

Abstract
A pyridine-functionalized sol-gel film has been formed by electrodeposition at a glassy carbon electrode surface. When this protonated film is exposed to a Cr(VI) solution, the Cr(VI) anions are preconcentrated at the electrode surface. Using square wave voltammetry, the Cr(VI) species are reduced to Cr(III), and a peak current corresponding to this reduction is generated at 0.17 V. The peak currents can be correlated with the Cr(VI) concentration. The functionalized sol-gel films demonstrated an enhanced sensitivity for Cr(VI) in aqueous solutions, providing a limit of detection at the low ppb level. Interference studies also displayed the high selectivity of the films for Cr(VI), and the system was able to tolerate a large excess of Cr(III) with no adverse affects. The reported electrodeposition method of film formation uses commercially available reagents and yields films quickly and reproducibly. The growth of these sol-gel films was monitored using an electrochemical quartz crystal microbalance (EQCM), and they were characterized by X-ray photoelectron spectroscopy (XPS) and scanning electron microscopy (SEM). The reported work shows the promise of such an electrode for use in Cr(VI) sensing applications.
Keywords: Sol-gel, Chromium(VI), Electrodeposition
1. Introduction
As a suspected carcinogenic agent and toxic pollutant, Cr(VI) poses a threat when present even at trace levels. Several methods have been reported for the successful determination and quantification of Cr(VI) in solution [1-16]. Some of these techniques rely on spectroscopic methods [1-4] while others depend on mass-sensitive devices [5-8]. Electrochemical detection of Cr(VI) species has attracted much interest [9-16] due to its high sensitivity, portability, and ability to distinguish Cr(VI) from Cr(III). Cox and Kulesza investigated Cr(VI) determination at a poly(4-vinylpryidine)-coated platinum electrode. They were able to demonstrate the preconcentration of Cr(VI) in the polymer film and show the lack of interference by metal cations [9]. Using diphenylcarbazide, Paniagua and coworkers showed the potential of their modified carbon paste electrode for enhanced Cr(VI) determination [10]. Svancara and coworkers also investigated Cr(VI) determination at a carbon paste electrode modified with quarternary ammonium salts [11]. More recently, Compton and coworkers have probed Cr(VI) detection at bare gold, glassy carbon, and boron-doped diamond electrodes [12]. Detection limits at the low ppt level using electrochemical methods have been reported. Yang and Huang have reported the use of a polyaniline/polystyrene composite electrode as a detector in a flow injection analysis system that provides a Cr(VI) detection limit of 4 ppt [13], while Wang and coworkers have developed a novel method of Cr(VI) determination at a bismuth film electrode using catalytic adsorptive stripping voltammetry [14]. Turyan and Mandler have also investigated the determination of Cr(VI) at a self-assembled monolayer electrode, achieving a detection limit of about 1 ppt and a high degree of selectivity over other metal ions [15].
One attractive method for analytical sensing is the use of sol-gel materials in conjunction with electrochemistry [17-28]. Typically, sol-gels have been deposited on electrode surfaces using spin-coating techniques [17,21,25,26]. Films formed in this manner are usually acid-catalyzed and, as a result, have a compact structure with low porosity [17,19]. Base-catalyzed sol-gels, formed by the reactions shown in Eq. 1-3, are usually of higher porosity [28,29], a critical issue in many sol-gel sensing applications. A very promising method that has recently emerged for the formation of sol-gel films of high porosity is the electrodeposition of sol-gels at electrode surfaces [18-20,22,23]. Initially investigated by Shacham et al. [23] and later elaborated on by Collinson et al. [18,19] and Walcarius and coworkers [20,22], this method of sol-gel formation relies on the application of a negative potential to increase the pH at the electrode surface, causing the immediate condensation of the sol-gel. The unique aspect of this procedure is that gelation and drying proceed independently of each other, allowing for the formation of films with greater porosity [19].
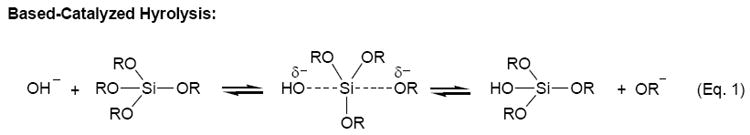 |
(Eq. 1) |
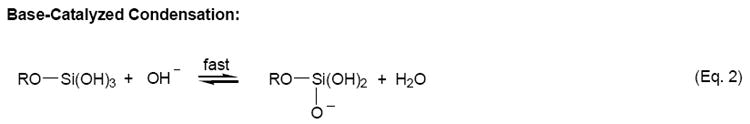 |
(Eq. 2) |
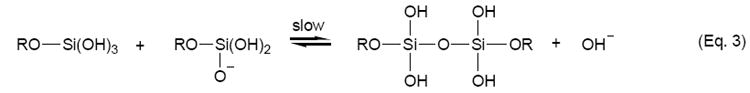 |
(Eq. 3) |
In this work, the electrodeposition technique is used to form a functionalized sol-gel film at the surface of a glassy carbon electrode for the enhanced determination of Cr(VI) anions. Most metal ions are cations. Chromium at its highest, VI oxidation state, however, exists mainly as anions ( , and in acidic and in basic solutions [30]), providing a unique opportunity for Cr(VI) preconcentration and detection [31]. Previous reports have demonstrated the use of sol-gels for the detection and separation of Cr(VI) from aqueous solutions [1,32]. We have designed and deposited a sol-gel thin film containing grafted pyridinium on a glassy carbon electrode, and the thin film was found to preconcentrate Cr(VI) anions, leading to Cr(VI) detection with enhanced sensitivity and selectivity (Scheme 1). This approach is based on the electrostatic interaction between the positively charged pyridinium groups in the sol-gel matrix and the negatively charged Cr(VI) anions [15, 33-37]. Reduction of the preconcentrated Cr(VI) anions converts them into Cr3+ cations, regenerating the electrode surface for subsequent preconcentration and analysis. Our studies are reported here.
Scheme 1.
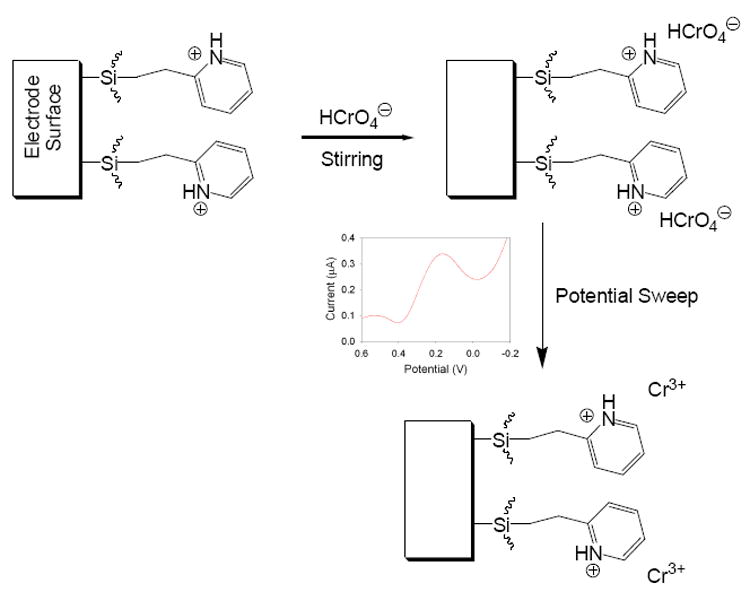
Schematic of the electroanalysis process at pyridinium-functionalized electrode. is used to represent the Cr(VI) anionic species.
2. Experimental
2.1 Chemical reagents and materials
Tetramethyl orthosilicate (TMOS, Si(OMe)4, 98%, Sigma-Aldrich), 2-[2- (trimethoxysilyl)ethyl]-pyridine (Gelest), hydrochloric acid (HCl, certified A.C.S., Fisher), ethanol (EtOH, 95%, Fisher), and potassium chloride (KCl, certified A.C.S., Mallinckrodt) were used as received. Standard solutions of Cr(VI) were prepared by serial dilution of a 1017 μg/mL AA standard (Sigma-Aldrich). All compounds used for interference studies were of the highest purity available. Solutions and standards were prepared using deionized (DI) water (18 MΩ-cm) from a Barnstead International E-pure 4-holder deionization system. The working electrodes typically used during the experiments were glass-encased glassy carbon electrodes (3 mm diameter, Cypress Systems, Inc.). Before each coating process, these electrodes were polished using 0.05 micron alumina powder. Each electrode was then rinsed with water and sonnicated for 20 minutes, followed by soaking in piranha solution for an additional 15 minutes (Caution: piranha solution will react violently with organics and proper caution should be taken when handling). For the electrodeposition experiments, a Ag/AgCl wire electrode, prepared by soaking Ag wire in bleach, was used. For all other applications, a typical, fritted reference Ag/AgCl electrode was employed. Platinum wire was used as an auxiliary electrode. For the electrochemical quartz crystal microbalance (EQCM) experiments, a Teflon cell was used to house the crystals. Polished, mounted, and bonded gold-coated quartz crystals (International Crystal Manufacturing Company, Inc.) were used as received. Each crystal had a fundamental frequency of approximately 7.995 MHz and an electrode diameter of 0.546 cm, consisting of a 1000 Ǻ film of gold deposited on a 100 Ǻ layer of chromium.
2.2 Instrumentation
A CH Instruments model 400A potentiostat with electrochemcial quartz crystal microbalance (EQCM) capability and corresponding software were used for the electrochemical studies. X-ray photoelectron spectroscopy (XPS) was carried out using a Phi 5100LS spectrometer with an Al Kα source at 300 W. Scanning electron microscopy (SEM) images were obtained using a Hitachi S4300-E SEM with the field-emission gun operating at 3 kV.
2.3 Coating procedure
A sol solution typically consisting of 2 mL of 0.2 M KCl, 2 mL of EtOH, 250 μL of TMOS, and 250 μL of 2-[2-(trimethoxysilyl)ethyl]-pyridine was prepared and stirred thoroughly for several minutes to ensure a homogeneous mixture. After mixing, the working electrode was exposed to the sol solution, and a potential of -0.9 V was applied for 60 s. The working electrode was then rinsed with several aliquots of a 50:50 mixture of EtOH and DI water. It was then placed in an oven at 68 °C for 12 h and subsequently cured further at room temperature for an additional 12 h.
2.4 Analysis procedure
Before each analysis procedure, the sol-gel coated electrode was placed in 0.1 M HCl (pH = 1) for 10 min to ensure protonation of the pyridinium [38] and silanol [29] groups within the film. The electrodes were then exposed to a solution consisting of 0.1 M KCl, 0.1 M HCl, and a variable amount of Cr(VI). After 10 min of solution stirring, a square wave voltammogram was collected, typically from 0.6 V to -0.2 V with a frequency of 15 Hz, amplitude of 0.025 V, and incremental potential of 0.004 V. The electrodes were then rinsed with electrolyte solution consisting of 0.1 M KCl and 0.1 M HCl, and several CV scans were run in only electrolyte solution to ensure that any residual Cr(VI) present was eliminated. Subsequent analyses of various Cr(VI) concentrations could then be carried out.
3. Results and Discussion
3.1 Film electrodeposition and characterization
The electrodeposition process is similar to those reported previously [18,19]. Briefly, a negative potential applied to the working electrode creates an increase in pH at the electrode surface due to the generation of hydroxide ions (Eq. 4-6). When TMOS and 2-[2-(trimethoxysilyl)ethyl]-pyridine are present, the increase in OH− concentration base catalyzes the hydrolysis and condensation (Eq. 1-3) of the sol at the electrode surface and produces a film functionalized with pyridine. Exposing the glassy carbon electrode to piranha solution prior to carrying out the electrodeposition process is expected to produce numerous surface oxides that allow for greater film adhesion to the electrode. The applied time and magnitude of the electrodeposition potential and the ratio of TMOS to 2-[2-(trimethoxysilyl)ethyl]-pyridine all affect the thickness and porosity of the film [37].
| (Eq. 4) |
| (Eq. 5) |
| (Eq. 6) |
3.1.1 EQCM study
In order to probe the electrodeposition process and to characterize the functionalized sol-gel films by XPS and SEM, Au-coated quartz crystal microbalance (QCM) crystals were employed to prepare sol-gel films under otherwise identical conditions as those used to coat sol-gel films on glassy carbon electrodes. Measurements using an electrochemical quartz crystal microbalance (EQCM), a device sensitive to minute mass changes at the electrode surface, were carried out to monitor in-situ the sol-gel formation. Figure 1 shows the change in current and frequency as a function of time at a Au-coated, 8.0 MHz QCM crystal when a potential of -0.9 V was applied to the standard sol solution (Experimental Section). As expected, the frequency drops as the potential is applied and the sol-gel film forms at the electrode surface. After 60 s of deposition, a relatively thin film is formed on the surface. Longer deposition times were found to result in thick films that eventually prevent the crystal from further oscillation [37]. In such cases, the deposited sol-gel mass had become so large that the crystal frequency had drifted significantly from its initial fundamental frequency and was no longer compatible with the EQCM oscillator. Increasing the amount of 2-[2-(trimethoxysilyl)ethyl]-pyridine in the sol solution hindered film formation and produced films with reduced stability that easily flaked off the electrode surfaces. A deposition time of 60 s was also used to make sol-gel coatings on glassy carbon electrodes. As discussed below, the Cr(VI) preconcentration in the pyridium-grafted sol-gel coatings and detection are likely a mass-transport control process, and the deposition time of 60 s gives coatings that provide optimum detection limits [37].
Fig. 1.
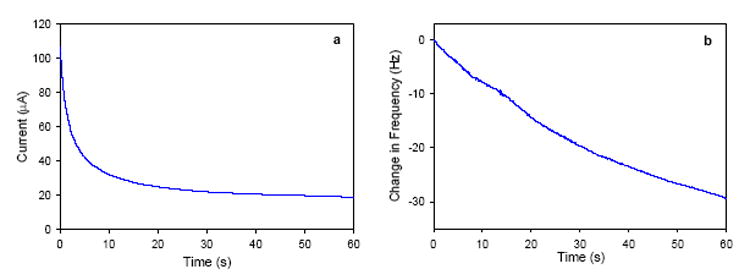
(a) Current-time and (b) frequency-time plots at a gold QCM electrode in a TMOS/2-[2-(trimethoxysilyl)ethyl]-pyridine sol solution when a potential of -0.9 V is applied.
3.1.2 Analysis of films using SEM
SEM studies were carried out to reveal the morphology of the sol-gel coatings electrodeposited on the Au QCM crystals (Figure 2). The images obtained at higher magnifications (Figure 2a and 2b) show islands of particles that are likely nucleation centers for film growth [22]. Lower magnifications (Figure 2c and 2d) show particles on the nanometer scale separated from one another. The vacant areas between the particles are likely the gold QCM surface, as Au peaks were observed in some of the XPS spectra that are discussed below [37]. Such vacancies can likely be attributed to the evolution of hydrogen gas during the electrodeposition process [22]. Also, considering the short deposition time of 60 s that provides the optimum detection limit, to be discussed below, it is perhaps not surprising that there are the vacant areas. Traditional sol-gel deposition processes usually require a much longer reaction time prior to, e.g., spin- or dip-coating on substrates [29,39]. The particulate nature of the films is expected as the sol formations are base-catalyzed [29], and coated glassy carbon electrodes viewed under an optical microscope revealed an analogous surface morphology. Similar structures of electrodeposited sol-gel films have been reported [19,22].
Fig. 2.

SEM images of a pyridine-functionalized sol-gel deposited on a gold QCM surface Images taken are at magnifications of: (a) 1 mm; (b) 100 μm; (c) 25 μm; (d) 5 μm.
3.1.3 XPS film analysis
XPS spectra were obtained of films deposited on gold QCM crystals [37]. Peaks of Si, C, O, and N as well as Au [37] were observed. Figure 3 shows the results obtained when one such film was analyzed. The Si, C, O, and N peaks are consistent with the formation of a sol-gel film functionalized with pyridine on the QCM electrode surface. The Au peaks [37] are likely from the vacant areas between islands of sol-gel particles. Additional studies showed that longer film deposition times led to increased peak intensity, as expected [37].
Fig. 3.
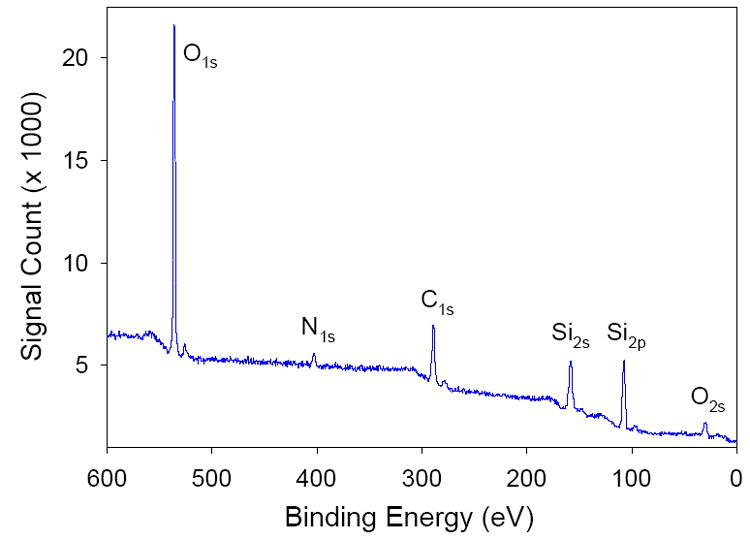
XPS spectrum obtained at a functionalized sol-gel film deposited on a QCM crystal.
3.2 Cr(VI) analysis
The general process occurring at the modified glassy carbon electrode during Cr(VI) analysis is illustrated in Scheme 1. The pyridinium groups present in the sol-gel are protonated by prior soaking of the film in 0.1 M HCl. The electrode is then exposed to a Cr(VI) solution where the analyte anions are preconcentrated at the electrode surface through the electrostatic interaction between the positively-charged pyridinium groups and the negatively-charged Cr(VI) ions. After a period of time has passed and the analyte species has had sufficient time to diffuse to the electrode surface, a square wave voltammogram is collected. A peak occurs at approximately 0.17 V corresponding to the reduction of Cr(VI) to Cr(III), as has been reported previously [9,11,15]. The produced Cr(III) cations are then expelled from the sol-gel film containing the pyridinium cations. To ensure that that Cr species are removed, however, the electrodes are rinsed with electrolyte solution and several potential cycles are performed [40].
Attempts in the current work at electrochemically protonating the pyridinium groups via an applied positive potential, similar to the procedure carried out by Walcarius and Sibottier in which a negative potential was applied for the deprotonation of their amine-functionalized silica film [22], did not result in an enhanced peak current for the Cr(VI) reduction. It was therefore assumed that prior film exposure to a pH 1 acidic solution provided sufficient protonation of both the pyridinium and silanol groups present in the sol-gel. Given the fact that the isoelectric point of silica sol-gel is close to pH = 2 [29] and the pKa value of 2-methylpyridine has been reported as 5.96 [38a], this assumption seems accurate [38b]. It should also be pointed out that most electrodeposited sol-gel films formed using similar procedures have been used for the analysis of cations and neutral species [18,19,22]. None of these films has been used for the analysis of anions, as the negatively charged silicate films have excluded the possibility of doing so [18,19]. However, by carrying our studies out under acidic conditions, it should be possible to analyze negatively-charged Cr(VI) ions.
3.3 as a redox probe
was used as a redox probe to investigate the electrodeposited sol-gel film and its anion-exchange capability. It was chosen as a model analyte for its negative charge and the fact that it is a well-established redox system [41]. Previous studies have demonstrated the use of anion-exchange selective layers for the partitioning of the couple [42-45]. In particular, Heineman, Seliskar, and coworkers have shown that sol-gel-derived poly(dimethyldiallylammonium chloride)–SiO2 composite films have been effective in this task [43-45]. We expect our pyridinium-containing sol-gel films to behave in a similar manner with respect to preconcentration. Figure 4 shows cyclic votammograms that were collected at a glassy carbon electrode before and after coating with a sol-gel film. There is a small, noticeable decrease (6%) in the peak current of the CV obtained with the sol-gel coated electrode, indicating that there is likely increased resistance to mass transport through the film at the electrode surface. Much thicker films result in larger decreases in peak current when compared to the bare glassy carbon electrode. A CV was also collected after the sol-gel electrode had been preconditioned by soaking in 0.1 M HCl for 10 min followed by stirring in the solution for 5 min (Figure 4). The resultant peak current showed an increase over that generated at either the coated or uncoated electrodes, indicating that preconcentration of the negatively-charged analyte species in the sol-gel film had taken place. This correlates well with previous reports showing the enhanced sensitivity achieved when anionic-exchange films are used for the determination of [42,43-45]. These results may suggest that the functionalized sol-gel films are adequate for the preconcentration and analysis of Cr(VI) anions, although the mechanisms of analyte transport for the two systems [mass transport for the irreversible Cr system, diffusion and charge-transport processes for the reversible system] are greatly different.
Fig. 4.
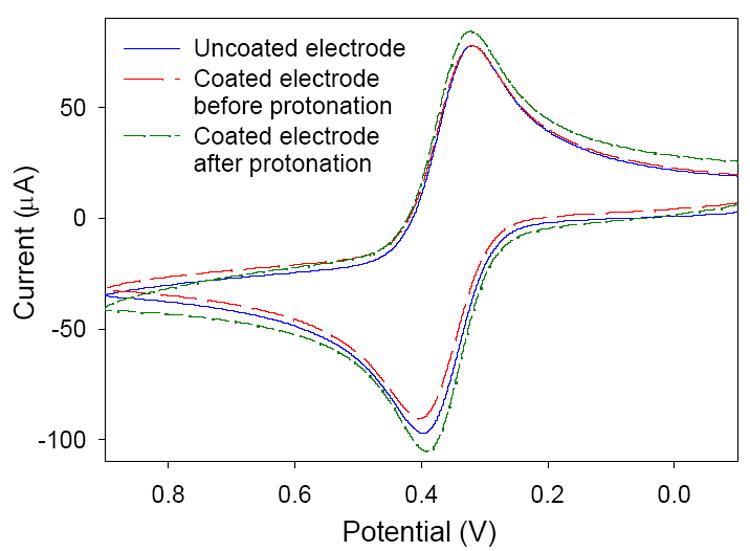
Cyclic voltammograms of 5 mM at a glassy carbon electrode before and after coating with a pyridium-functionalized sol-gel.
3.4 Cr(VI) quantification
Studies were carried out in which the electrode response was monitored as a function of Cr(VI) concentration. The square wave voltammograms and the corresponding calibration plot are given in Figures 5 and 6, respectively. There is a linear response (R2 = 0.992) between the concentration of Cr(VI) and the peak current at the electrode, indicating the electrode would be useful for quantifying the concentration of Cr(VI) in an unknown sample.
Fig. 5.
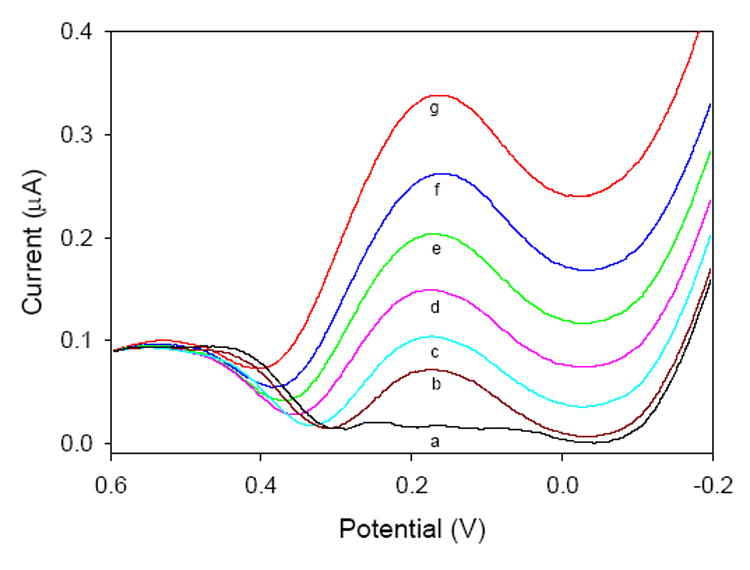
Square-wave voltammograms of various Cr(VI) concentrations collected at a pyridine-functionalized sol-gel electrode: (a) 0 ppb; (b) 11.7 ppb; (c) 50.1 ppb; (d) 104 ppb; (e) 203 ppb; (f) 308 ppb; (g) 400 ppb Cr (VI).
Fig. 6.
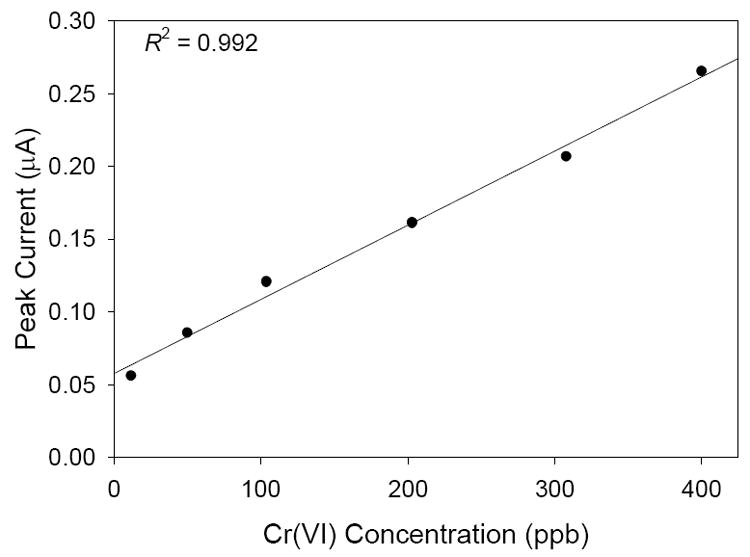
Calibration plot for the measurements in Figure 5.
3.5 Reproducibility and limit of detection
The reproducibility of the measurements obtained using the pyridinium-functionalized electrode was investigated. Four successive square wave voltammograms of solutions consisting of 53.3 ppb Cr(VI) were carried out (Figure 7). The various scans were highly reproducible with an average standard deviation of 1.0 × 10−3 μA for the peak currents from each scan. The detection limit of the proposed system is 4.6 ppb Cr(VI) [46]. In addition, over 40 scans were conducted before a substantial decrease in sensitivity, presumably due to deterioration of the sol-gel film, was noticed.
Fig. 7.
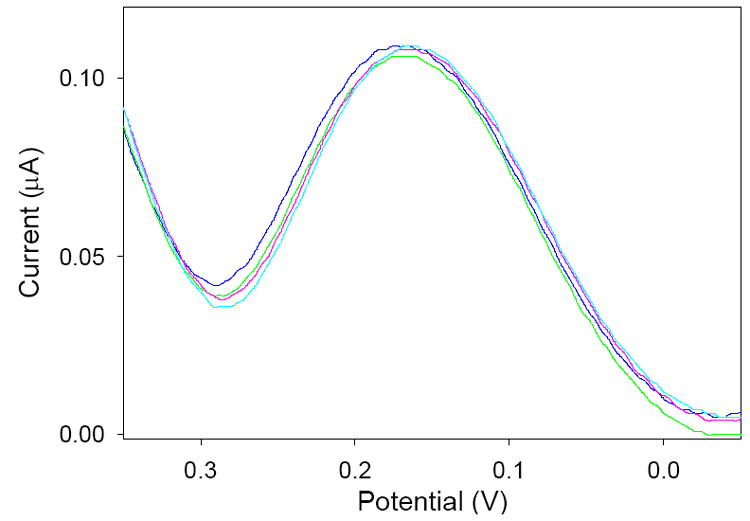
Square wave voltammgrams of separate 53.3 ppb Cr(VI) solutions. Average standard deviation of the peak currents: 1.0 × 10−3 μA.
3.6 Effect of preconcentration time
In a typical analysis procedure, the functionalized electrode is exposed to the Cr(VI) solution for 10 min while stirring. However, the length of preconcentration time affects the measured peak current. Figure 8 shows the relationship between the preconcentration time and the peak current obtained at the electrode surface. Initially, the slope of the plot is rather steep as Cr(VI) anions have more time to diffuse to the vacant pyridinium sites at the electrode surface. Later the slope of the plot begins to flatten out as more pyridinium sites are occupied by Cr(VI) anions. This is an indication that the electrode is becoming saturated with the analyte ions. However, even after 60 min, the peak current is continuing to increase slightly. This continual increase in peak current, even after the long preconcentration, suggest that the mass transport of the Cr(VI) ions inside the sol-gel film is a fairly slow process. Longer preconcentration times may yield improved limits of detection. We chose a 10 min preconcentration time in the current studies.
Fig. 8.
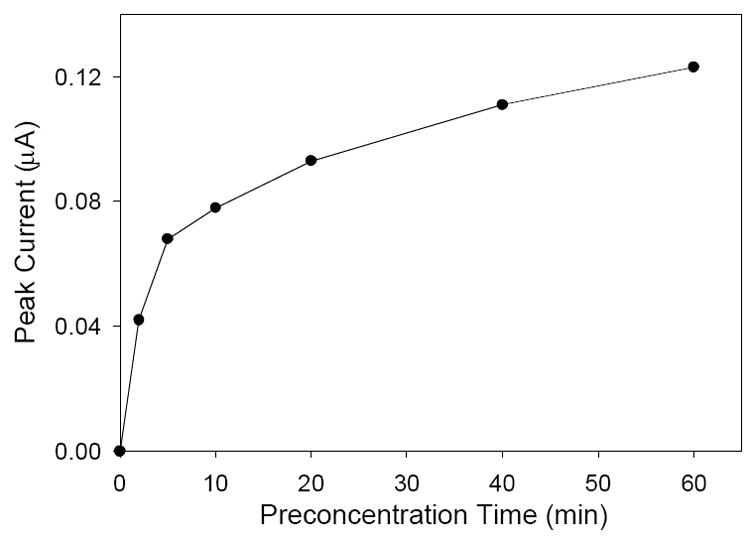
Effect of Cr(VI) preconcentration time at the electrode surface on the generated peak current. Analyte solutions consisted of 22.4 ppb Cr(VI), 0.1 M KCl, and 0.1 M HCl.
3.7 Interference study
The fact that Cr(VI) is one of few metallic anions makes it very likely that this electrode should be highly selective with regard to Cr(VI) since preconcentration relies on the interaction between the positively-charged pyridinium groups and the negatively-charged anions (Scheme 1). As a result, measurements should suffer little interference from other metallic species. Results from an interference study are shown in Figure 9. Even at relatively high interferent concentrations, the peak current associated with the Cr(VI) concentration changes very little. In particular, a 105 excess of Cr3+ can be tolerated with no adverse affects to the peak current. This is of particular importance, as Cr(III) has been shown to be an essential trace element for mammals [47] while Cr(VI) is a suspected carcinogen and highly toxic to humans. In addition, the relatively small interference of other anions indicates that the interaction between Cr(VI) and pyridine is more than a simple ion-exchange process, as has been shown previously [15].
Fig. 9.
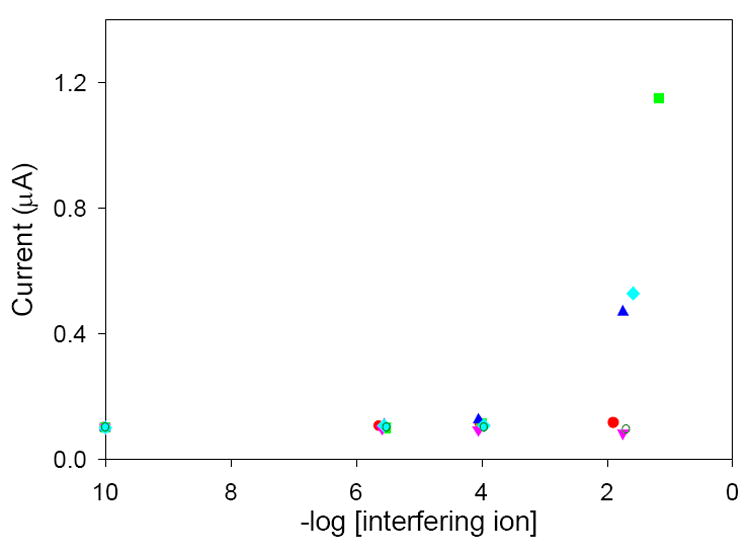
Results of the interference study. Peak currents of square wave voltammograms are obtained in solutions consisting of 51.6 ppb Cr(VI) and varying concentrations of the following: (
 ) Cr3+; (
) Cr3+; (
 ) Fe3+; (
) Fe3+; (
 )
(
)
(
 ) CH3COO−; (
) CH3COO−; (
 ) Cu2+; (
) Cu2+; (
 )
.
)
.
4. Conclusions
Pyridine-functionalized sol-gel films have been electrodeposited on glassy carbon electrodes for the enhanced determination of Cr(VI). The proposed coating technique uses commercially available reagents and can be carried out relatively quickly and reproducibly. Although previously reported sol-gel coatings prepared in this manner have been used for the determination of cations and neutral species, the reported work uses the film for enhanced determination of anions. The films were characterized using as a redox probe, XPS, and SEM. Interaction of the pyridinium groups in the sol-gel film with Cr(VI) anions in solution results in increased selectivity and sensitivity for the analyte species. The analytical process here tolerates significant concentrations of interferent species, including Cr(III), with little affect on the expected peak current. The calculated limit of detection for the system was 4.6 ppb Cr(VI), although this may be lowered if longer preconcentration times are used. The reported work has demonstrated the success of electrodeposition for the formation of pyridine-functionalized sol-gel films and exhibits their potential in Cr(VI) sensing applications.
Acknowledgments
The authors gratefully acknowledge the U.S. National Institutes of Health (1 R21 DK068107-01) for financial support. We also thank Prof. James Q. Chambers for helpful discussions, Johnny C. Jones for XPS assistance, and Jenny M. Oran for SEM assistance.
References
- 1.Zevin M, Reisfeld R, Oehme I, Wolfbeis OS. Sens Actuators. 1997;B38-39:235. [Google Scholar]
- 2.Balasubramanian S, Pugalenthi V. Talanta. 1999;50:457. doi: 10.1016/s0039-9140(99)00135-6. [DOI] [PubMed] [Google Scholar]
- 3.Posta J, Berndt H, Luo SK, Schaldach G. Anal Chem. 1993;65:2590. [Google Scholar]
- 4.Scindia YM, Pandey AK, Reddy AVR, Manohar SB. Anal Chim Acta. 2004;515:311. [Google Scholar]
- 5.Ji H-F, Thundat T, Dabestani R, Brown GM, Britt PF, Bonnesen PV. Anal Chem. 2001;73:1572. doi: 10.1021/ac0013103. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Ji H-F, Brown GM, Thundat T. Anal Chem. 2003;75:4773. doi: 10.1021/ac0343026. [DOI] [PubMed] [Google Scholar]
- 7.Pinnaduwage LA, Boiadjiev VI, Brown GM, Thundat T, Petersen SW. Sens Lett. 2004;2:25. [Google Scholar]
- 8.Tian F, Boiadjiev VI, Pinnaduwage LA, Brown GM, Thundat T. J Vac Sci Technol. 2005;A 23:1022. [Google Scholar]
- 9.Cox JA, Kulesza PJ. Anal Chim Acta. 1983;154:71. [Google Scholar]
- 10.Paniagua AR, Vazquez MD, Tascon ML, Sanchez Batanero P. Electroanalysis. 1993;5:155. [Google Scholar]
- 11.Svancara I, Foret P, Vytras K. Talanta. 2004;64:844. doi: 10.1016/j.talanta.2004.03.062. [DOI] [PubMed] [Google Scholar]
- 12.Welch MW, Nekrassova O, Compton RG. Talanta. 2005;65:74. doi: 10.1016/j.talanta.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y-J, Huang H-J. Anal Chem. 2001;73:1377. [Google Scholar]
- 14.Lin L, Lawrence NS, Thongngamdee S, Wang J, Lin Y. Talanta. 2005;65:144. doi: 10.1016/j.talanta.2004.05.044. [DOI] [PubMed] [Google Scholar]
- 15.Turyan I, Mandler D. Anal Chem. 1997;69:894. doi: 10.1021/ac9607525. [DOI] [PubMed] [Google Scholar]
- 16.Ge H, Zhang J, Wallace GG. Anal Lett. 1992;25:429. [Google Scholar]
- 17.Collinson MM, Rausch CG, Voigt A. Langmuir. 1997;13:7245. [Google Scholar]
- 18.Collinson MM, Moore N, Deepa PN, Kanungo M. Langmuir. 2003;19:7669. [Google Scholar]
- 19.Deepa PN, Kanungo M, Claycomb G, Sherwood PMA, Collinson MM. Anal Chem. 2003;75:5399. doi: 10.1021/ac026459o. [DOI] [PubMed] [Google Scholar]
- 20.Sayen S, Walcarius A. Electrochem Comm. 2003;5:341. [Google Scholar]
- 21.Etienne M, Walcarius A. Electrochem Commun. 2005;7:1449. [Google Scholar]
- 22.Walcarius A, Sibottier E. Electroanal. 2005;17:1716. [Google Scholar]
- 23.Shacham R, Avnir D, Mandler D. Adv Mater. 1999;11:384. [Google Scholar]
- 24.Marx S, Zaltsman A, Turyan I, Mandler D. Anal Chem. 2004;76:120. [Google Scholar]
- 25.Fireman-Shoresh S, Turyan I, Mandler D, Avnir D, Marx S. Langmuir. 2005;21:7842. doi: 10.1021/la050240y. [DOI] [PubMed] [Google Scholar]
- 26.Kane SA, Iwuoha EI, Smyth MR. Analyst. 1998;123:2001. [Google Scholar]
- 27.Walcarius A, Mandler D, Cox JA, Collinson M, Lev O. J Mater Chem. 2005;15:3663. [Google Scholar]
- 28.Buckley AM, Greenblat M. J Chem Ed. 1994;71:599. [Google Scholar]
- 29.Brinker J, Scherer G. Sol-Gel Science. Academic Press; New York: 1989. [Google Scholar]
- 30.Greenwood NN, Earnshaw A. Chemistry of the Elements. second ed. Elsevier Science; London: 1997. pp. 1002–1040. [Google Scholar]
- 31.Bichromate () is believed to be the predominant Cr(VI) species in dilute, acidic solution at pH < 6.5. However, there have been controversies recently about whether bichromate (HCrO4-) exists. See, e.g. (a) House DA. Adv Inorg Chem. 1997;44:341.Poulopoulou VG, Vrachnou E, Koinis S, Katakis D. Polyhedron. 1997;16:521.Ramsey JD, Xia L, Kendig MW, McCreery RL. Corrosion Sci. 2001;43:1557.
- 32.Deshpande K, Cheung S, Rao MS, Dave BC. J Mater Chem. 2005;15:2997. [Google Scholar]
- 33.Camelot M. Rev Chim Miner. 1969;6:853. [Google Scholar]
- 34.Corey EJ, Schmidt G. Tetrahderon Lett. 1979;5:399. [Google Scholar]
- 35.Martin-Zarza P, Gili P, Rodriguez-Romero FV, Ruiz-Perez C, Solans X. Polyhedron. 1995;14:2907. [Google Scholar]
- 36.Sisler H, Ming WChL, Metter E, Hurley FR. J Am Chem Soc. 1953;75:446. [Google Scholar]
- 37.See supplementary data.
- 38.Gero A, Markham JJ. J Org Chem. 1951;16:1835. [Google Scholar]; Given the equilibrium constant log K = [Hpy+] / [H+] [py] = 5.217(1) for the protonation of pyridine( Israeli M, Laing DK, Pettit LD. J Chem Soc Dalton Trans. 1974:2194.) and stability constant for the formation of Cr3+-pyridine complex ( Relan PS, Bhattacharya PK. J Indian Chem Soc. 1969;46:534.), it is unlikely that pyridinium groups in the sol-gel films form a complex with Cr(III) ions. Pre-concentration of Cr(VI) anions on a self-assembled pyridinium monolayer electrode, followed by the reduction of Cr(VI) anions to Cr(III) cations, was also used to detect chromium [15].
- 39.Allain LR., PhD . Thesis. Chapter 2 The University of Tennessee; 1999. [Google Scholar]; Canada TA., PhD . Thesis. Chapter 2 The University of Tennessee; 2002. [Google Scholar]
- 40.The scans were highly reproducible with an average standard deviation of 1.0 × 10−3 μA for the peak currents (Section 3.5), suggesting that, if there are Cr(III) cations left in the film after the washing, they perhaps do not interfere with the Cr(VI) anion uptake and analysis in the subsequent cycles.
- 41.Pauliukaite R, Hocevar SB, Ogorevc B, Wang J. Electroanal. 2004;16:719. [Google Scholar]
- 42.Harrison DJ, Daube KA, Wrighton MS. J Electroanal Chem. 1984;163:93. [Google Scholar]
- 43.Petit-Dominguez MD, Shen H, Heineman WR, Seliskar CJ. Anal Chem. 1997;69:703. [Google Scholar]
- 44.Shi Y, Slaterbeck AF, Seliskar CJ, Heineman WR. Anal Chem. 1997;69:3679. doi: 10.1021/ac970520l. [DOI] [PubMed] [Google Scholar]
- 45.Ross SE, Shi Y, Seliskar CJ, Heineman WR. Electrochim Acta. 2003;48:3313. [Google Scholar]
- 46.The calculated detection limit here is based on three times the standard deviation of the noise level and the calibration plot established in Figure 6.
- 47.Schwarz K, Mertz W. Arch Biochem Biophys. 1959;85:292. doi: 10.1016/0003-9861(59)90479-5. [DOI] [PubMed] [Google Scholar]; Mertz W, Schwartz K. J Physiol. 1959;196:614. doi: 10.1152/ajplegacy.1959.196.3.614. [DOI] [PubMed] [Google Scholar]