Sons; New York: 1987. [Google Scholar]
- Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 2002;148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- Barekzi N, Beinlich K, Hoang TT, Pham XQ, Karkhoff-Schweizer R, Schweizer HP. High-frequency flp recombinase-mediated inversions of the oriC- containing region of the pseudomonas aeruginosa genome. J Bacteriol. 2000;182:7070–7074. doi: 10.1128/jb.182.24.7070-7074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone TL, Stevens RD, Oas TG. High-level misincorporation of lysine for arginine at AGA codons in a fusion protein expressed in Escherichia coli. J Mol Biol. 1996;262:407–412. doi: 10.1006/jmbi.1996.0524. [DOI] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Deana A, Ehrlich R, Reiss C. Silent mutations in the Escherichia coli ompA leader peptide region strongly affect transcription and translation in vivo. Nucleic Acids Res. 1998;26:4778–4782. doi: 10.1093/nar/26.20.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol. 1999;181:3981–3993. doi: 10.1128/jb.181.13.3981-3993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22:346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Hasan N, Koob M, Szybalski W. Escherichia coli genome targeting, I. Cre-lox-mediated in vitro generation of ori- plasmids and their in vivo chromosomal integration and retrieval. Gene. 1994;150:51–56. doi: 10.1016/0378-1119(94)90856-7. [DOI] [PubMed] [Google Scholar]
- Heffron F, McCarthy BJ, Ohtsubo H, Ohtsubo E. DNA sequence analysis of the transposon Tn3: three genes and three sites involved in transposition of Tn3. Cell. 1979;18:1153–1163. doi: 10.1016/0092-8674(79)90228-9. [DOI] [PubMed] [Google Scholar]
- Kana BD, Mizrahi V. Molecular genetics of Mycobacterium tuberculosis in relation to the discovery of novel drugs and vaccines. Tuberculosis (Edinb) 2004;84:63–75. doi: 10.1016/j.tube.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Kane JF. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Opin Biotechnol. 1995;6:494–500. doi: 10.1016/0958-1669(95)80082-4. [DOI] [PubMed] [Google Scholar]
- Kanekiyo M, Matsuo K, Hamatake M, Hamano T, Ohsu T, Matsumoto S, Yamada T, Yamazaki S, Hasegawa A, Yamamoto N, Honda M. Mycobacterial codon optimization enhances antigen expression and virus-specific immune responses in recombinant Mycobacterium bovis bacille Calmette-Guerin expressing human immunodeficiency virus type 1 Gag. J Virol. 2005;79:8716–8723. doi: 10.1128/JVI.79.14.8716-8723.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaps I, Ehrt S, Seeber S, Schnappinger D, Martin C, Riley LW, Niederweis M. Energy transfer between fluorescent proteins using a co-expression system in Mycobacterium smegmatis. Gene. 2001;278:115–124. doi: 10.1016/s0378-1119(01)00712-0. [DOI] [PubMed] [Google Scholar]
- Kitts PA, Symington LS, Dyson P, Sherratt DJ. Transposon-encoded site-specific recombination: nature of the Tn3 DNA sequences which constitute the recombination site res. Embo J. 1983;2:1055–1060. doi: 10.1002/j.1460-2075.1983.tb01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland C, Gallant J. Errors of heterologous protein expression. Curr Opin Biotechnol. 1996;7:489–493. doi: 10.1016/s0958-1669(96)80050-4. [DOI] [PubMed] [Google Scholar]
- Lee MH, Pascopella L, Jacobs WR, Jr, Hatfull GF. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc Natl Acad Sci U S A. 1991;88:3111–3115. doi: 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaga W, Perez E, Guilhot C. Production of unmarked mutations in mycobacteria using site-specific recombination. FEMS Microbiol Lett. 2003;219:261–268. doi: 10.1016/S0378-1097(03)00003-X. [DOI] [PubMed] [Google Scholar]
- McLeod M, Craft S, Broach JR. Identification of the crossover site during FLP-mediated recombination in the Saccharomyces cerevisiae plasmid 2 microns circle. Mol Cell Biol. 1986;6:3357–3367. doi: 10.1128/mcb.6.10.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin C, McAteer S, Masters M. Tools for characterization of Escherichia coli genes of unknown function. J Bacteriol. 2002;184:4573–4581. doi: 10.1128/JB.184.16.4573-4581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris TD, Miller LK. Promoter influence on baculovirus-mediated gene expression in permissive and nonpermissive insect cell lines. J Virol. 1992;66:7397–7405. doi: 10.1128/jvi.66.12.7397-7405.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicic V, Jackson M, Reyrat JM, Jacobs WR, Jr, Gicquel B, Guilhot C. Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1997;94:10955–10960. doi: 10.1073/pnas.94.20.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicic V, Reyrat JM, Gicquel B. Genetic advances for studying Mycobacterium tuberculosis pathogenicity. Mol Microbiol. 1998;28:413–420. doi: 10.1046/j.1365-2958.1998.00807.x. [DOI] [PubMed] [Google Scholar]
- Sander P, Meier A, Boettger EC. rpsL+: a dominant selectable marker for gene replacement in mycobacteria. Mol Microbiol. 1995;16:991–1000. doi: 10.1111/j.1365-2958.1995.tb02324.x. [DOI] [PubMed] [Google Scholar]
- Scholz O, Thiel A, Hillen W, Niederweis M. Quantitative analysis of gene expression with an improved green fluorescent protein. Eur J Biochem. 2000;267:1565–1570. doi: 10.1046/j.1432-1327.2000.01170.x. [DOI] [PubMed] [Google Scholar]
- Schweizer HP. Applications of the Saccharomyces cerevisiae Flp-FRT system in bacterial genetics. J Mol Microbiol Biotechnol. 2003;5:67–77. doi: 10.1159/000069976. [DOI] [PubMed] [Google Scholar]
- Springer B, Sander P, Sedlacek L, Ellrott K, Böttger EC. Instability and site-specific excision of integration-proficient mycobacteriophage L5 plasmids: development of stably maintained integrative vectors. Int J Med Microbiol. 2001;290:669–675. doi: 10.1016/S1438-4221(01)80004-7. [DOI] [PubMed] [Google Scholar]
- Stenstrom CM, Isaksson LA. Influences on translation initiation and early elongation by the messenger RNA region flanking the initiation codon at the 3′ side. Gene. 2002;288:1–8. doi: 10.1016/s0378-1119(02)00501-2. [DOI] [PubMed] [Google Scholar]
- Stephan J, Bender J, Wolschendorf F, Hoffmann C, Roth E, Mailander C, Engelhardt H, Niederweis M. The growth rate of Mycobacterium smegmatis depends on sufficient porin-mediated influx of nutrients. Mol Microbiol. 2005;58:714–730. doi: 10.1111/j.1365-2958.2005.04878.x. [DOI] [PubMed] [Google Scholar]
- Stephan J, Stemmer V, Niederweis M. Consecutive gene deletions in Mycobacterium smegmatis using the yeast FLP recombinase. Gene. 2004;343:181–190. doi: 10.1016/j.gene.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Sternberg N, Hamilton D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J Mol Biol. 1981;150:467–486. doi: 10.1016/0022-2836(81)90375-2. [DOI] [PubMed] [Google Scholar]
- Tsuda M. Use of a transposon-encoded site-specific resolution system for construction of large and defined deletion mutations in bacterial chromosome. Gene. 1998;207:33–41. doi: 10.1016/s0378-1119(97)00601-x. [DOI] [PubMed] [Google Scholar]
- Varaldo PB, Miyaji EN, Vilar MM, Campos AS, Dias WO, Armoa GR, Tendler M, Leite LC, McIntosh D. Mycobacterial codon optimization of the gene encoding the Sm14 antigen of Schistosoma mansoni in recombinant Mycobacterium bovis Bacille Calmette-Guerin enhances protein expression but not protection against cercarial challenge in mice. FEMS Immunol Med Microbiol. 2006;48:132–139. doi: 10.1111/j.1574-695X.2006.00133.x. [DOI] [PubMed] [Google Scholar]
- Withers-Martinez C, Carpenter EP, Hackett F, Ely B, Sajid M, Grainger M, Blackman MJ. PCR-based gene synthesis as an efficient approach for expression of the A+T-rich malaria genome. Protein Eng. 1999;12:1113–1120. doi: 10.1093/protein/12.12.1113. [DOI] [PubMed] [Google Scholar]
- Wolschendorf F, Mahfoud M, Niederweis M. Porins are required for uptake of phosphates by Mycobacterium smegmatis. J Bacteriol. 2007;189:2435–2442. doi: 10.1128/JB.01600-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: Alignment of the flpe and flpm genes. The sequence alterations in the flpm gene compared to the flpe gene are marked by boxes. The encoded amino acid sequence is shown under the nucleotide sequence in one letter code.
Table S1: Replacement of rare codons in the flpe gene The codon usage of 1,272 bpflpe gene and the occurrence of encoded 424 amino acids are indicated in this table. The synthesizedflpm gene was designed according to the codon usage ofMtb (bolded codon), which was obtained from 4,067 CDS (1,368,699 codons) of H37Rv. The codon usages offlpe and the genome ofM. tuberculosis H37Rv were analyzed using the Codon Usage Database at http://www.kazusa.or.jp/codon/. Codons used byflpe with a frequency of 5% or less inMtb are marked with a star.