

Summary
The phosphatase calcineurin (Cn) plays pivotal roles in many physiological processes, including cell proliferation, development, and apoptosis. Most prominently, Cn dephosphorylates multiple residues of nuclear factors of activated T-cells (NFATs). It jointly translocates with dephosphorylated NFAT to the nucleus where it activates transcription of cytokines. This process relies on the interaction between the catalytic domain of Cn (CnCat) and a conserved PxIxIT motif within a region distinct from NFAT’s dephosphorylation sites. Here, we present the structure of the complex between the 39 kDa CnCat and a 14-residue peptide containing the PxIxIT motif. The structure of the complex was determined using NMR-derived structural constraints, and the X-ray coordinates of free CnCat. The peptide binds in a hydrophobic cleft adding a short parallel strand to CnCat’s central β-sheet. The structure reveals that the orientation of bound PxIxIT directs the phosphorylation sites in NFAT’s regulatory domain towards the Cn catalytic site.
Keywords: calcineurin, NFAT, nuclear magnetic resonance, complex structure, phosphatase
Introduction
Calcineurin (Cn) is a heterodimeric phosphoprotein serine/threonine phosphatase that is activated by Ca2+/calmodulin (Figure 1A). The active site of calcineurin is located in the CnA subunit, which is 57-59 kDa in mammals, and is regulated by interaction with the 19kDa CnB subunit (Rusnak and Mertz, 2000). CnA has one Zn and one Fe ion in the catalytic core and both metals are indispensable for catalytic activity (Mondragon et al., 1997). Cn was originally identified as a major calmodulin binding protein in the brain (Klee et al., 1979; Stewart et al., 1982) but has since been shown to play pivotal roles in a wide variety of other biological contexts, such as regulation and development of the immune, nervous, cardiovascular and musculoskeletal systems, and be involved in apoptosis and necrosis (Aramburu et al., 2004; Aramburu et al., 2000; Clipstone and Crabtree, 1992; Hemenway and Heitman, 1999; Hogan and Li, 2005; Rusnak and Mertz, 2000).
Figure. 1.

Schematic representation of the domain structure of human calcineurin (A) and human NFAT2 (B). (A) For CnA, the locations of the binding sites for CnB, calmodulin (CaM), and the auto-inhibitory domain (AI) are indicated with gray boxes. For CnB, four EF hand motifs are indicated. (B) The location of the calcineurin binding site, the serine-rich region 1 (SRR1), the SPxx-repeat motifs 2 and 3 (SP2 and SP3), and nuclear localization and export signals of hNFAT2 are indicated.
The most-studied role of Cn is its role in T-cell signaling. Increased intracellular calcium levels activate Cn which causes it to dephosphorylate multiple residues in nuclear factors of activated T-cells (NFATs) (Jain et al., 1993). Phosphorylation sites are located in NFAT’s regulatory domain in three different serine rich motifs, termed SRR1, SP2 and SP3 (Figure 1B). Dephosphorylation of these serine residues is thought to cause exposure of nuclear localization signal sequences triggering translocation of the dephosphorylated NFAT-Cn complex to the nucleus, where NFAT activates target gene expression (e.g. IL2)(Rao et al., 1997). The DNA binding domain of NFAT is located in the REL-homology region (RHR), which also contains the binding sites for activator protein 1 (AP1) proteins, such as FOS and JUN. The structures of RHR alone, and the RHR-DNA and RHR-DNA-FOS-JUN complexes, which have been determined with NMR and/or X-ray crystallography, revealed the cooperativity aspects of transcriptional activation by NFAT (Chen et al., 1998; Wolfe et al., 1997; Zhou et al., 1998). Due to its role in T-cell activation, Cn has been targeted for the development of immune-suppressant drugs. The two most successful Cn inhibitors discovered so far, cyclosporin A (CsA) and tacrolimus (FK506), block Cn’s phosphatase activity by obscuring its catalytic site. They are presented as stable trimeric complexes with the intracellular proteins cyclophilin and FKBP12, respectively (Griffith et al., 1995; Huai et al., 2002; Kissinger et al., 1995). Although highly successful, CsA and FK506 have severe potential side effects, which may be due to intrinsic toxicity; more likely, however, these effect result from the general inhibition of the enzymatic activity of Cn, which plays other biologically important roles besides NFAT activation (Kiani et al., 2000).
It is known that Cn-NFAT signaling depends on an interaction between the Cn catalytic domain (CnCat) and the conserved PxIxIT motif, which is located N-terminal to the phosphorylation sites in NFAT’s regulatory domain (Aramburu et al., 1998) (Figure 1B). Interfering with the Cn-NFAT interaction by mutating the conserved sequence impairs NFAT’s activation capabilities (Aramburu et al., 1998). While the interaction between CnCat and the PxIxIT motif of NFAT is indispensable for NFAT activation, it does not depend on the polypeptide segment containing the phosphorylation sites (Aramburu et al., 1998; Garcia-Cozar et al., 1998). Based on the topologies of this interaction, an alternative strategy for more specific inhibition of Cn-NFAT signaling has been proposed by targeting the NFAT interaction site rather than the enzyme’s catalytic site, in order to achieve selectivity in suppressing T-cell activation (Roehrl et al., 2004a; Roehrl et al., 2004b). Affinity selection from a consensus PxIxIT sequence-based combinatorial peptide library showed that Cn is able to accommodate a variety of sequences without significantly losing their affinity and some of the selected peptides had a higher Cn binding affinity than the original NFAT sequences (Aramburu et al., 1999). While the native targeting sequences have relatively low affinity for Cn (Kd ∼20 μM for SPRIEIT sequence in NFAT1), the best selected peptide with the sequence VIVIT at the center, exhibits a 50-fold higher affinity for Cn (Aramburu et al., 1999). Efforts to identify the VIVIT peptide binding site with X-ray crystallography have failed so far because crystals cracked after the soaking with the peptide in various Cn crystal forms (Roehrl et al., 2004a; Roehrl et al., 2004b). This observation suggested that the VIVIT peptide might bind to a conserved crystal contact site, which maps to a conserved hydrophobic surface around β-strand 13 (319-325) and β-strand 14 (328-334), and the idea was recently supported by a combination study of photoaffinity crosslinking, mutation, and in-silico docking simulations (Li et al., 2004).
To obtain a more detailed picture of the Cn-NFAT interaction, we have determined the structure of Cn-VIVIT peptide complex using solution NMR techniques. The VIVIT peptide binds to the exposed β-strand 14 while forming a short parallel β-sheet. The conserved hydrophobic sidechains of the PxIxIT motif are located in a hydrophobic cleft formed by β-strands 11 and 14, and the β11-β12 loop (i.e. the loop between β-strand 11 and 12). On the other hand, non-conserved residues in the PxIxIT motif are largely exposed to solvent. These results provide a detailed view of NFAT recognition by Cn and reveal the mechanism by which Cn specifically recognizes the conserved PxIxIT sequence. The information is potentially useful for developing new bioactive compounds that target Cn-NFAT interaction.
Results and discussion
Identification of the VIVIT binding site on calcineurin
The 40kDa catalytic domain of calcineurin which includes residues 2-347 was prepared based on the method developed by Aramburu et al. with minor modifications (Aramburu et al., 1999). As shown in Figure 2, the TROSY 1H-15N HSQC spectrum of CnCat is well dispersed, reflecting the α-β mixed structure of the CnCat domain (Griffith et al., 1995; Huai et al., 2002; Jin and Harrison, 2002; Kissinger et al., 1995). About 210 resonances out of 326 expected resonances are identified in the TROSY 1H-15N HSQC spectrum of perdeuterated CnCat. One explanation for the absence of some of the resonances is insufficient D/H exchange after expression of the protein in D2O. The other possibility is the paramagnetic effect, since CnCat possesses a paramagnetic iron (Fe3+ or Fe2+) in its catalytic center. To ascertain the main cause for the missing resonances, we utilized a partial deuteration strategy where the protein was cultured on deuterated media but in H2O to avoid the back exchange problem (Lohr et al., 2003). This strategy, however, rescued only 20 additional resonances out of the ∼110 missing signals, indicating that paramagnetic effect, rather than insufficient back exchange, is the primary reason for the missing resonances. Indeed, the results of our mainchain assignments of the protein revealed that resonances originating from any hydrogens within a 10 Å distance from the iron in the catalytic center of CnCat could not be observed.
Figure. 2.

1H-15N TROSY HSQC spectra of 2H15N-labeled catalytic domain of Cn (CnCat) without (red) and with (black) the 14mer VIVIT peptide (1.2 fold excess).
Upon the addition of VIVIT peptide, CnCat resonances showed chemical shift perturbations indicative of the slow-exchange kinetics, confirming that the VIVIT peptide forms a stable complex with CnCat. The chemical shift changes were saturated at a 1:1 stoichiometric amount of peptide to protein indicating a 1:1 complex between CnCat and the VIVIT peptide. Main chain resonances of CnCat in complex with VIVIT peptide were assigned using the data from standard TROSY triple resonance experiments and information from selective labeling. Comparing the spectra of free and bound CnCat, we identified the resonances shifted upon binding of the VIVIT peptide (peaks shifted more than half peak width). Figure 3B and 3C show the distribution of the residues affected by VIVIT binding. Chemical shift changes were observed for >30 resonances, which reside in the C-terminus of Helix 3 (residue number 72-74), β-strand 1 (79), the C-terminal side of Helix4 (103-106), the H4-β3 loop (i.e. the loop between Helix 4 and β-strand 3) (111), β-strand 3 (114), the β5-β6 turn (192-194), the β7-β8 loop (240 and 242), the C-terminus of Helix 12 (271 and 272), β-strand 10 (277), the β11-β12 loop (293,294, 296, 298, and 299), and β-strands 13 and 14 (324-335). Most of these residues are mapped into a contiguous surface, which includes two (lower and upper) clefts on the CnCat structure (colored in magenta in Figure 3C). The lower cleft includes the C-terminus of Helix 3 and β-strand 13, while the upper cleft includes the β-strands 11 and 14, and the β11-β12 loop. The types of amino acids that line these two clefts are significantly different. The upper cleft consists of hydrophobic or aromatic residues, while the lower cleft is lined primarily by hydrophilic and charged residues. The total solvent accessible surface area for the shifted residues is 1400 Å, which is much larger than what a single 14-mer peptide can cover. Given the fact that the peptide and the protein form a 1:1 complex, it seems that CnCat has a secondary site that allosterically changes its structure upon the binding of the NFAT peptide to its cognate site. The overall structure of CnCat, however, doesn’t seem to change significantly since the global features of the spectra do not alter dramatically upon VIVIT binding. To identify the direct VIVIT-binding site, a cross-saturation experiment was performed for 2H15N CnCat in complex with unlabeled VIVIT peptide. Selective irradiation to the methyl resonances of VIVIT caused a significant signal intensity loss for the resonances of Met329 and Ile331 on β-strand 14 (Figure 3A). The data strongly indicate that the upper cleft rather than lower cleft is the direct binding site of VIVIT (Figure 3C). Moreover, since the upper cleft consists mostly of hydrophobic residues, the nature of the cleft is also favorable for accommodating the hydrophobic VIVIT peptide.
Figure 3.

Summary of VIVIT peptide binding experiments for CnCat. (A) Intensity reduction ratio for each resonance in the cross-saturation experiment. A saturation time of 300 ms was applied to the methyl proton resonances of the VIVIT peptide. The residues that experienced a significant intensity reduction are colored in red. (B and C) Sites perturbed by the binding of VIVIT are mapped on the ribbon (B), and surface (C), of the CnCat structure. Red spheres or surfaces indicate the sites that experienced both cross-saturation and chemical shift perturbations. Yellow indicates the sites that showed chemical shift changes upon VIVIT binding. Blue residues were not affected in both experiments. White surfaces were not assigned. Magenta shadows in (C) indicate the positions of the two clefts on the CnCat structure.
Identification of the Cn binding site on the VIVIT peptide
For precise identification of the CnCat binding sequence on the VIVIT peptide, the mainchain resonance assignment of the free VIVIT peptide and in complex with CnCat was accomplished with triple-resonance experiments (Figure 4). The VIVIT peptide binds with slow-exchange kinetics, which is consistent with the features observed on the CnCat side. As shown in Figure 4A, the region from Ile6 to Gly10 experiences a significant downfield shift upon binding, indicating that this segment forms an extended secondary structure in the complex. This observation was also supported by the upfield shift of Cα resonances upon binding to CnCat (data not shown). Val5 exhibited a significant downfield chemical shift change. Figure 4B displays the normalized chemicals shift for amide resonances ((Δδ H)2 + (Δδ N / 5)2)1/2) of VIVIT peptide upon binding to CnCat (upper). The lower panel reports the result of cross saturation for the same complex. The region from Val5 to Gly10 showed >0.2 ppm chemical shift changes and was also strongly saturated in the cross saturation experiment, indicating the region binds tightly to the CnCat. His12 also showed a modest chemical shift perturbation, while the regions before His3 or after Glu13 do not seem to interact with the protein.
Figure 4.

Summary of CnCat binding experiments for VIVIT peptide. (A) 1H-15N TROSY HSQC spectra of 2H15N-labled VIVIT peptide without (red) and with (blue) CnCat. The free and bound resonances are connected by a dotted line with the assignments. (B) Upper panel: normalized chemical shift perturbation upon binding to CnCat. Lower panel: intensity reduction ratio for each resonance in cross-saturation experiment. A saturation time of 300 ms was applied for the methyl proton resonances of CnCat.
Structure of CnCat-VIVIT complex
To obtain a detailed view of the Cn-NFAT interaction, we then set out to determine the structure of the Cn-VIVIT peptide complex by simulated annealing of the VIVIT peptide in the presence of the X-ray structure of free CnCat (PDB code: 1AIU) using structural information derived from NMR spectroscopy. In total we used 22 intermolecular distance restraints, 52 intramolecular distance restraints within the VIVIT peptide, and 9 mainchain angular constraints for I6-G10 in the VIVIT peptide. Taking advantage of the knowledge of the VIVIT binding site on CnCat, the sidechains of the residues forming the VIVIT peptide binding site in CnCat were allowed to be flexible during the simulation. Other parts of CnCat were tightly restrained by distance and angular constraints calculated from the X-ray structure of free CnCat.
Figure 5A shows a set of converged structures of the Cn-VIVIT complex based on the structural restraints derived from NMR experiments. The VIVIT binding site is located on the side of the CnCat molecule. There is no VIVIT peptide atom within 18 Å distance from the catalytic core iron, which may explains how the peptide inhibits NFAT binding without affecting the catalytic activity of CnCat (Aramburu et al., 1998). The total buried surface area of the peptide is 698 ± 70 Å2, which corresponds to 32 ± 3 % of the total exposed surface of the peptide. The conserved PxIxIT sequence of the VIVIT peptide is particularly well ordered in the structure. The root-mean-square deviation values are 0.68 ± 0.32 Å and 1.15 ± 0.39 Å for mainchain and all heavy atoms, respectively in this region, due to the extensive number of experimental constraints applied (see supplemental figure 1). The VIVIT peptide binds on the exposed β strand 14 of CnCat forming a short 4 residue parallel β-sheet (Ile6 to Thr9, Figures 5B and 5C). The parallel β-sheet structure is supported by inter-strand NOEs shown in Figure 5C. In the calculated structures, intermolecular mainchain hydrogen bonds are observed for M329 (CO)-V7 (HN) (7/10), I331 (HN)-V7 (CO) (10/10), and I331 (CO)-T9 (HN) (10/10). The VIVIT peptide forms a bend at the N-terminus of the conserved sequence between Pro4 and Val5. This bend allows for contact between the N-terminus of the peptide and the β11-β12 loop. Figure 6A shows the surface of CnCat with the color representation derived from NMR binding experiments. The results of cross saturation experiment reliably identify the region corresponding to the core VIVIT binding site, where a tight interaction between the peptide and the protein occurs. In contrast, chemical shift perturbation experiments also report weak interacting regions such as the β11-β12 loop, where the N-terminus of the peptide interacts, or the lower cleft shown in Figure 3C, which apparently senses allosteric effects. It is also known that mutations to the linker region joining the catalytic domain and the regulatory domain of Cn (S337P, H339L, or L343S) disrupt the binding of Cn to NFAT1 (Rodriguez et al., 2005). Since there is no atom from the bound VIVIT peptide within 6 Å from these mutated residues, there appears to be no direct interaction between the linker region and the VIVIT peptide. However, because the mutated residues are C-terminus to the VIVIT binding β strand 14 of CnCat, these mutations might allosterically affect the structure of the VIVIT binding site in CnCat and disrupt the binding between Cn and NFAT.
Figure 5.

NMR model of the CnCat-VIVIT peptide complex. (A) Stereo view of an ensemble of 10 final NMR structures of the CnCat-VIVIT peptide complex. Blue lines show the backbone trace of CnCat. Red lines show the backbone trace of the VIVIT peptide with the sidechain heavy atoms for the PxIxIT motif (i.e. Pro4-Thr9). (B) Ribbon representation of the CnCat-VIVIT peptide complex with the lowest energy function. CnCat is shown in light blue and VIVIT peptide is shown in red with sidechains for the PxIxIT motif. In (A) and (B), the two metal ions are shown as spheres (yellow: Fe, purple: Zn). (C) A schematic representation of the secondary structure elements in the CnCat-VIVIT peptide complex. The hydrogen bonds observed in the structure are marked by black dotted lines. Blue dotted arrows indicate the observed interstrand NOEs.
Figure 6.
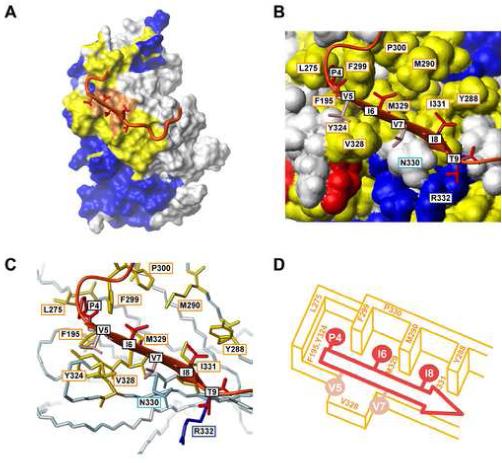
Interface of the CnCat-VIVIT peptide interaction. (A) Sites that are perturbed by the binding of the VIVIT peptide are mapped on the CnCat surface in the CnCat-VIVIT peptide complex. Red surfaces indicate sites that experienced both cross-saturation and chemical shift perturbation. Yellow indicates the sites that showed chemical shift changes upon binding of the VIVIT peptide. Blue residues were not affected in both experiments. White surfaces were not assigned. The VIVIT peptide is shown as a ribbon representation with the sidechains for the PxIxIT motif. (B) and (C) show the interface of CnCat and the conserved PxIxIT motif. In (B), the conserved residues in CnCat are shown with a colored surface representation based on the properties of each amino acid. Hydrophobic, acidic, basic, and hydrophilic residues are shown in yellow, red, blue and white, respectively. The VIVIT peptide is shown as a ribbon representation with stick representation for the sidechain heavy atoms of the PxIxIT motif. The conserved Pro4, Ile6, Ile8, Thr9 are shown in dark red, while non-conserved Val6 and Val8 are colored in pink. In (C), the conserved residues in CnCat are shown as a stick model with color representation based on the properties of each amino acid. Hydrophobic, acidic, basic, and hydrophilic residues are shown in yellow, red, blue and light blue, respectively. VIVIT is shown in the same representation as in (B). (D) Schematic representation for the interaction between the PxIxI sequence and hydrophobic pockets of CnCat. The VIVIT peptide binds on the hydrophobic floor formed by Phe195, Tyr324, Val328, Met329, and Ile331. The conserved PxIxI sequence is accommodated in the hydrophobic pockets separated by Leu275, Tyr288, Met290, and Phe299. The peptide backbone is represented by the red arrow and conserved and non-conserved sidechains are shown in dark and light red, respectively.
Architecture of the PxIxIT/Cn interface
Figures 6B and 6C show enlarged views of the binding site of the conserved PxIxIT motif with a color representation based on the property of residues. The VIVIT binding site on Cn is very hydrophobic and forms several hydrophobic pockets in CnCat structures. As shown in Figure 6B and the cartoon of Figure 6D, Phe195 in β-strand 6, Tyr324 in β-strand 13, and M329 and Ile331 in β-strand 14 of CnCat form the base of the hydrophobic pockets. Leu275 in the H12-β11 loop, and Pro300 in the β11-β12 loop define the boundary of the hydrophobic regions. Tyr288 and Met 290 in β-strand 11 and Phe299 in the β11-β12 loop separate this section into three hydrophobic pockets. All residues that form the hydrophobic pockets are conserved among different species. Each of the conserved hydrophobic pockets accommodates one of the residues from the conserved motif in the VIVIT peptide. Pro4 in the VIVIT peptide fits into a hydrophobic cleft between Leu275 and Phe299 of CnCat. Ile6 is the least exposed residue (10% of the total surface is exposed) in the conserved motif and a hydrophobic pocket formed by Met290, Phe299, Pro300, and Met328 in CnCat accommodates the sidechain of Ile6. Ile8 is sandwiched between Tyr288 and Met290 and forms an extensive interaction with Ile331 in a nested rearrangement (Wouters and Curmi, 1995). Mutation of Ile331 to Ala is known to reduce the affinity of the peptide to protein by more than 20-fold, indicating that the sidechain hydrophobic interaction between these residues is energetically important (Li et al., 2004). Although, Ile6 and Ile8 are conserved within NFATs, these interaction sites can potentially accommodate a different type of amino acid residue with hydrophobic properties. Indeed, it has been shown that even the least exposed Ile6 can be mutated to Phe, Val, or Leu without significantly loosing the affinity to CnCat (Aramburu et al., 1999). Two valine residues (Val5 and Val7) in between these conserved residues are oriented towards the solvent, allowing extensive substitution of these residues. The positions corresponding to Val5/Val7 is Arg/Glu in hNFAT1 and hNFAT2, Ser/Arg in hNFAT3, and Ser/Gln in hNFAT4. In a similar recognition motif in the calcineurin inhibitor Cabin1, the corresponding residues are Glu/Thr. However, since Val328 in β-strand 14 of CnCat makes hydrophobic contacts with Val5 and Val7, hydrophobic residues may be more preferable compared to the native hydrophilic sequences. This might be one of the reasons why the VIVIT sequence has a higher affinity relative to native sequences. Thr9 of the VIVIT peptide is exposed despite being conserved within NFATs. As shown in Figure 6C, Thr9 is in close proximity to Asn330 and Arg332, both of which are conserved in CnCat. Indeed, the energetic interaction between the sidechains of Thr9 and Asn330 is shown by mutational analysis (Li et al., 2004). Since the γ-hydroxyl group of Thr9 is observed in some of our structures to form hydrogen bond with the sidechain amide of Asn330 (30% of structures) and the guanidine group of Arg332 (40% of structures), hydrogen-bonding interaction appears to can be the source of the energetic interaction.
To confirm our complex model and rule out the possibility that the hydrophobic binding pockets for Ile6 and Ile8 may instead accommodate Val5 and Val7 in a different flipped orientation, selective NOE difference experiments were performed with a complex of [ul-2H, FIMY-1H] Cn and [ul-2H15N, Iδ1LδVγ -1H13C] VIVIT (Reibarkh et al., 2006; Takahashi et al., 2006). Figure 7A shows the 1H-13C HSQC spectra of [ul-2H, ILV-Me-1H] VIVIT with and without [ul-2H15N, Iδ1LδVγ -1H13C] Cn. Chemical shift changes upon CnCat binding were more significant for Ile6 and Ile8 compared to Val5 and Val7, and the resonances of the isoleucines were less intense than that of the valine resonances. These results indicate a stronger preference of Cn for Ile6 and Ile8 over Val5 and Val7, which is consistent with our structural model. Selective irradiation of the aromatic proton resonances of either Tyr288 or Phe299 selectively saturated the Ile6 and Ile8 methyl resonances without affecting Val5 and Val7 resonances (Figure 7B). The quantitative reduction in intensity also showed a good correlation with the distance between irradiated and saturated residues. The irradiation of Tyr288 caused stronger saturation for Ile8 than Ile6, while that of Phe299 decreased the intensity of the Ile6 resonance more than the Ile8 resonance. These results confirmed the orientation observed in the determined complex between CnCat and the VIVIT peptide. A conserved interaction involving the Thr9 sidechain as well as the relative preference of the hydrophobic pockets for the Ile residues may be the determining factors for the unique orientation between CnCat and the VIVIT peptide. In fact, the Ile-Ile pair (e.g. Ile8-Ile331) is one of the most preferred amino pairings in a parallel β-sheet (Fooks et al., 2006). In contrast, the IleHB-ArgnHB pair (i.e. Ile8-Arg332), which would occur in an alternative flipped interaction, is one of the most unfavorable pairing of residues in a parallel β-sheet.
Figure 7.

(A) HSQC spectra of [ul-2H15N, Iδ1LδVγ -1H13C] VIVIT peptide with (red) and without (blue) CnCat. Assignment for each resonance is also shown. (B) Selective cross-saturation experiment using [ul-2H15N, Iδ1LδVγ -1H13C]] VIVIT peptide in complex with [U-2H, FIMY-1H] CnCat. Intensity reduction ratios for each methyl resonance with 250 ms of continuous wave irradiation to aromatic resonances of Tyr288 and Phe299 are shown. Average intensity reduction ratios of two γ-methyl resonances are shown for Val resonances.
PxIxIT interaction orients NFAT’s phosphorylated residues towards the Cn catalytic site
The topology of the PxIxIT interaction with CnCat raises the question of how this relates to dephosphorylation of the regulatory region (Figure 1). All of NFAT’s phosphorylation sites are C-terminal to the Cn-binding sequence. The Cn binding sequence of hNFAT2, for example, is 118-PRIEIT-123 and the dephosphorylation regions are 172-SPASSLSSRSCNSE-185 (serine-rich region 1 (SRR1), dephosphorylation residues are underlined and bolded), 233-SPRHSPSTSPRA-244 (SPXX-repeat motif 2 (SP2)), and 278-SPHHSPTPSPHGSP-291 (SP3) (Macian, 2005). The orientation of the C-terminus of the bound VIVIT peptide (Figure 5) directs the phosphorylated residues towards the catalytic site of Cn. NFAT dephosphorylation is thought to trigger exposing of the nuclear localization sequence (NLS), 265-KRK-267, which is N-terminal to SP3. Although close to the dephosphorylation sites, the mechanism by which the NLS is activated is not clear without additional structural information. A second potential second NLS, 682-KRK-684, is located in the C-terminal domain of NFAT2, but it is unclear how it would be activated by dephosphorylation of the regulatory domain.
Conclusion
In summary, the structure of CnCat-VIVIT complex was determined by using NMR-derived constraints and the known crystal structure of CnCat. The VIVIT peptide forms a parallel β-sheet with the exposed β-strand 14 in Cn. The structure reveals that the bound peptide is oriented to direct the phosphorylation sites towards the Cn catalytic site. The conserved PxIxIT sequence is either recognized by hydrophobic pockets or by forming sidechain-sidechain interaction with conserved residues in Cn. Thsee results provide a detailed view of NFAT recognition by Cn, providing insight on how Cn specifically recognizes the conserved PxIxIT motif. The information obtained here may be useful for developing small compounds that specifically inhibit the Cn-NFAT interaction. The hydrophobic pockets of the PVIVIT binding site we characterized in this study define a target site that could accommodate inhibitors, of the CnCat/NFAT interaction. The NMR assignments established here will provide the means for future characterization of complexes between CnCat and inhibitors, such as described in (Roehrl et al., 2004a)
Experimental procedures
All chemicals were purchased from Sigma unless otherwise noted. 1-13C amino acids were purchased from Cambridge Isotope laboratories. Celtone® base media were purchased from Spectra Stable Isotopes.
Expression and purification of the catalytic domain of human calcineurin
The catalytic domain of human calcineurin Aα (CnCat), comprising residues 2-347 with substitutions Y341S, L343A, and M347D (Aramburu et al., 1999), was produced as a cleavable GST fusion protein in the E.coli strain Rosetta™ (DE3). Cleavage of the fusion protein with PreScission™ protease produces CnCat (2-347) with an additional GPLG sequence at its N-terminus. The CnCat domain obtained is enzymatically active as described in our previous publication (Roehrl et al., 2004b). A crystal structure of the protein prepared in this way was solved and revealed a structure nearly identical to that of the catalytic domain in full-length calcineurin A including the Fe-Zn cluster (Michael, 2004). The CnCat protein, uniformly labeled with 2H, 13C and 15N, was obtained by growing the E. coli in modified M9 Celtone medium, which consists of 1 kg/L 99.8% D2O, 8.5 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 40 mg/L carbenicillin, 15 mg/L chloramphenicol, 1 g/L 15NH4Cl (99.9% enriched), 2 g/L 2H613C-glucose (97% enriched), 1g/L 2H (>97% enriched), 13C (>97% enriched), 15N (>98% enriched) Celtone® Base Powder, 2 mM MgSO4, 0.1 mM CaCl2, 10 mg/L ZnSO4, and 10 mg/L FeCl3. To obtain the partially 2H and 15N labeled CnCat protein, modified M9 Celtone medium consisting of 1L H2O, 8.5 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 40 mg/L carbenicillin, 15 mg/L chloramphenicol, 1 g/L 15NH4Cl (99.9% enriched), 2 g/L 2H6-glucose (97% enriched), 1g/L 2H (>97% enriched), 15N (>98% enriched) Celtone® Base Powder, 2 mM MgSO4, 0.1 mM CaCl2, 10 mg/L ZnSO4, and 10 mg/L FeCl3 was used. To obtain selectively labeled CnCat, E.coli was grown in modified M9 Celtone medium, and targeted amino acids are added after induction of protein expression at 7-10 times the amount present in the Celtone media (Spectra Stable Isotopes).
All expression experiments were performed at 37 °C; after reaching an OD600 = 0.6, protein expression was induced by the addition of 1 mM IPTG. The cells were harvested after 36-48 hr of induction for uniformly labeled samples, and after 24 hr for selectively labeled samples. The harvested cells were resuspended in 40 ml of PBS with 2mM dithiothreitol (DTT), and 0.2 mg mL-1 phenylmethylsufonylfuruolide (PMSF) at 4 °C. The suspended cells were then disrupted by sonication and the insoluble fraction was removed by centrifugation for 20 min at 15000 g. The supernatant was applied to a 5 ml column of Glutathione Sepharose 4 Fast Flow. After washing the resin with 40 ml of PBS with 2mM DTT, the CnCat protein was eluted with 40 ml of PBS containing 2 mM DTT and 25 mM of reduced glutathione. The elution fraction was concentrated to 20 ml using 10,000-MWC membrane ultrafiltration. PreScission protease (0.5 units / 20 μg CnCat) was added to the concentrated elution fraction, and the solution was dialyzed against 1L of PBS with 2 mM DTT for 15 hrs. The digested solution was concentrated to 2 ml and subjected to PD-10 desalting column to remove remaining glutathione. The fractions containing CnCat were then applied to a 1.5 ml bed volume of Glutathione Sepharose 4 Fast Flow to remove PreScission protease and GST. The elution protein was further purified with Superedex75 column. Traces of GST in the Superedex75 elution fractions were then removed by passing the eluate through 1.5 ml bed volume of Glutathione Sepharose 4 Fast Flow. A typical final yield of CnCat was 2 mg/L culture.
Preparation of the VIVIT peptide
A synthetic VIVIT 14-mer peptide (H4N-GPHPVIVITGPHEE-COOH, Tufts-New England Medical Center peptide synthesis facility, Boston, MA) was used as the non-labeled form of peptide. The 2H15N13C-labeled peptide was produced as a GB1 fusion protein in an E.coli strain, BL21 (DE3). The GB1 fusion protein has the PreScission™ protease recognition site between GB1 and VIVIT peptide, which produces VIVIT (14-mer) with an additional LEHHHHHH purification tag at its C-terminus. For the expression of uniformly 2H15N13C labeled VIVIT peptide, E.coli harboring the GB1-VIVIT fusion vector was grown in M9 Celtone medium, which consists of 1 kg/L 99.8% D2O, 8.5 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 50 mg/L kanamycin, 1 g/L 15NH4Cl (99.9% enriched), 2 g/L 2H613C-glucose (97% enriched), 1g/L 2H (>97% enriched), 13C (>97% enriched), 15N (>98% enriched) Celtone® Base Powder, 2 mM MgSO4, 0.1 mM CaCl2. The uniformly 2H15N, ILV-methyl 1H13C labeled ([ul-2H15N, ILV-1H13C-Methyl]) VIVIT peptide was expressed in M9 minimum medium following the literature using [4-1H13C] α -ketobutyrate and [4,4-1H13C] α-ketoisovalerate (Goto et al., 1999).
The cells were incubated at 37 °C, and after reaching an OD600 = 0.6, protein expression was induced by the addition of 1 mM of IPTG. The cells were harvested after 4-6 hr of induction, and were resuspended in 40 ml of buffer A (50 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 20 mM imidazole) with 0.2 mg / mL PMSF at 4 °C. The suspended cells were disrupted by sonication and the insoluble fraction was removed by centrifugation at 15000 g for 20 min. The supernatant was applied to a 5 ml column of Ni-NTA agarose. After washing the resin with 40 ml of buffer A, the GB1-VIVIT protein was eluted with 40 ml of buffer consisting of 50 mM Tris-HCl (pH 7.5), 300 mM NaCl, and 300 mM imidazole. The elution fraction was concentrated to 2 ml by 5,000-MWC-membrane ultrafiltration. PreScission protease and 1 mM DTT were added to the concentrated elution fraction. The digested solution was purified on a Superedex75 column. The final yield of the VIVIT peptide was 20 mg/L culture.
NMR spectroscopy
All experiments were performed on a Bruker Avance 750 spectrometer equipped with a cryogenic probe. All spectra were collected using 0.4 - 0.6 mM protein in 10 mM sodium phosphate buffer (pH 6.8) containing 150 mM NaCl, 2mM DTT and 90% H2O / 10% D2O at 298 K. Spectra were processed using XWINNMR and analyzed with Sparky (Goddard and Kneller, 2006). The backbone assignment of CnCat in complexed with the VIVIT peptide was accomplished by using standard TROSY triple resonance experiments (Ferentz and Wagner, 2000). A 500 μM sample of uniformly 2H, 13C and 15N-labeled CnCat mixed with 1.2 molar excess of the non-labeled VIVIT peptide was prepared. To confirm the assignments, four samples of perdeuterated 15N labeled CnCat amino-acid-specifically labeled with unlabeled Arg, Lys, or 1-13C Val and 1-13C Leu were prepared. The backbone assignment of the VIVIT peptide in free form and in complexed with CnCat was accomplished by using standard triple resonance experiment without and with TROSY pulse scheme, respectively (Ferentz and Wagner, 2000). For the observation of free VIVIT spectra, a 500 μM concentration of 2H, 13C and 15N-labeled VIVIT was prepared. To record spectra of VIVIT in complex with CnCat, 2H, 13C and 15N-labeled peptide was complexed with 1.2 molar excess of non-labeled CnCat. For the assignment of the proton resonances of the VIVIT peptide, 1H-TOCSY and 1H-NOESY (100 ms mixing time) spectra were recorded for the non-labeled VIVIT peptide in complex with [U-2H15N13C] CnCat. To obtain the mainchain-mainchain and mainchain-sidechain intermolecular NOE between VIVIT and CnCat, 15N-NOESY-HSQC spectra with 200 ms mixing time were recorded for 2H15N13C labeled VIVIT peptide or CnCat in the presence of a slight excess amount of non-labeled binding partner (Gross et al., 2003). Sidechain-sidechain NOEs were obtained from recording a 13C-NOESY-HSQC spectrum with 200 ms mixing time of a complex of [ul-2H15N, Iδ1LδVγ -1H13C] VIVIT peptide and [2H, FIMY-1H] CnCat,. Cross saturation experiments were performed in accordance with previous literature (Takahashi et al., 2006; Takahashi et al., 2000).
Structure determination of CnCat-VIVIT complex
The structure of the CnCat-VIVIT complex was determined by simulated annealing of the VIVIT peptide in the presence of the X-ray structure of free CnCat ((Kissinger et al., 1995), PDB code: 1AIU) by using structural information derived from NMR spectroscopy. The experimental restraints include 22 intermolecular distance restraints between CnCat and VIVIT peptide, 52 intramolecular distance restraints within VIVIT peptide, and 9 mainchain angular constraints for I6-G10 in VIVIT peptide. While calculating the structure, CnCat was modeled by mainchain and sidechain angular restraints as well as amide-amide, amide-alpha, and alpha-alpha intramolecular proton-proton distance restraints and hydrogen bonding restraints calculated from X-ray structure of free CnCat (21-347). The sidechains of the residues near the VIVIT peptide binding sites in CnCat were left flexible, by not applying the angular restraints. The final structures, with statistical results shown in Table 1, were calculated using CYANA. We ran 100 sets of the final structure calculations with all of the determined structural constraints, and chose the 10 best structures based on lowest energy function, no NOE violations >0.5 Å and no angular violations >5°. The structures were analyzed using the PROCHECK-NMR (Laskowski et al., 1996) and MOLMOL (Koradi et al., 1996) programs. The solvent accessible surface area for amino acid residues was calculated with a solvent radius of 1.4 Å using the MOLMOL program. Structural figures were generated using the MOLMOL program.
Table 1.
Statistics for the final 10 NMR structures of VIVIT peptide in complex with CnCat a
| Experimental NOE distance restraints | 74 |
| Intra-molecule (VIVIT peptide): | 52 |
| Inter-molecule: | 22 |
| Backbone angular restraints | 9 |
| Ramachandran Plot (Pro4-Thr9)b | |
| Most favored region (%) | 85.0 |
| Additionally allowed region(%) | 15.0 |
| Generously allowed region (%) | 0.0 |
| Disallowed region(%) | 0.0 |
| Average RMS differences to mean structure (Å) | |
| Backbone (N, Cα, C) (Pro4-Thr9) | 0.68 ± 0.32 |
| All heavy atoms (Pro4-Thr9) | 1.15 ± 0.39 |
None of these 10 structures exhibited distance violations >0.5 Å or dihedral angle violations >5°. There is no bad contact in the VIVIT peptide as well as the interface of Cn-VIVIT interaction. The RMSD for covalent bonds and angles relative to the standard dictionary is 0.001 Å and 0.2 degree, respectively, with all covalent bonds and angles within 6.0*RMSD for VIVIT peptide as well as the interface of Cn-VIVIT.
The program PROCHECK-NMR (Laskowski et al., 1996) was used to assess the stereochemical quality of the structures.
Artificial constraints are applied to CnCat (see text more detail)
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (grants AI37581, GM47467 and EB 002026). K.T. is supported by Japan Society of Promotion of Science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aramburu J, Garcia-Cozar F, Raghavan A, Okamura H, Rao A, Hogan PG. Selective Inhibition of NFAT Activation by a Peptide Spanning the Calcineurin Targeting Site of NFAT. Molecular Cell. 1998;1:627–637. doi: 10.1016/s1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- Aramburu J, Heitman J, Crabtree GR. Calcineurin: a central controller of signalling in eukaryotes. EMBO Rep. 2004;5:343–348. doi: 10.1038/sj.embor.7400133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu J, Rao A, Klee CB. Calcineurin: from structure to function. Curr Top Cell Regul. 2000;36:237–295. doi: 10.1016/s0070-2137(01)80011-x. [DOI] [PubMed] [Google Scholar]
- Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–48. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- Ferentz AE, Wagner G. NMR spectroscopy: a multifaceted approach to macromolecular structure. Q Rev Biophys. 2000;33:29–65. doi: 10.1017/s0033583500003589. [DOI] [PubMed] [Google Scholar]
- Fooks HM, Martin ACR, Woolfson DN, Sessions RB, Hutchinson EG. Amino Acid Pairing Preferences in Parallel [beta]-Sheets in Proteins. Journal of Molecular Biology. 2006;356:32–44. doi: 10.1016/j.jmb.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Garcia-Cozar FJ, Okamura H, Aramburu JF, Shaw KTY, Pelletier L, Showalter R, Villafranca E, Rao A. Two-site Interaction of Nuclear Factor of Activated T Cells with Activated Calcineurin. J Biol Chem. 1998;273:23877–23883. doi: 10.1074/jbc.273.37.23877. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY 3 - NMR Assignment and Integration Software. University of California; San Francisco: 2006. [Google Scholar]
- Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J Biomol NMR. 1999;13:369–374. doi: 10.1023/a:1008393201236. [DOI] [PubMed] [Google Scholar]
- Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell. 1995;82:507–522. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- Gross JD, Gelev VM, Wagner G. A sensitive and robust method for obtaining intermolecular NOEs between side chains in large protein complexes. J Biomol NMR. 2003;25:235–242. doi: 10.1023/a:1022890112109. [DOI] [PubMed] [Google Scholar]
- Hemenway CS, Heitman J. Calcineurin. Structure, function, and inhibition. Cell Biochem Biophys. 1999;30:115–151. doi: 10.1007/BF02737887. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Li H. Calcineurin. Curr Biol. 2005;15:R442–443. doi: 10.1016/j.cub.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Huai Q, Kim HY, Liu Y, Zhao Y, Mondragon A, Liu JO, Ke H. Crystal structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. Proc Natl Acad Sci U S A. 2002;99:12037–12042. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, Rao A. The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature. 1993;365:352–355. doi: 10.1038/365352a0. [DOI] [PubMed] [Google Scholar]
- Jin L, Harrison SC. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc Natl Acad Sci U S A. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani A, Rao A, Aramburu J. Manipulating immune responses with immunosuppressive agents that target NFAT. Immunity. 2000;12:359–372. doi: 10.1016/s1074-7613(00)80188-0. [DOI] [PubMed] [Google Scholar]
- Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature. 1995;378:641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- Klee CB, Crouch TH, Krinks MH. Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci U S A. 1979;76:6270–6273. doi: 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14 doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Li H, Rao A, Hogan PG. Structural delineation of the calcineurin-NFAT interaction and its parallels to PP1 targeting interactions. J Mol Biol. 2004;342:1659–1674. doi: 10.1016/j.jmb.2004.07.068. [DOI] [PubMed] [Google Scholar]
- Lohr F, Katsemi V, Hartleib J, Gunther U, Ruterjans H. A strategy to obtain backbone resonance assignments of deuterated proteins in the presence of incomplete amide 2H/1H back-exchange. J Biomol NMR. 2003;25:291–311. doi: 10.1023/a:1023084605308. [DOI] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Michael HAR. Structural and Chemical Genetic Insights into Protein-Protein Interaction of Phosphate-Dependent Cell Signaling: The Cases of Calcineurin and Protein Kinase C Iota. Harvard University; Boston: 2004. [Google Scholar]
- Mondragon A, Griffith EC, Sun L, Xiong F, Armstrong C, Liu JO. Overexpression and purification of human calcineurin alpha from Escherichia coli and assessment of catalytic functions of residues surrounding the binuclear metal center. Biochemistry. 1997;36:4934–4942. doi: 10.1021/bi9631935. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Reibarkh M, Malia TJ, Hopkins BT, Wagner G. Identification of individual protein-ligand NOEs in the limit of intermediate exchange. J Biomol NMR. 2006;36:1–11. doi: 10.1007/s10858-006-9028-7. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Martinez-Martinez S, Lopez-Maderuelo MD, Ortega-Perez I, Redondo JM. The linker region joining the catalytic and the regulatory domains of CnA is essential for binding to NFAT. J Biol Chem. 2005;280:9980–9984. doi: 10.1074/jbc.C400401200. [DOI] [PubMed] [Google Scholar]
- Roehrl MH, Kang S, Aramburu J, Wagner G, Rao A, Hogan PG. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc Natl Acad Sci U S A. 2004a;101:7554–7559. doi: 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrl MHA, Wang JY, Wagner G. Discovery of Small-Molecule Inhibitors of the NFAT-Calcineurin Interaction by Competitive High-Throughput Fluorescence Polarization Screening. Biochemistry. 2004b;43:16067–16075. doi: 10.1021/bi048232o. [DOI] [PubMed] [Google Scholar]
- Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- Stewart AA, Ingebritsen TS, Manalan A, Klee CB, Cohen P. Discovery of a Ca2+- and calmodulin-dependent protein phosphatase: probable identity with calcineurin (CaM-BP80) FEBS Lett. 1982;137:80–84. doi: 10.1016/0014-5793(82)80319-0. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Miyazawa M, Ina Y, Fukunishi Y, Mizukoshi Y, Nakamura H, Shimada I. Utilization of methyl proton resonances in cross-saturation measurement for determining the interfaces of large protein-protein complexes. J Biomol NMR. 2006;34:167–177. doi: 10.1007/s10858-006-0008-8. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Nakanishi T, Kami K, Arata Y, Shimada I. A novel NMR method for determining the interfaces of large protein-protein complexes. Nat Struct Biol. 2000;7:220–223. doi: 10.1038/73331. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Zhou P, Dotsch V, Chen L, You A, Ho SN, Crabtree GR, Wagner G, Verdine GL. Unusual Rel-like architecture in the DNA-binding domain of the transcription factor NFATc. Nature. 1997;385:172–176. doi: 10.1038/385172a0. [DOI] [PubMed] [Google Scholar]
- Wouters MA, Curmi PMG. An analysis of side chain interactions and pair correlations within antiparallel?-sheets: The differences between backbone hydrogen-bonded and non-hydrogen-bonded residue pairs. Proteins: Structure, Function, and Genetics. 1995;22:119–131. doi: 10.1002/prot.340220205. [DOI] [PubMed] [Google Scholar]
- Zhou P, Sun LJ, Dotsch V, Wagner G, Verdine GL. Solution structure of the core NFATC1/DNA complex. Cell. 1998;92:687–696. doi: 10.1016/s0092-8674(00)81136-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.