

Abstract
Peroxynitrite anion (ONOO−) is a reactive species of increasingly recognized biological relevance that contributes to oxidative tissue damage. At present, however, there is limited knowledge about the mechanisms of peroxynitrite diffusion through biological compartments. In this work we have studied the diffusion of peroxynitrite across erythrocyte membranes. In solution, peroxynitrite rapidly reacts with oxyhemoglobin to yield methemoglobin, with k2 = (10.4 ± 0.3) × 103 M−1⋅s−1 at pH 7.4 and 25°C. Addition of peroxynitrite to intact erythrocytes caused oxidation of intracellular oxyhemoglobin to methemoglobin. Oxidation yields in red blood cells at pH 7.0 were approximately 40% of those obtained in solution, which results mostly from competition of other cytosolic components for peroxynitrite. Indeed, rather small differences were observed between oxidation yields in lysates compared with intact erythrocytes, in particular at acidic and neutral pH values, indicating that membrane was not precluding peroxynitrite diffusion. Incubation of erythrocytes at pH 7.0 with 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), a specific inhibitor of anion exchange, resulted in up to 50% inhibition of oxyhemoglobin oxidation by peroxynitrite. More protection by DIDS was achieved at alkaline pH, while no effect was observed at pH 5.5, where 95% of peroxynitrite is in the acidic form, ONOOH (pKa = 6.8). In addition, peroxynitrite caused nitration of intracellular hemoglobin, in a process that was enhanced in thiol-depleted erythrocytes. Our results indicate that peroxynitrite is able to cross the erythrocyte membrane by two different mechanisms: in the anionic form through the DIDS-inhibitable anion channel, and in the protonated form by passive diffusion.
Keywords: nitric oxide; superoxide; 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid; anion transport; hemoglobin
Peroxynitrite anion (ONOO−), the product of the reaction between superoxide anion (O2⨪) and nitric oxide (·NO) and its conjugated acid, peroxynitrous acid (ONOOH) (pKa = 6.8; refs. 1 and 2), are potent oxidants known to be formed in vivo (3–5).¶ At physiological pH, 80% of peroxynitrite is present in the anionic form, but once protonated it rapidly rearranges to nitrate (kobs = 0.9 s−1 at 37°C and pH 7.4; ref. 2). The biological half-life of peroxynitrite is low (<0.1 s) because of the proton-catalyzed isomerization to nitrate but mainly because of reactions with target molecules.
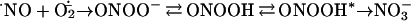 |
The chemistry of peroxynitrite is complex and strongly pH dependent (6, 7). Peroxynitrite can directly oxidize a variety of biomolecules (1, 8–14) by one- or two-electron oxidations or rearrange after protonation to a highly oxidizing species (ONOOH*, E0 = +2.1 V) with a reactivity close to that of hydroxyl radical. In addition, peroxynitrite is capable of nitrating aromatics (15, 16) in a process that can be enhanced by metal centers and CO2 (17). The reaction between peroxynitrite and CO2 leads to the formation of nitrosoperoxocarbonate (ONO2CO2−), a reactive intermediate that represents one of the major routes of peroxynitrite consumption in biological systems (17, 18).
Because peroxynitrite is a stronger oxidant than both of its precursors, O2⨪ and ·NO, it has been suggested to be the species responsible for some of the pathological conditions associated with an overproduction of these radicals (9). Its participation in ischemia/reperfusion injury, neurodegenerative diseases, cardiovascular disorders, atherosclerosis, and severe inflammation conditions, among others, has been reported (3, 19–24).
The reaction between ·NO and O2⨪ is almost diffusion controlled, k2 = 6.9 × 109 M−1⋅s−1 (25); therefore nearly every collision between ·NO and O2⨪ results in the formation of peroxynitrite. ·NO is a small hydrophobic and relatively unreactive free radical that freely diffuses across membranes (26, 27). On the other hand, O2⨪ has a shorter biological half-life than ·NO and, because it is negatively charged at physiological pH (pKa = 4.8), its diffusion across membranes depends on the presence of anion channels (28). It is reasonable, therefore, to consider that in biological systems peroxynitrite will be preferentially formed close to the O2⨪ generation sites with the arrival of ·NO molecules produced at more distant areas (29). Now the question arises whether peroxynitrite is able to diffuse through cells and interact with critical targets located far from its site of production.
Indirect evidence of peroxynitrite transmembrane diffusion has been observed by various authors, including ourselves (29–33). However, in these studies, the intracellular effects observed after exposure to externally added peroxynitrite could also be due to secondary oxidants derived from peroxynitrite (i.e., nitrogen dioxide, ·NO2) or even intracellular migration of secondary membrane lipid oxidation products. Up to now, the biological fate of peroxynitrite has been predicted mainly on kinetic grounds (34), but diffusional considerations become another key factor as it is apparent that peroxynitrite-producing cells mediate toxic effects in target cells and extracellular compartments (3, 35).
In this report we study the diffusion of peroxynitrite from the extra- to the intracellular compartment, using red blood cells (RBC) as a model system, and we define the mechanisms by which peroxynitrite can cross cell membranes.
MATERIALS AND METHODS
Chemicals.
The following reagents were purchased from Sigma: 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), nigericin sodium salt, hydrogen peroxide, sodium nitrite, bovine serum albumin (BSA), and N-ethylmaleimide (NEM). The anion-exchanger DEAE-Sepharose CL-6B was obtained from Pharmacia. 2′,7′-Bis-(2-carboxyethyl)-5-carboxyfluorescein (BCECF) and its acetoxymethyl ester (BCECF-AM) were purchased from Molecular Probes. All other reagents were of research grade quality.
Peroxynitrite was synthesized in a quenched-flow reactor as previously described (1, 9) with minor modifications (30) and stored at −80°C until use. The concentration of nitrite in the peroxynitrite preparations was determined by the method of Griess (36) and was typically ≈50% of the peroxynitrite concentration. The absence of hydrogen peroxide was confirmed by decomposing peroxynitrite in buffer before assay with horseradish peroxidase and iodide as the second substrate (ɛ353 = 25,000 M−1⋅cm−1; ref. 37). Peroxynitrite concentrations were determined spectroscopically at 302 nm (ɛ = 1, 670 M−1⋅cm−1), and dilutions in water were made immediately before use to achieve the desired concentrations. A rabbit polyclonal antibody against nitrotyrosine was raised with nitrated keyhole limpet hemocyanin and purified in our laboratory by affinity chromatography as described elsewhere (38).
Preparation of Oxyhemoglobin.
Oxyhemoglobin solution was prepared from human erythrocytes (RBC) as previously described (39). Briefly, cells were obtained by centrifugation (800 × g, 10 min) of freshly drawn blood from a healthy donor with heparin as the anticoagulant. After the RBC had been washed with 0.15 M NaCl/5 mM Tris⋅HCl, pH 8.0, lysis was achieved by diluting with 1.5 vol of distilled water. After 15 min at room temperature the lysate was centrifuged and the supernatant was dialyzed against 50 mM Tris⋅HCl, pH 8.3/0.1 mM edta for 2 h. The dialyzed lysate was loaded onto a DEAE-Sepharose column (2.5 × 19 cm) previously equilibrated with 50 mM Tris⋅HCl, pH 8.3/0.1 mM edta. Oxyhemoglobin was eluted by applying a pH gradient from 8.3 to 7.0 in the same buffer. The purity of the hemoglobin preparation was assessed by SDS/15% polyacrylamide gel electrophoresis, yielding a wide single protein band around 15.5 kDa corresponding to the α and β chains of human hemoglobin (15.1 and 15.8 kDa, respectively). The solution of hemoglobin obtained was 95% in the oxy form, and neither superoxide dismutase (40) nor catalase (41) activity was detected.
Concentration of Hemoglobin.
The concentration of oxy- and methemoglobin was determined spectrophotometrically by measuring the absorbance at 577 and 630 nm as described by Winterbourn (39): oxyhemoglobin (concentration of heme, μM) = 66 A577 − 80 A630; methemoglobin (concentration of heme, μM) = 279 A630 − 3 A577.
Kinetic Experiments.
Stopped-flow measurements were performed in an Applied Photophysics SF-17MV spectrophotometer (Leatherhead, England) with a mixing time of less than 2 ms. Spectral changes during oxyhemoglobin–peroxynitrite interactions were studied by using the photo diode array accessory, SX-18MV (200–750 nm). Oxyhemoglobin (25 μM) was mixed with peroxynitrite (180 μM) in 100 mM sodium phosphate buffer, pH 7.5, at 37°C, and spectra (450–700 nm) were taken every 250 ms for 2 s. When peroxynitrite was decomposed in buffer before mixing with oxyhemoglobin, no change in the spectra was observed even after 10 s. The second-order rate constant for the reaction of oxyhemoglobin with peroxynitrite was determined by the method of initial velocities (34, 42) following the disappearance of oxyhemoglobin with time at 577 nm (ɛ = 11,000 M−1⋅cm−1; ref. 39). The kinetics of peroxynitrite-mediated oxidation of oxyhemoglobin was studied at various pH values, 5.0–8.0 (±0.01) in 100 mM phosphate buffer/0.05 mM diethylenetriaminepentaacetic acid (dtpa). The temperature was kept constant at 25 ± 0.2°C and the pH values of the reaction mixtures were determined at the outlet.
Preparation of RBC.
RBC were obtained by centrifugation (800 × g, 10 min) of freshly drawn heparinized human blood. After removal of plasma and buffy coat, the cells were resuspended and washed in 0.15 M NaCl/5 mM phosphate buffer, pH 7.2, and kept at 4°C until used on the same day.
Oxyhemoglobin Oxidation by Peroxynitrite.
(i) Oxidation yields in solution. Oxyhemoglobin (200 μM) in 200 mM phosphate buffer was exposed to increasing concentrations of peroxynitrite (0–250 μM). The reaction was completed in less than 2 s, and immediately after mixing, any secondary reaction was stopped by dilution 1/10 with water containing 0.1% Triton X-100. The absorbances at 577 and 630 nm were measured to determine the amount of methemoglobin formed. From these values, the oxidation yields in buffer, at three different pH values (5.5, 7.0, and 8.2) were calculated.
(ii) Oxidation yields in intact RBC. A suspension of RBC (200 μM oxyhemoglobin)‖ in isotonic buffer A (80 mM sodium phosphate buffer/40 mM NaCl/10 mM KCl) was incubated 10 min at room temperature with nigericin (10 μg/ml) before addition of peroxynitrite (0–600 μM), immediately mixed, and centrifuged (8,000 × g, 10 s). The supernatant was set aside to measure pH and the pellet was resuspended in the same volume with distilled water. The whole procedure after addition of the oxidant was completed in 2 ± 0.5 min. The absorbance at 577 and 630 nm of the lysate was measured, and the yields of intracellular hemoglobin oxidation at three different pH values (5.5, 7.0, and 8.2) were calculated.
(iii) Oxidation yields in RBC lysates. To determine the oxidation yields in solution but in the presence of all the cytosolic components of the erythrocyte, the cells were lysed by osmotic shock and centrifuged, and an aliquot of the supernatant (lysate) equivalent to 200 μM oxyhemoglobin was resuspended in 200 mM phosphate buffer at the pH of study (5.5, 7.0, and 8.2). The reaction with peroxynitrite was conducted and followed in the same way as described above (i).
Intracellular pH Measurements.
The emission spectra of entrapped carboxyfluorescein derivative (BCECF) were used to measure internal pH of RBC as previously described (43) with minor modifications. RBC (300 μM oxyhemoglobin) were incubated with BCECF-AM (10 μM) for 30 min at pH 7.2 and 4°C. The cells were then centrifuged and washed to eliminate any excess of BCECF-AM. A calibration curve was constructed by using isotonic buffer A (130 mM in K+) at different pH values (from 5.5 to 8.5) and nigericin (10 μg/ml, 10 min at room temperature) to equilibrate external and internal pH. Fluorescence intensities at each pH were measured in a Kontron SFM-25 spectrofluorimeter exciting at two wavelengths: 500 nm (absorption maximum) and 440 nm (isosbestic point), and following emission at 535 nm. Then RBC with entrapped BCECF were exposed to three different external pH values (5.5, 7.0, or 8.2) by resuspending the cells in isotonic buffer A, in the absence and presence of 200 μM DIDS. Intracellular pH in each case was assessed by measuring the fluorescence emission as described above.
Effect of DIDS on Intracellular Oxidation of Oxyhemoglobin by External Addition of Peroxynitrite.
Intact RBC (60 μM heme in 5 mM phosphate buffer, pH 7.2/0.15 M NaCl) were incubated with increasing concentrations of DIDS (0–300 μM) for 30 min at room temperature. After the cells had been washed to remove any excess DIDS, peroxynitrite was added (300 μM) and the oxidation of intracellular oxyhemoglobin was determined spectrophotometrically as described above. The effect of pH was assessed by previously incubating RBC with 200 μM DIDS (30 min, room temperature), then centrifugation and resuspension in the same volume with buffer A at different pHs and 10 μg/ml nigericin. The yields of intracellular oxyhemoglobin oxidation by externally added peroxynitrite were determined as described above.
Western Blot Analysis.
Purified human hemoglobin and RBC, with and without peroxynitrite treatment, were analyzed on SDS/15% polyacrylamide gels. Peroxynitrite treatment consisted of exposure of pure oxyhemoglobin (210 μM) or intact RBC (420 μM oxyhemoglobin) to 2 mM peroxynitrite. Depletion of erythrocyte thiols was achieved by incubating intact RBC (420 μM oxyhemoglobin) with 1 mM NEM at room temperature for 30 min, and the excess of NEM was removed by washing with isotonic buffer. The proteins were separated on a gel and electrophoretically transferred (240 mA, 2 h) to nitrocellulose membranes (0.2 μm pore size, Hybond-C extra, Amersham), and the nonspecific binding sites were blocked for 1 h with blocking buffer (5% BSA in 50 mM Tris⋅HCl/150 mM NaCl, pH 7.4) and 0.3% Tween 20. Nitrocellulose filters were probed with 0.2 mg/ml rabbit polyclonal antibody to nitrotyrosine (1:1000 dilution) in blocking buffer containing 0.6% Tween 20 for 1 h. After extensive washings in the same buffer, the immunocomplexed membranes were further incubated (1 h) with a horseradish peroxidase-conjugated secondary antibody [donkey polyclonal anti-rabbit IgG (1:5000 dilution; Amersham)]. Probed membranes were washed in blocking buffer containing 0.3% Tween 20, and immunoreactive proteins were detected by using the luminol-enhanced chemiluminescence detection system (ECL; Amersham).
General Procedures.
All spectrophotometric measurements were performed in a Milton Roy Spectronic 3000 Array spectrophotometer. Protein concentration was determined by the Bradford method (44), using BSA as standard. Whenever the effect of peroxynitrite was studied, runs were performed under the same conditions with peroxynitrite that had been previously decomposed in buffer (reverse addition experiments; ref. 34). These runs allow us to account, in the time course of our experiments, for any contribution to the oxidation and nitration of oxyhemoglobin by any contaminant of the peroxynitrite preparation and decomposition products, in particular nitrite. In addition, experiments with nitrite at the concentrations found during peroxynitrite exposures were performed.
RESULTS
Oxidation of Oxyhemoglobin by Peroxynitrite.
Fig. 1 shows the rapid visible spectral changes associated with the reaction of human oxyhemoglobin with peroxynitrite at pH 7.4 and 37°C. In the 450- to 700-nm region of the spectrum, in the 2-s study period, the absorbances at 544 and 577 nm decrease, while an absorption at 630 nm appears and increases, indicating the oxidation of oxyhemoglobin to methemoglobin. Even though nitrite is able to perform the same oxidation (45), addition of 500 μM nitrite to oxyhemoglobin (50 μM) caused no significant oxidation after 3 min, indicating that nitrite-dependent oxidation of oxyhemoglobin is too slow to account for any of our observations. In addition, no change in the spectrum of oxyhemoglobin was observed in the 2-min period of study when peroxynitrite previously decomposed in buffer was used.
Figure 1.

Oxidation of oxyhemoglobin by peroxynitrite. Time-resolved (0–2 s) visible absorption spectra for stopped-flow reaction of human oxyhemoglobin (25 μM) and peroxynitrite (100 μM) in 100 mM phosphate buffer/0.1 mM dtpa, pH 7.4, 37°C. The dotted line represents the absorbance of control oxyhemoglobin.
Initial rates of oxyhemoglobin oxidation expressed as μM⋅s−1 (ɛ577 = 11,000 M−1⋅cm−1; ref. 39) linearly increased with initial peroxynitrite concentration (Fig. 2) and from this plot, an apparent second-order rate constant of (10.4 ± 0.3) × 103 M−1⋅s−1 was determined for the reaction, at pH 7.4 and 25°C. The pH profile (Fig. 2 Inset) shows a pKa value of 7.0, close to the one reported for cis-peroxynitrite (1, 2), indicating that peroxynitrous acid is the predominant species reacting with oxyhemoglobin.
Figure 2.

Initial velocities for the reaction between oxyhemoglobin and peroxynitrite as a function of peroxynitrite concentration. The reactions were conducted at 25°C in 100 mM phosphate buffer, pH 7.4/0.1 mM dtpa with 25 μM oxyhemoglobin. (Inset) Dependence of the determined second-order rate constant on pH.
Oxidation of Intracellular Oxyhemoglobin by Externally Added Peroxynitrite.
Addition of peroxynitrite to intact RBC caused a rapid, dose-dependent, oxidation of intracellular oxyhemoglobin (Fig. 3). The amount of methemoglobin linearly increased with increasing concentrations of added peroxynitrite until all the hemoglobin was consumed (ONOO− > 230 μM for 50 μM oxyhemoglobin). The yield of methemoglobin from 100 μM peroxynitrite as a function of total hemoglobin increased hyperbolically (Fig. 3 Inset), and from these values an oxidation yield of 45% was determined at pH 6.8. The experiments were performed in less than 2 min to avoid any contribution of nitrite to the oxidation process (see reverse-order addition runs). Therefore, the result suggests that peroxynitrite is the oxidant species responsible for the oxidation of intracellular oxyhemoglobin after having diffused across the erythrocyte membrane. The oxidation yield in intact RBC at pH 7.0 was significantly lower than the one determined in solution, and that determined in lysates had an intermediate value (Table 1), which indicates that both kinetic and diffusional factors were affecting oxidation yields in intact cells. As anionic and protonated forms of peroxynitrite (ONOO− and ONOOH) have different reactivities and net charge, it was likely that the two forms also differed in membrane permeance. Therefore, we studied the effect of pH on oxyhemoglobin oxidation yields, in solution, intact RBC, and lysates. The results are summarized in Table 1. For each condition studied, the dependence on pH had the same tendency—i.e., oxidation yields were higher at low pH values and decreased with pH. Significant differences were observed between lysate and intact RBC at pH 8.2, where 96% of peroxynitrite is in the anionic form and only 4% of the conjugated acid is present. However, at pH 5.5 (95% ONOOH), the oxidation yields in intact RBC and lysates were the same, indicating that the membrane does not represent a diffusional barrier for peroxynitrous acid.
Figure 3.

Oxidation of intracellular oxyhemoglobin by externally added peroxynitrite. Peroxynitrite (0–0.5 mM) was added to intact RBC (50 μM oxyhemoglobin) suspended in isotonic buffer A, pH 6.8. ○, Same experiment but decomposing peroxynitrite in buffer before addition to RBC. (Inset) Yield of methemoglobin from 100 μM peroxynitrite at pH 6.8.
Table 1.
Yields of oxyhemoglobin oxidation by peroxynitrite
| pH | Yield, %
|
||
|---|---|---|---|
| Solution | Lysate | RBC | |
| 5.5 | 170 ± 8 | 90 ± 5 | 90 ± 6 |
| 7.0 | 77 ± 4 | 40 ± 3 | 30 ± 2 |
| 8.2 | 20 ± 1 | 20 ± 2 | 10 ± 1 |
Yields were calculated from the slope of the line obtained by linear regression fitting of the experimental data (methemoglobin formed from 200 μM hemoglobin vs. peroxynitrite concentration). The reaction times and concentration ranges used were such that reverse-order-addition experiments yielded negligible amounts of methemoglobin in the three systems studied.
Effect of DIDS on Peroxynitrite Diffusion.
The diffusion of peroxynitrite anion through anion channels was investigated by using the HCO3−/Cl− exchanger inhibitor DIDS. Fig. 4 shows that in intact RBC at pH 7.0, DIDS inhibited the oxidation up to 50%. Importantly, the presence of DIDS (up to 300 μM) did not alter the oxidation yields of oxyhemoglobin in solution (data not shown), confirming that there is not a direct reaction between peroxynitrite and DIDS. The results indicate that DIDS, in agreement with its known effect on the exchange of stable anions, was blocking the entrance of peroxynitrite through the anion channel. Therefore, it was of interest to see how extracellular pH could affect diffusion of the oxidant in its anionic or acid forms. Intact RBC were resuspended in isotonic phosphate buffer at three different pH values: 5.5, 7.0, and 8.2. Intracellular pH was followed by loading the cells with the acetoxymethyl ester of the pH-sensitive fluorophore, BCECF, as described in Materials and Methods. Because the HCO3−/Cl− transporter participates in intracellular pH (pHi) maintenance, the final pHi observed in the presence of DIDS was different than the one achieved in its absence (ΔpH = 0.7). To avoid this, nigericin was added to equilibrate external with internal pH by a different mechanism: exchange of H+ for K+. In this case, the difference between external and internal pH after equilibration with nigericin was less than 0.1 pH unit, and the presence of DIDS made no difference. As oxidation yields decrease with pH (see Table 1), the concentration of external peroxynitrite added was increased with pH to obtain similar total oxidation under the three pH values studied. The results obtained are summarized in Table 2. At pH 5.5, where 95% of peroxynitrite is in the protonated form, the presence of DIDS did not affect the yield of intracellular hemoglobin oxidation. On the other hand, at pH 7.0, 60% of the peroxynitrite is in the anionic form, and the blockade of the anion channels by DIDS reduced the oxidation yields by 50%. Moreover, at pH 8.2 the protection by DIDS was higher, reaching 65%.
Figure 4.

Effect of DIDS on intracellular oxyhemoglobin oxidation. Erythrocytes (60 μM oxyhemoglobin) resuspended in isotonic buffer A, pH 6.8, were incubated in the presence of the indicated amount of DIDS (30 min, room temperature) and washed before addition of 300 μM peroxynitrite. Methemoglobin formed in the reverse-order-addition experiments was no higher than in the controls.
Table 2.
Effect of pH on DIDS-dependent inhibition of intracellular hemoglobin oxidation
| pH | Methemoglobin, %
|
% inhibition by DIDS | |
|---|---|---|---|
| Without DIDS | With DIDS | ||
| 5.5 | 55 ± 2 | 57 ± 3 | 0 |
| 7.0 | 48 ± 2 | 24 ± 2 | 50 |
| 8.2 | 37 ± 2 | 13 ± 1 | 65 |
Peroxynitrite concentrations used were 50 μM at pH 5.5, 150 μM at pH 7.0, and 650 μM at pH 8.2 to achieve similar oxidation yields.
Nitration of Intracellular Hemoglobin by Peroxynitrite.
Under reducing and denaturing conditions (SDS/15% PAGE), human hemoglobin electrophoretically moved as a monomer of ≈15.5 kDa (Coomassie blue staining for proteins) and was not recognized by the anti-nitrotyrosine antibody (Fig. 5, lane 1). After exposure to peroxynitrite, it was nitrated as revealed by the anti-nitrotyrosine antibody (lane 2). It was also observed that a nitrated dimer (mobility corresponding to 31 kDa) was formed in the peroxynitrite treatment. Analysis of intact RBC exposed to peroxynitrite demonstrated nitration of some of the protein bands (lane 6). The concentration of peroxynitrite used for the nitration experiments (2 mM) was higher than the concentrations used in the oxidation experiments (≤0.5 mM, see Fig. 3). This was necessary for obtaining a significant nitrotyrosine signal, as peroxynitrite-mediated nitrations occur at much lower yields than direct oxidations (15). When RBC were pre-treated with NEM, to block the reactivity of peroxynitrite toward sulfhydryls (1), the signal corresponding to the nitrated proteins was significantly intensified (lane 5 vs. lane 6). The main nitrated band in RBC lysates seemed to correspond with the dimeric nitrated hemoglobin. Reverse-order-addition experiments (lane 3 and 4) showed some nitrotyrosine formation that is not observed in the control sample (lane 7 and 8). Exposure of RBC to 4 mM nitrite for 2 min resulted in moderate nitration similar in extent to that observed in the reverse-order-addition experiments (data not shown).
Figure 5.

Effect of peroxynitrite on tyrosine nitration of human hemoglobin and RBC. Samples were separated on an SDS/15% polyacrylamide gel and examined by Western blot analysis with the polyclonal antibody against nitrotyrosine. Lanes correspond to the following: 1,12 μg of human hemoglobin; 2, 12 μg of peroxynitrite-treated hemoglobin; 6, intact RBC treated with peroxynitrite; 5, RBC treated with 1 mM NEM before peroxynitrite addition; 4, reverse-order-addition experiment of lane 6; 3, reverse-order-addition experiment of lane 5; 8, intact RBC; and 7, RBC pretreated with 1 mM NEM. Lanes 3–8 contained RBC equivalent to 24 μg of protein. Molecular mass markers were BSA (66), ovalbumin (45), trypsinogen (24), and α-lactalbumin (14.2) and are expressed in kDa to the left.
DISCUSSION
Peroxynitrite reacts fast with oxyhemoglobin to yield methemoglobin (Fig. 1), and this reaction was utilized as an indicator of intracellular peroxynitrite diffusion in intact erythrocytes. A second-order rate constant of (10.4 ± 0.3) × 103 M−1⋅s−1 was determined for the reaction of human oxyhemoglobin with peroxynitrite at pH 7.4 and 25°C. The rate constant and oxidation yields decreased with pH with a pKa of 7.0 (Fig. 2, Table 1) which indicates that the predominant species reacting with hemoglobin is the protonated form of peroxynitrite, peroxynitrous acid. The oxidation of hemoglobin is not specific for peroxynitrite, since the same oxidation product can be obtained by reactions of oxyhemoglobin with peroxynitrite decomposition products such as nitrite (NO2−) and nitrogen dioxide (·NO2) (45, 46). However, the peroxynitrite-mediated oxidation of hemoglobin is significantly faster than the reaction with nitrite, which is kinetically characterized by a lag period followed by an autocatalytic phase (45). Moreover, at the concentrations of peroxynitrite used for causing oxidation of oxy- to methemoglobin (≤500 μM ONOO−) neither decomposed peroxynitrite nor up to 500 μM nitrite were able to perform the process within the 2-min period of this study. In addition, the reaction of oxymyoglobin with ·NO2 was also reported to be very slow, with k = 9 ± 5 M−1⋅s−1 (46). Oxidation yields based on molecules of oxidized oxyhemoglobin per molecule of added peroxynitrite were higher than 100% at pH 5.5 (170%), similar to what was previously observed for thiol oxidation yields with peroxynitrite anion (1). This yield reflects the fact that each molecule of peroxynitrite is capable of abstracting up to two electrons from a given target, either directly or in sequential steps.
Addition of peroxynitrite to RBC also rapidly transformed the intracellular oxyhemoglobin into methemoglobin in a dose-dependent manner (Fig. 3). The oxidation yield for free hemoglobin in phosphate buffer at pH 7.0 was 77% and decreased to 30% for intact RBC (Table 1). These differences are partially due to the presence of erythrocyte cytosolic components that compete with hemoglobin for peroxynitrite (for example glutathione, which is 2–3 mM inside the RBC; ref. 47), as can be concluded from the experiments with cell lysates (Table 1). When the oxidation yields with lysate and intact RBC (Table 1) are compared, it can be seen that at pH 5.5, where most of peroxynitrite is in the protonated form (95% ONOOH), the presence of a membrane did not alter the oxidation yields, indicating that peroxynitrous acid can freely diffuse across the lipid bilayer. On the other hand, at pH 8.2, 96% of peroxynitrite is in the anionic form, and the yields of oxyhemoglobin oxidation decreased by 2-fold because of the diffusional barrier imposed by the erythrocyte membrane. Thus, the possibility of peroxynitrite anion diffusing inside the cell by anion channels was investigated by using specific inhibitors of these anion transporters.
DIDS belongs to a family of sulfonated stilbenes that inhibits the exchange of anions across the erythrocyte cell. The mechanism of inhibition is through binding to band 3 (the HCO3−/Cl− transporter, major component of the RBC membrane) in a two-step process: initial noncovalent attachment followed by covalent reaction with an amino group of a lysine in the N-terminal third of the membrane domain (48). At pH 7.0, DIDS inhibited up to 50% the oxidation of hemoglobin by external addition of peroxynitrite (Fig. 4). When the cells were equilibrated at acidic pH, where most of the oxidant is in the protonated form, no protection by DIDS was afforded (Table 2). Moreover, the inhibition by the stilbene disulfonate was higher at alkaline pH than at pH 7.0. These results indicate there is a transmembrane diffusion of peroxynitrite anion by way of the erythrocytic anion channel, band 3.
To further confirm peroxynitrite diffusion inside the cell, Western blot analysis of the relatively more specific peroxynitrite reaction product, nitrotyrosine, was performed. Nitrotyrosine residues were detected on intracellular proteins after external addition of 2 mM peroxynitrite to intact RBC (Fig. 5, lane 6), whereas decomposed peroxynitrite (lane 4) as well as 4 mM nitrite caused much lower levels of nitration. The low protein nitration observed in the reverse-order-addition experiment and in the exposure to nitrite must be due to ·NO2 formation from hemoglobin oxidation by nitrite coming from peroxynitrite stock solutions and as a byproduct of its decomposition (35). Indeed, as the reaction with nitrite is autocatalytic (45), we found that 2–4 mM nitrite caused significant oxyhemoglobin oxidation (>50%) within 2 min, implying ·NO2 formation. It is important to note that ·NO2 formation from nitrite requires the presence of oxyhemoglobin but would not occur with methemoglobin. Because in the peroxynitrite-treated samples (i.e., Fig. 5, lanes 5 and 6) all oxyhemoglobin was rapidly oxidized to methemoglobin, it is safe to assume that most, if not all, of the nitration observed during the peroxynitrite exposure was exclusively due to peroxynitrite and not to secondary products. When intracellular thiols were covalently blocked by NEM, nitration of intracellular proteins by external peroxynitrite was significantly increased (Fig. 5, lane 5), demonstrating an effective competition between cytosolic hemoglobin and thiols for the oxidant. This result correlates well with the observed reduction on the yields of oxyhemoglobin oxidation in buffer solution compared with erythrocyte lysates (Table 1). The more intense signal observed for the reverse-order-addition experiment with NEM-treated RBC (Fig. 5, lane 3 vs. lane 4) can also be explained considering the reaction of ·NO2 with glutathione (k2 ≈ 108 M−1⋅s−1; ref. 49), therefore, part of the nitrating agent, ·NO2, in lane 4 is consumed in the reaction with reduced glutathione.
An important point when considering the likelihood of peroxynitrite diffusion relates to its fate in the presence of extracellular targets (50, 51). In this context, it is particularly important to consider the reaction of peroxynitrite with CO2 (k = 5.7 × 104 M−1⋅s−1 at 37°C; ref. 17) that leads to the formation of an adduct (nitrosoperoxocarbonate, ONO2CO2−), which in turn has a very short half-life (≤1 ms). In extracellular fluids, the reaction with CO2 (1–1.5 mM in equilibrium with bicarbonate) determines a half-life of peroxynitrite in the order of ≈20 ms. Still, assuming a diffusion coefficient for peroxynitrite anion similar to that of nitrate (1,500 μm2⋅s−1; ref. 52) and a mean diffusion distance to an RBC (hematocrit approximately 45%) of 4 μm, the average diffusion time, applying Fick’s second law, will be ≈6 ms, significantly less than that of the reaction with CO2. Thus, even in the presence of CO2, peroxynitrite anion will be able to reach inside the erythrocyte. It is worth noting that peroxynitrous acid is expected to have an even higher diffusion coefficient than the anion.
In summary, our results indicate that peroxynitrite is able to cross the erythrocyte membrane by two different mechanisms: (i) the anionic form through the HCO3−/Cl− exchanger (specifically inhibited by DIDS), and (ii) the protonated form by passive diffusion. Diffusion of O2⨪ by the erythrocyte anion channel was previously reported (28); however, due to the low pKa of the pair, 4.8, diffusion of the protonated form is unlikely to be biologically relevant. On the contrary, the pKa for the pair ONOO−/ONOOH of 6.8, close to physiological pH, implies that both mechanisms of peroxynitrite diffusion could operate in vivo. The diffusion of peroxynitrite anion could be different in cells other than the erythrocyte, depending on the presence of membrane stilbene-inhibitable anion channels. The erythrocyte membrane is particularly abundant in band-3 protein (1 × 106 copies per cell; ref. 48), but there are proteins homologous to band 3 in nonerythroid cells with similar function, promoting anion transport (48), that might serve to transport peroxynitrite anion as well. Passive diffusion of peroxynitrous acid becomes more relevant under acidic conditions or in membrane systems with limited presence of anion channels. Therefore, our results help to understand the mechanisms by which peroxynitrite diffuses across membranes and also provide evidence indicating that peroxynitrite could react at a distance from its site of production (i.e., 1–2 cell diameters), even in the presence of excess target molecules.
Acknowledgments
The authors thank Dr. A. Cayota for his assistance on the Western blot analysis and Dr. G. Ferrer-Sueta for collaborating on the stopped-flow scans. This work was supported by Consejo Nacional de Investigaciones Científicas y Técnicas (Uruguay) Grant 138 and a grant from the Swedish Agency of Research Cooperation with Developing Countries to R.R. and by Consejo Nacional de Investigaciones Científicas y Técnicas (Uruguay) Grant 313 to A.D.
Note Added in Proof
During the review process two manuscripts affirming the general concept of membrane penetrability by peroxynitrite were published (53, 54).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: ONOO−, peroxynitrite anion; ONOOH, peroxynitrous acid; DIDS, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid; ·NO, nitric oxide; O2⨪, superoxide anion; RBC, red blood cells; BCECF, 2′,7′-bis-(2-carboxyethyl)-5-carboxyfluorescein; NEM, N-ethylmaleimide; dtpa, diethylenetriaminepentaacetic acid.
The International Union of Pure and Applied Chemistry recommended names for peroxynitrite anion (ONOO−), peroxynitrous acid (ONOOH), and nitric oxide (·NO) are oxoperoxonitrate(1−), hydrogen oxoperoxonitrate, and nitrogen monoxide, respectively. The term peroxynitrite is used to refer to the sum of ONOO− and ONOOH.
The concentration of hemoglobin used in intact erythrocyte suspension experiments will be expressed as the concentration of oxyhemoglobin (concentration of heme, μM) released after lysis, which is achieved by pelleting the cells and resuspending them in the same volume of distilled water.
References
- 1.Radi R, Beckman J S, Bush K M, Freeman B A. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 2.Koppenol W H, Moreno J J, Pryor W, Ischiropoulos H, Beckman J S. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 3.Beckman J S, Ye Y Z, Anderson P G, Chen J, Accavitti M A, Tarpey M M, White C R. Biol Chem Hoppe-Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 4.Beckman J S, Chen J, Crow J P, Ye Y Z. Prog Brain Res. 1994;103:371–380. doi: 10.1016/s0079-6123(08)61151-6. [DOI] [PubMed] [Google Scholar]
- 5.Royall J A, Kooy N W, Beckman J S. New Horiz. 1995;3:113–122. [PubMed] [Google Scholar]
- 6.Pryor W A, Squadrito G L. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 7.Crow J P, Spruell C, Chen J, Gunn C, Ischiropoulos H, Tsai M, Smith C D, Radi R, Koppenol W H, Beckman J S. Free Radical Biol Med. 1994;16:331–338. doi: 10.1016/0891-5849(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 8.Radi R, Beckman J S, Bush K M, Freeman B A. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- 9.Beckman J S, Beckman T W, Chen J, Marshall P M, Freeman B A. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King P A, Anderson V E, Edwards J O, Gustafson G, Plumb R C, Suggs J W. J Am Chem Soc. 1992;114:5430–5432. [Google Scholar]
- 11.Pryor W A, Jin X, Squadrito G L. Proc Natl Acad Sci USA. 1995;91:11173–11177. doi: 10.1073/pnas.91.23.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomson L, Trujillo M, Telleri R, Radi R. Arch Biochem Biophys. 1995;319:491–507. doi: 10.1006/abbi.1995.1321. [DOI] [PubMed] [Google Scholar]
- 13.Bartlett D, Church D F, Bounds P L, Koppenol W H. Free Radical Biol Med. 1994;18:85–92. doi: 10.1016/0891-5849(94)e0133-4. [DOI] [PubMed] [Google Scholar]
- 14.Vasquez-Vivar J, Denicola A, Radi R, Augusto O. Chem Res Toxicol. 1997;10:786–794. doi: 10.1021/tx970031g. [DOI] [PubMed] [Google Scholar]
- 15.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin J C, Smith C D, Beckman J S. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 16.Alvarez B, Rubbo H, Kirk M, Barnes S, Freeman B A, Radi R. Chem Res Toxicol. 1996;9:390–396. doi: 10.1021/tx950133b. [DOI] [PubMed] [Google Scholar]
- 17.Denicola A, Freeman B A, Trujillo M, Radi R. Arch Biochem Biophys. 1996;333:49–58. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- 18.Lymar S V, Hurst K. J Am Chem Soc. 1995;117:8867–8868. [Google Scholar]
- 19.Leeuwenburgh C, Hardy M M, Hazen S L, Wagner P, Oh-ishi S, Steinbrecher U P, Heinecke J W. J Biol Chem. 1997;272:1433–1436. doi: 10.1074/jbc.272.3.1433. [DOI] [PubMed] [Google Scholar]
- 20.Ischiropoulos H, Al-Mehdi A B, Fisher A B. Am J Physiol. 1995;269:L158–L164. doi: 10.1152/ajplung.1995.269.2.L158. [DOI] [PubMed] [Google Scholar]
- 21.Wang P, Zweier J L. J Biol Chem. 1996;271:29223–29230. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- 22.MacMillan-Crow L A, Crow J P, Kerby J D, Beckman J S, Thompson J A. Proc Natl Acad Sci USA. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beckman J S, Carson M, Smith C D, Koppenol W H. Nature (London) 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 24.Mulligan M S, Hevel J M, Marletta M A, Ward P A. Proc Natl Acad Sci USA. 1991;88:6338–6342. doi: 10.1073/pnas.88.14.6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huie R E, Padmaja S. Free Radical Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 26.Denicola A, Souza J M, Radi R, Lissi E. Arch Biochem Biophys. 1996;328:208–212. doi: 10.1006/abbi.1996.0162. [DOI] [PubMed] [Google Scholar]
- 27.Lancaster J R., Jr Proc Natl Acad Sci USA. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lynch R E, Fridovich I. J Biol Chem. 1978;253:4697–4699. [PubMed] [Google Scholar]
- 29.Radi R, Rodríguez M, Castro L, Telleri R. Arch Biochem Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- 30.Rubbo H, Denicola A, Radi R. Arch Biochem Biophys. 1994;308:96–102. doi: 10.1006/abbi.1994.1014. [DOI] [PubMed] [Google Scholar]
- 31.Hu P, Ischiropoulos H, Beckman J S, Matalon S. Am J Physiol. 1994;266:L628–L634. doi: 10.1152/ajplung.1994.266.6.L628. [DOI] [PubMed] [Google Scholar]
- 32.Szabo C, Zingarelli B, O’Connor M, Salzman A L. Proc Natl Acad Sci USA. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soszynski M, Bartosz G. Biochim Biophys Acta. 1996;1291:107–114. doi: 10.1016/0304-4165(96)00052-9. [DOI] [PubMed] [Google Scholar]
- 34.Radi R. Methods Enzymol. 1996;269:354–365. doi: 10.1016/s0076-6879(96)69036-3. [DOI] [PubMed] [Google Scholar]
- 35.Evans T J, Buttery L D, Carpenter A, Springall D R, Polak J M, Cohen J. Proc Natl Acad Sci USA. 1996;93:9553–9558. doi: 10.1073/pnas.93.18.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt H H W, Kelm M. In: Methods in Nitric Oxide Research. Feelisch M, Stamler J, editors. New York: Wiley; 1996. pp. 491–498. [Google Scholar]
- 37.Cotton M L, Dunford H B. Can J Chem. 1973;51:582–587. [Google Scholar]
- 38.Ye Y Z, Strong M, Huang Z, Beckman J S. Methods Enzymol. 1996;269:201–209. doi: 10.1016/s0076-6879(96)69022-3. [DOI] [PubMed] [Google Scholar]
- 39.Winterbourn C C. Methods Enzymol. 1990;186:265–272. doi: 10.1016/0076-6879(90)86118-f. [DOI] [PubMed] [Google Scholar]
- 40.Flohe L, Otting F. Methods Enzymol. 1984;105:93–104. doi: 10.1016/s0076-6879(84)05013-8. [DOI] [PubMed] [Google Scholar]
- 41.Aebi H. Methods Enzymol. 1984;105:121–125. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 42.Espenson J H. Chemical Kinetics and Reaction Mechanisms. 2nd Ed. New York: McGraw–Hill; 1995. [Google Scholar]
- 43.Thomas J A, Buchsbaum N, Zimniak A, Racker E. Biochemistry. 1979;18:2210–2218. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- 44.Bradford M. Anal Biochem. 1976;72:248–256. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 45.Kosaka H, Imaizumi K, Tyuma I. Biochim Biophys Acta. 1981;702:234–241. doi: 10.1016/0167-4838(82)90508-8. [DOI] [PubMed] [Google Scholar]
- 46.Wade R S, Castro C E. Chem Res Toxicol. 1996;9:1382–1390. doi: 10.1021/tx9600457. [DOI] [PubMed] [Google Scholar]
- 47.Tietze F. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 48.Jennings M L. Annu Rev Biophys Biophys Chem. 1989;18:397–430. doi: 10.1146/annurev.bb.18.060189.002145. [DOI] [PubMed] [Google Scholar]
- 49.Prütz W A, Moening H, Butler J, Land E J. Arch Biochem Biophys. 1985;243:125–134. doi: 10.1016/0003-9861(85)90780-5. [DOI] [PubMed] [Google Scholar]
- 50.Beckman J S. In: Nitric Oxide: Principles and Actions. Lancaster J R Jr, editor. New York: Academic; 1996. pp. 1–82. [Google Scholar]
- 51.Lymar S V, Hurst J K. Chem Res Toxicol. 1996;9:845–850. doi: 10.1021/tx960046z. [DOI] [PubMed] [Google Scholar]
- 52.Lide D R, editor. Handbook of Chemistry and Physics. 71st Ed. Boca Raton, FL: CRC; 1990. pp. 6–151. [Google Scholar]
- 53.Marla S S, Lee J, Groves J T. Proc Natl Acad Sci USA. 1997;94:14243–14248. doi: 10.1073/pnas.94.26.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mallozzi C, Di Stasi A M M, Minetti M. FASEB J. 1997;11:1281–1290. doi: 10.1096/fasebj.11.14.9409547. [DOI] [PubMed] [Google Scholar]