

Summary
The Adult forms of Tay-Sachs (ATSD) and Sandhoff (ASD) diseases result when the activity levels of human β-hexosaminidase A (Hex) fall below ~10% of normal, due to mutations that destabilize the native folded form of the enzyme in the endoplasmic reticulum (ER). The native fold not only conveys activity, but is also required for the transport of the enzyme out of the ER to the lysosome. We have shown that conventional carbohydrate-based competitive inhibitors of purified Hex also act as pharmacological chaperones (PC) in ATSD or ASD cells, overcoming to some extent the destabilizing effects of the mutation, increasing the levels of mutant Hex protein and activity in the lysosome 3–6 fold. We now report the development of a fluorescence-based real-time enzyme assay suitable for high throughput screening of the Maybridge library of 50,000 drug-like compounds to identify novel inhibitors (hits) of purified human Hex. Each hit was then evaluated as a PC in a cell-based assay. Three structurally distinct compounds, a bisnaphthalimide, a nitro-indan-1-one, and a pyrrolo[3,4-d]pyridazin-1-one were identified as micromolar competitive inhibitors of the enzyme that also specifically increased the levels of lysosomal Hex protein and activity in patient fibroblasts.
Introduction
Pharmacological chaperones (PC) as small molecule therapeutics represent a novel paradigm for the treatment of disorders arising from mutations that destabilize and thus reduced protein-levels [1–3]. In these cases the mutation affects the equilibrium between the folded and unfolded states of the protein, shifting it away from the functional (folded) conformation. Improperly folded mutant (or wild type) proteins are then cleared by the “protein quality control systems” (QC) associated with the synthetic machinery in the cytosol and endoplasmic reticulum (ER) [4]. By preferentially binding to the native-like structure of the mutant protein, PCs, typically compounds acting as antagonists/inhibitors, shift the equilibrium back towards the functional conformation, which is deemed “competent for release” by the protein cells’ QC [5]. Depending on the target protein, this treatment has been shown to result in increased levels of the functional mutant protein in the cytosol [6], specific organelles, e.g. the lysosome [2], or the cell surface [7].
The PC approach has been shown to successfully enhance the enzyme levels of five different mutant lysosomal enzymes that lead to the chronic form of the lysosomal storage disorders; GM2 gangliosidosis [8], Fabry [9], Gaucher [10], and Morquio B diseases [11]. GM2 gangliosidosis, arising from the neuronal storage of GM2 ganglioside (GM2), occurs in three variants; Tay-Sachs disease (TSD), Sandhoff disease (SD) and the AB-variant. The former two result from mutations in the evolutionarily related HEXA or HEXB genes, encoding the α or β subunits, respectively, of heterodimeric β-N-acetyl-hexosaminidase A (Hex A, αβ) [12]. Two other homodimeric Hex isozymes exist, i.e. Hex B (ββ) and Hex S (αα), but can not utilize GM2 gangliosidase as a substrate.
Whereas both the α- and/or β-active sites of dimeric Hex can hydrolyze neutral synthetic N-acetyl hexosamine-terminal substrates, only the α-site of Hex A and S can efficiently utilize negatively charged substrates, e.g. 6-sulphated GlcNAc [13, 14]. Therefore, total Hex activity can be measured using 4-methylumbelliferyl-β-N-acetylglucosamine (MUG); whereas, 4-methylumbelliferyl-β-N-acetylglucosamine-6-sulfate (MUGS) is used to measure Hex A and Hex S activity [14]. Substrates based on 4-methylumbelliferone (MU) are available and used to diagnose enzyme deficiencies in the other LSD.
The more common infantile TSD (ITSD) variant of GM2 gangliosidosis, results from absent α subunits, and elevated amounts of Hex B, such that levels of total Hex activity (MUG) are near normal. Less common is infantile SD (ISD), resulting from an absence of β-subunits and very low levels of Hex activity associated with the unstable Hex S isozyme. In contrast to the infantile forms, Adult TSD (ATSD) and SD (ASD) are chronic, slowly progressive, neurodegenerative diseases that vary in age-onset. In many cases they are associated with missense mutations, usually producing thermolabile Hex A with residual activity (MUGS) and protein levels that are <10%, but >2% of normal. The correlation between clinical phenotypes and residual activity indicates that there is a surprisingly low critical threshold level of Hex A activity, i.e. the level of Hex A needed to prevent GM2 ganglioside storage, of ~10% of normal [15].
The majority of patients with ATSD possess a missense mutation in exon 7 of the α-subunit gene i.e. αG269S [12]. This and similar point mutations do not directly affect the α-active site of Hex A or the interface between its α-and β-subunits [13] [16], but are believed to result in increased amounts of misfolded α-protein in the endoplasmic reticulum (ER), which are in turn retained by its QC and degraded [17]. Since only a small proportion of the newly synthesized mutant α-precursor can adopt the proper conformation necessary to form heterodimers and become transport-competent, there are reduced levels of both Hex A activity and protein in the lysosome.
Previously, we used conventional carbohydrate-based Hex inhibitors, e.g. N-Acetyl Glucosamine Thiazoline (NGT), to enhance the stability, protein levels and enzyme activity of mutant Hex A in ATSD fibroblasts, and Hex S levels in ISD patient fibroblasts [8]. NGT is a stable thiazoline derivative of the oxazoline reaction intermediate formed by lysosomal Hex and other members of glycoside hydrolase family GH20 [18, 19]. The 3D-structures of GH20 family-members human Hex B [13, 20] and Hex A [16], Streptomyces plicatus Hex [21], and Serratia marcesans chitobiase [22], have been determined; the former two in complex with NGT. Although these enzymes have an overall identity of less than 25%, their active site residues share greater than 95% identity, and are superimposable to less than 1.5 A rmsd [13, 21].
In all examples to date, compounds acting as PCs in cells have also been competitive inhibitors of the target enzyme [2]. Their effectiveness often varying with the nature of the mutation [23], but generally correlating with their bioavailability and Ki [24]. Potential PC have been identified for only a minority of the 40 known gene products that when mutated, can result in a LSD. High throughput screening (HTS) of small molecule libraries has been used to identify specific drug-like inhibitory compounds for proteins with [25] or without [26, 27] known structures or mechanisms of action. It is proposed that HTS of a library of compounds for inhibitors of the target enzyme would also result in the identification of candidate PC, which could then be evaluated by a secondary live cell-based assay. Thus, we developed a real time MU-based enzyme assay and coupled it with HTS, utilizing purified human placental Hex as a target to identify several novel drug-like inhibitors. Subsequently, lysates from patient cells grown in the presence of these compounds were demonstrated to have enhanced levels of Hex A (ATSD) or Hex S (ISD) activity and protein. This approach represents a facile and efficient strategy for identification of candidate PCs in small molecule libraries for any lysosomal enzyme with an available MU-based substrate by first screening for inhibitory compounds.
Results and Discussion
Development of an assay to continuously monitor Hex activity
Conventional 4-Methylumbelliferyl (MU) based substrate assays for the known lysosomal glycosidases are end-point assays which are less convenient for HTS, because they are more prone to false positive (quenchers) or false negatives (fluorescent compounds) [28]. Consequently we developed a real time MU-based enzyme assay and coupled it with HTS of the 50,000 small molecule compound Maybridge library to identify novel drug-like inhibitory compounds against highly purified placental human lysosomal Hex.
As the MU-fluorophore released by Hex and other lysosomal enzymes, has a pKa = 8.5 that is higher than the acidic pH optimums of these enzymes, an endpoint assays has been used. Recently, it has been shown that the released MU fluorophore can be continuously and efficiently monitored at an acidic pH by decreasing the excitation wavelength from 365 nm to 330 nm [29], but with no change in emission optimum (450 nm). Using this assay the rate of MU hydrolysis was linear over 25 minutes, with an apparent Km of 0.35 mM at room temperature. For the HTS, a lower concentration of substrate was chosen (75 μM), to diminish the quenching effects of the substrate [30].
Since all of the test compounds were dissolved in DMSO, we checked for effects of DMSO on Hex A activity. DMSO inhibited Hex A activity by more than 50% at the concentrations at which the library compounds were screened (compounds tested at 10 μM gave the equivalent of 0.26 M DMSO in the assays) (Fig 1a.). Furthermore, since Vmax remained constant whereas Km increased with increasing DMSO concentrations, DMSO was found to be a competitive inhibitor of Hex (Fig. 1a.) with a Ki of 0.17 ± 0.02 M.
Fig. 1.
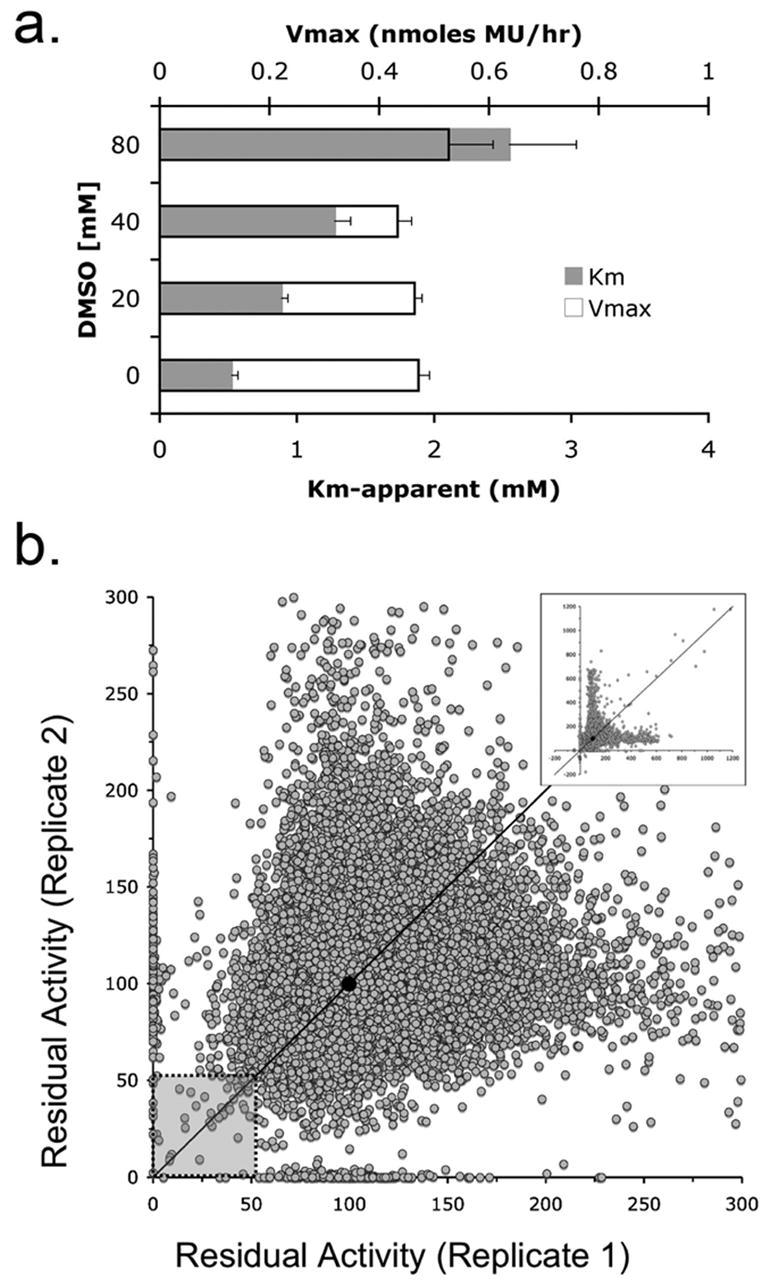
Primary screen of a 50,000 compound Maybridge library for Hex A inhibitors in the presence of DMSO, a competitive inhibitor of Hex A. Panel a. The change in Km (grey bars) and Vmax (bars outlined in black) versus increasing concentrations of DMSO (n=3). Panel b. A portion of the replicate plot of residual Hex A activity (0–300%) in the presence of individual compounds from the Maybridge library (inset: entire replicate plot (0–1200%) for all compounds). Diagonal line denotes compounds with identical activities in both replicates. The hit zone containing candidate inhibitory compounds and average residual activity is denoted by the shaded box and filled circle, respectively. Residual activity values for each compound is provided in supplemental data.
Primary biochemical screen: identification of candidate Hex inhibitors
To identify inhibitors, the activity of purified human Hex A was evaluated in the presence of each of 50,000 compounds from the Maybridge small-molecule library. Inhibitory compounds in the library were expected to reduce the residual activity of Hex A relative to the residual activity of the enzyme treated with DMSO (High Controls). Despite the fact that DMSO was a weak competitive inhibitor of Hex A, the HTS was successful in identifying Hex inhibitory compounds.
Replicate assays for each compound were plotted as a single (X,Y) co-ordinate (Fig. 1b). The majority (>75%) of these values differed by less than 1.4 fold, and fell near the X=Y diagonal, i.e. the replicates were generally reproducible. The average Z′ statistic [31] of 0.75 for the two replicates was well within the acceptable range for a robust and interpretable screen [32]. The statistical cut-off for the “hit”-threshold, based on the standard deviations of the high (DMSO only) and low (NGT and DMSO) control values, was determined to be 69%. Since this resulted in greater than 600 hits, the hit threshold was empirically set to 50% residual activity, which lowered the number of compounds to be evaluated to a more manageable 64 (0.13% hit rate).
Secondary screen: validation of candidate inhibitors from primary screen
By virtue of their semilogarithmic sigmoidal dose response curve, which is typical of an inhibitor, only 24 of the 64 hits were confirmed as bona fide inhibitors (Table 1). Interestingly, the 24 confirmed hits were also compounds that replicated best in the primary screen. The inhibitory activity of these compounds (Table 1) span three-orders of magnitude; i.e., one compound had an IC50 of 0.2 μM, two were >6 μM, 16 ranged from 12–79 μM, and 6 from 105–385 μM. These results confirm the utility of coupling the real-time enzyme assay based on 4-MU substrates with HTS to identify inhibitory compounds for Hex and other lysosomal enzymes from a library of small molecule compounds.
Table 1.
List of inhibitory compounds verified in a secondary screen showing corresponding structure, identification number and IC50 (μM) values.
|
|
|
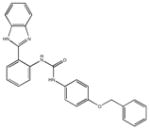
|
| M-00573 6.5±4.3 | M-00659 34±21 | M-02984 65±99 | M-19672 12±14 |
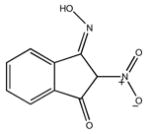
|
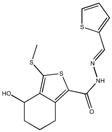
|
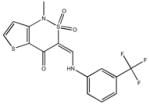
|
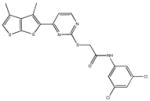
|
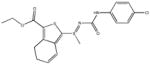
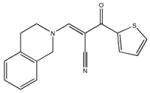
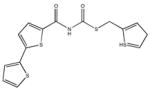
| M-22971 1.8±2.6 | M-26024 30±6.7 | M-26463 270±256 | M-26553 20±7.2 |
|
|
|
|
| M-27352 39±52 | M-27368 42±92 | M-28322 29±9.1 | M-28324 180±105 |
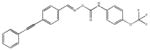
|
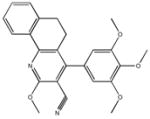
|
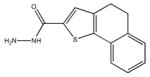
|
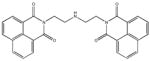
|
| M-28347 71±25 | M-31516 35±35 | M-31526 24±4.0 | M-31850 0.20±0.14 |
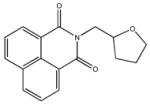
|
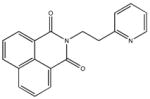
|
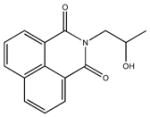
|
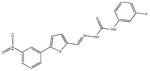
|
| M-31860 138±111 | M-31862 108±53 | M-31867 31±15 | M-32527 42±69 |
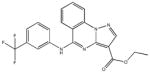
|
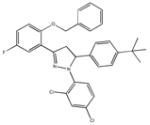
|
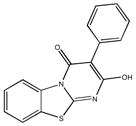
|
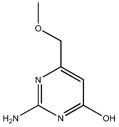
|
| M-34700 45±12 | M-35014 162±94 | M-37955 72±32 | M-38728 44±62 |
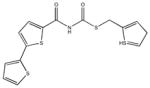
|
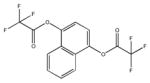
|
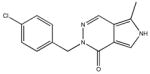
|
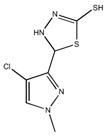
|
| M-42881 77±54 | M-44054 93±82 | M-45373 5.8±0.44 | M-49773 79±35 |
Singleton compounds which had an IC50 > 100 μM, or had reactive groups; e.g., compound M-49773, which bore a reactive thiol group (known to inactivate Hex), were not examined further. Similarly, cyclic thiol compounds, represented by M-27352 and M-26024, were eliminated from study, as they have been found to be promiscuous inhibitors, identified in other HTS using different enzymes. Compounds such as M-28324 and M-00659, that were difficult to dissolve and/or precipitated readily in aqueous solutions above 10 μM, also were not further examined.
The majority of compounds identified by HTS, that inhibited Hex were substituted bicyclic and tricyclic nitrogen containing heterocycles. The structures of these compounds differ greatly from known azasugar-based Hex inhibitors, and represent novel scaffolds that could be further optimized for use as specific Hex inhibitors.
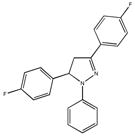
Three of the 24 compounds, M22971, M31850 and M45373, were chosen to be evaluated in greater detail. M-31850 had the lowest IC50 identified, 200 nM, and three additional, structurally related compounds, i.e. bearing the 1,8 naphthalimide moiety compounds, were also identified with IC50s > 10 μM. The other two compounds, M-22971, an indan-1-one, and M-45373, bearing a pyrrolo[3,4-d]pyridazin-1-one moiety, were found to have IC50s < 10 μM (1.8 and 5.8 μM IC50, respectively). Furthermore, compounds M-31850 and M-22971 contained scaffolds found in the drugs, Alrestatin [33] and Indacrinone [34], that have been approved for use in humans. Although compound M-22971 has a structure common to bidentate ligands, i.e. one capable of binding a metal ion, Hex does not require a metal ion for activity.
The inhibitory activities of the HTS inhibitors of Hex towards other members of glycosidase family GH20, and human O-GlcNAcase
In order to determine the selectivity of the HTS inhibitors, their activities were evaluated against human Hex A (hHexA) and Hex B (hHex B), Jack Bean Hex (JBHex), the plant ortholog of lysosomal human Hex, bacterial Hex from Streptomyces plicatus (SpHex), and chitobiase B from Serratia marcesens (SmCHB); all are members of the GH20 glycosidase family. Human O-GlcNacase (hOGN), a member of the GH84 family of glucosaminidases that has a reaction mechanism similar to Hex, was also tested with the HTS inhibitors (Table 2). Glycosidases in both of these families use substrate assisted catalysis, have conserved residues in their active site that are superimposable to within 1.5 A and are all inhibited by NGT to different degrees.
Table 2.
Summary of IC50 (mM) values for the confirmed inhibitory compounds against human Hex isozymes, a panel of Family 20 glucosaminidases and O-GlcNAcase (hOGN).
| Inhibitor1 | hHexA | hHexB | JBHex | SpHex | SmHex | hOGN |
|---|---|---|---|---|---|---|
| DMSO | 350 ± 31 | 98 ± 20 | 300 ± 13 | NI (380) | NI (380) | NA2 |
| NGT | 0.000073 | 0.000073 | 0.000284 | >3.05 | >3.0 | 0.000076 |
| M-229717 | 0.027 ± 0.006 | 0.071 ± 0.10 | 0.23 ± 0.07 | >1.0 | NI (1.0) | NI (2.0) |
| M-31850 | 0.0060 ± 0.0018 | 0.0031 ± 0.0015 | 0.28 ± 0.04 | > 0.5 | NI (0.3) | NI (1.0) |
| M-45373 | 0.020 ± 0.008 | 0.040 ± 0.05 | NI (0.6)8 | NI (0.6) | NI (0.6) | NI (2.0) |
Based on endpoint assay (1.6mM MUG)
Not Applicable (NA)
Ki (mM) was determined using placental Hexosaminidase which is a mixture of HexA and Hex B, From Macauley et al (2005) [35], Table II
Ki was determined using pNP-GlcNAc substrate [45]
Non-inhibitor Compound (NI); highest concentration (mM) at which compound was evaluated.
Ki (mM) as shown in Macauley et al (2005)[35], Table II
Based on 1.5 mM pNP-GlcNAc substrate
Incomplete dose response curve; estimated IC50 (mM)
Although all of the HTS inhibitors show activity towards both Hex A and B isozymes, there was a two-fold difference in the IC50 values for the two isozymes. Thus, DMSO and M-31850 have lower IC50 values for Hex B as compared to Hex A, while the reverse is true for both M-22971 and M-45373. M-45373 did not display any inhibitory activity towards JBHex, even at concentrations approaching 1mM. Although, the two HTS inhibitors, M-22971 and M-31850 showed some activity towards SpHEx, their IC50values decreased more than 30 and 100 fold, respectively, relative to hHex. Most significantly, none of the HTS compounds M-22971, M-31850 or M-45373 showed any inhibitory activity towards hOGN, even though this enzyme readily hydrolyses MUG (Km = 0.43 mM) and NGT functions as an effective competitive inhibitor (Ki = 70 nM) [35]. None of the three HTS inhibitors, even at concentrations approaching 1mM, showed any inhibitory activity towards an unrelated lysosomal glycosidase, glucocerebrosidase (Cerezyme).
NGT, the compound mimicking the reaction intermediate common to all members of Family 20 and 84 glycosidases, showed the broadest range of inhibitory activity against the enzymes listed in Table 2. This is not surprisingly as NGT interacts only with the conserved residues that directly participate in substrate binding and catalysis. The specificity shown for the human Hex isozymes by the compounds identified through HTS, suggests that they likely interact with only a few of the same conserved active-site residues, requiring interactions with other peripheral, non-conserved residues at or near the active site of hHexA/B for high affinity binding.
Evaluation of chaperoning potential of HTS Hex inhibitors in ISD patient fibroblasts
The ability of the HTS inhibitors to act as PCs was evaluated using a cell line from a patient with ISD, i.e. only expresses Hex S. This isozyme is intrinsically unstable and was shown previously to be very responsive to the chaperoning effects of NGT [8]; e.g., similar treatment of ISD or ATSD with NGT resulted in a 6-fold increase in Hex S activity, but only a 3-fold increase in Hex A activity [8]. All three compounds, M-31850, M-22971 and M-45373, produced a dose dependent increase in MUG hydrolysis (Hex S levels) in lysates from treated ISD cells (Fig. 2a). The increase was specific to Hex, as a similar increase was not observed in hydrolysis of MUbGal by lysosomal enzyme β-galactosidase (Fig 2b). Clearly the three inhibitory compounds function as specific PCs for Hex.
Fig. 2.
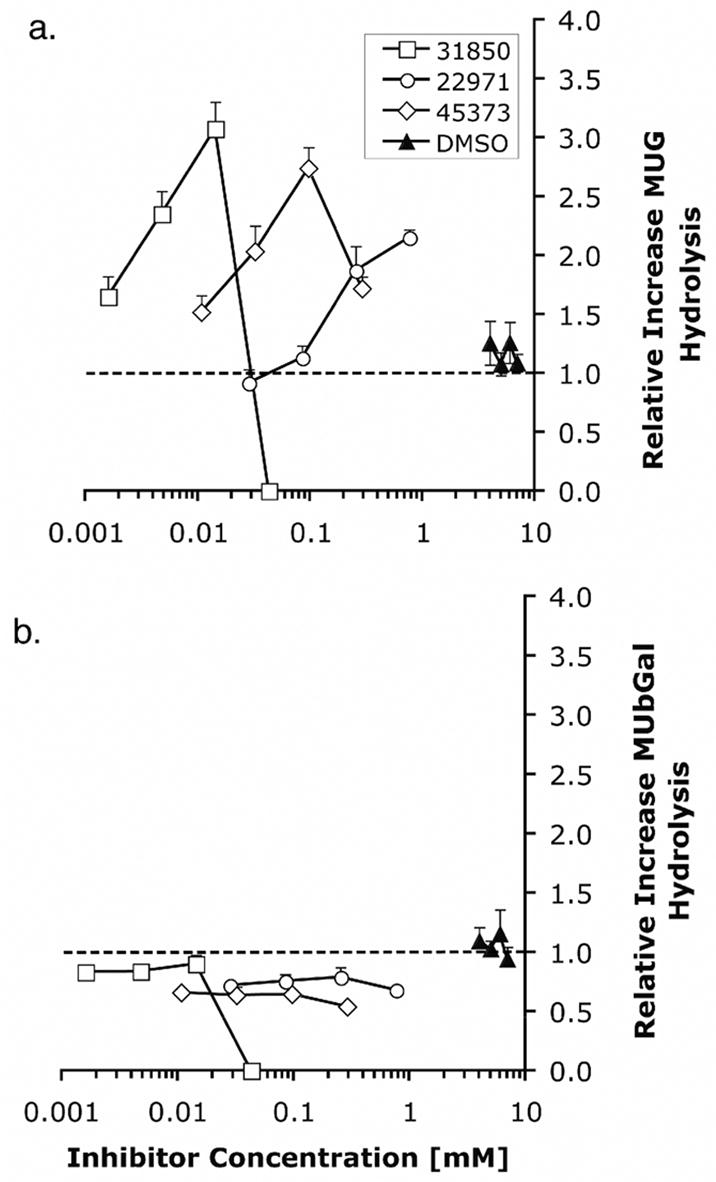
Dose dependent increase in MUG activity following growth of ISD fibroblasts in media containing HTS derived inhibitory compounds. ISD fibroblasts were grown in media containing different concentrations of M-22971, M-31850, M-45373 or DMSO for 5 days, lysed and MUGS or MUbGal activity determined (n=4). The relative increases in MUGS (Panel a) or MUbGal (Panel b) hydrolysis were determined as (MU fluorescence of the inhibitor-treated cells)/(MU fluorescence of mock (DMSO)-treated cells).
Compared to NGT, which is most effective in enhancing enzyme activities at 1 mM [8], both M-31850 and M-45373 are maximally effective as PCs at 100- and 10- fold lower concentrations, respectively. In contrast, M-22971 produces a maximal response near 1mM. Although effective as PCs, the parallel decrease in relative activity of both MUG and MUbGal seen at concentrations above 1mM are indicative of the cellular toxicity of M-31850 and M-22971 at elevated levels. Bisnapthalimide compounds are toxic to cells in nM to μM range depending on the number of amines and length of alkylamine linker [36].
Treatment of ATSD fibroblasts with HTS compounds results in increased levels of mature (proteolytically processed) α-subunit in lysosomes
To demonstrate that the compounds identified by HTS can enhance the functional levels of the G269S mutant α-subunit, ATSD patient fibroblasts were grown in the presence of M22971, M31850 and M45373 at concentrations shown to be optimal in ISD cells. Western blotting with an anti-Hex A antibody showed that treatment of ATSD fibroblasts with the HTS compounds resulted in increased levels of α-subunit (Fig. 3a). In comparison to untreated cells (media only), the DMSO and HTS-inhibitor treated cells showed increased levels of a 56 kDa band, corresponding to the processed, mature α-subunit (αm) found in WT fibroblasts. Densitometric quantitation of the αm band demonstrated a clear increase in levels of α-subunit protein that closely paralleled the increase in specific MUGS activity found in the lysates of inhibitor-treated cells (Fig. 3b). Not surprisingly, since DMSO is a weak competitive inhibitor of Hex, it also acted as a weak PC, increasing the amount of αm approximately 1.5 fold relative to untreated cells. However, one can not exclude the possibility that DMSO may enhance Hex activity by other mechanisms as it has been reported to enhance protein levels by acting as a non-specific chemical chaperone [37] and was found to increase mRNA levels of Il-1beta by augmenting promotor activity [38].
Fig. 3.
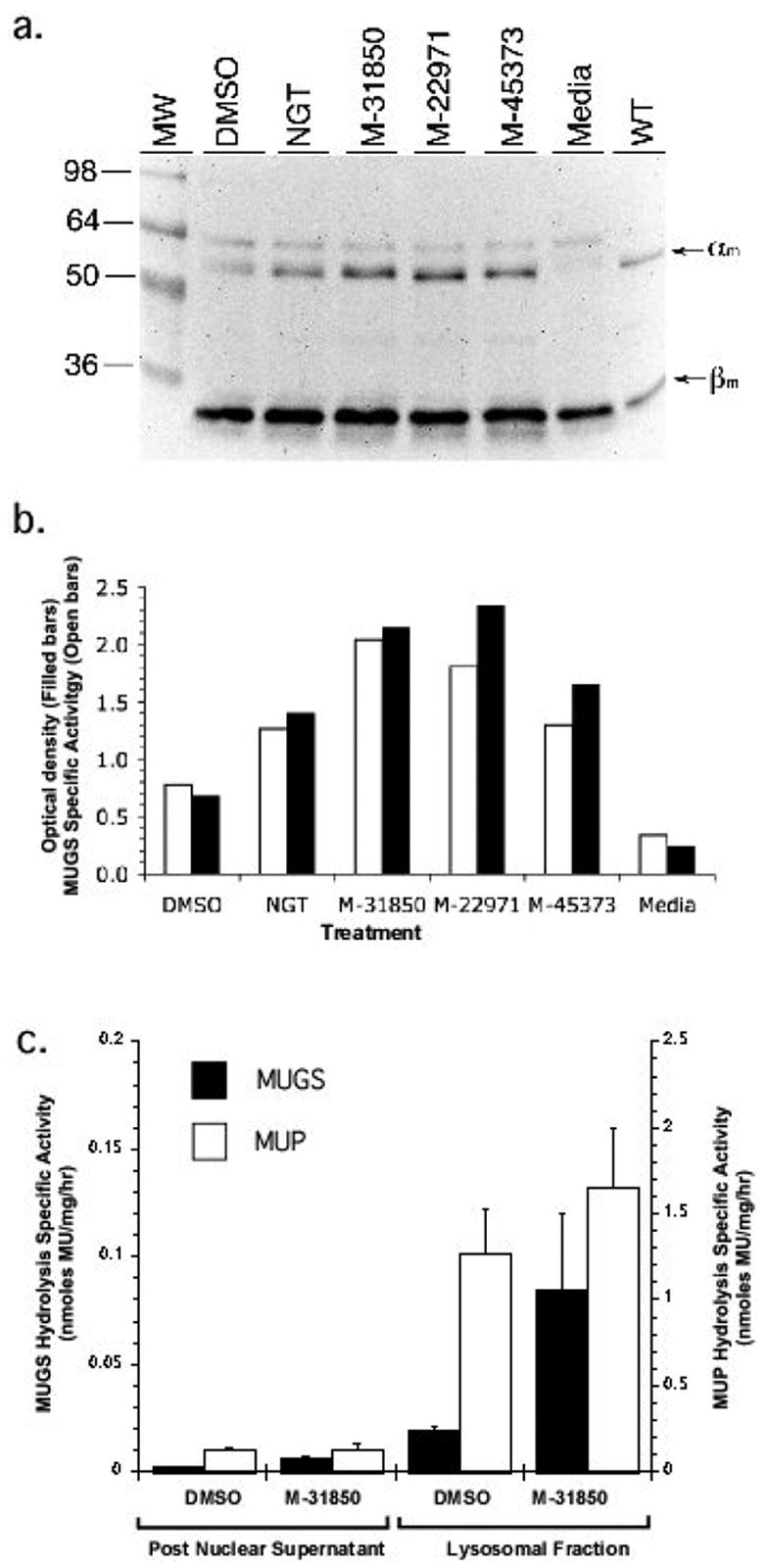
Increased MUGS activity in treated ATSD fibroblasts is associated with an increase in the mature α-subunit protein and levels of the active Hex A isozyme. Panel a. ATSD cells were treated for five days with DMSO (1%), NGT (1 mM), M-31850 (0.002 mM), M-22971 (1mM) or M-45373 (0.1mM). Following resolution of the α and β subunits by SDS-PAGE and subsequent transfer to nitrocellulose, the blot was probed with rabbit polyclonal anti-human Hex A antibody. Positions of the lysosomally processed, mature α-subunits (αm) and β-subunits (βm) in mock (media) or inhibitor-treated fibroblasts are denoted by arrows to the left of the blot. Position of Mr markers are shown to the left of the blot. Panel b. the increase in MUGS activity parallels the increase in amount of mature α-subunit from lysates of treated ATSD fibroblasts. The specific MUGS activity (nanomoles MU released/mg total cell protein/h) of lysates used to produce the Western blot in panel a. is shown plotted (open bars) adjacent to the optical density of the αm band (filled bars) from the Western blot in panel a. Panel c. Increased MUGS (Hex A) activity is found in the lysosomal fraction of M-31850 treated ATSD cells. Comparison of MUGS activity (nanomoles/mg of total cell protein/h) in the post nuclear supernatant (PNS) and lysosome (lyso) enriched fractions from M-31850 (0.002 mM) treated (filled bar) ATSD fibroblasts.
The proteolytic processing of lysosomal enzymes occurs post ER, i.e. in lysosomes or late endosomes [39, 40]. D’azzo et al (1984) [41] previously demonstrated that the αG269S Hex A is retained in the ER, resulting in barely detectable levels of αm. Thus, the increased levels of αm in the presence of the HTS-inhibitors, suggests that the compounds function as PCs, increasing the levels of α-subunit that can be transported from the ER to the lysosome.
To conclusively demonstrate that the inhibitor treated cells have increased levels of Hex A in the lysosomes, a lysosome enriched fraction was prepared from DMSO and M-31850-treated fibroblasts (Fig 4c). This method has been previously used to isolate an ER-depleted lysosomal fraction by magnetic chromatography following labeling of fibroblasts by iron-dextran colloid [8, 42]. There was an approximately ten-fold increase in Hex (MUGS) and acid phosphatase (MUP) specific activity in the lysosomal fraction. More importantly MUGS activity was increased three-fold both in the post nuclear supernatant and lysosome enriched fractions, consistent with the hypothesis that increased levels of Hex A in treated cells are found in the lysosome. The activity of lysosomal acid phosphatase was not significantly effected in either fractions from M-31850-treated cells. Fibroblast do not synthesis significant levels of the higher gangliosides, e.g. GM1 ganglioside [43]. Thus, the ATSD and ASD cells used in this study do not store appreciable amounts of GM2 ganglioside. However, our data strongly imply that patient cells that do synthesis higher gangliosides, i.e. neurons, would benefit from M-31850 treatment.
Fig. 4.
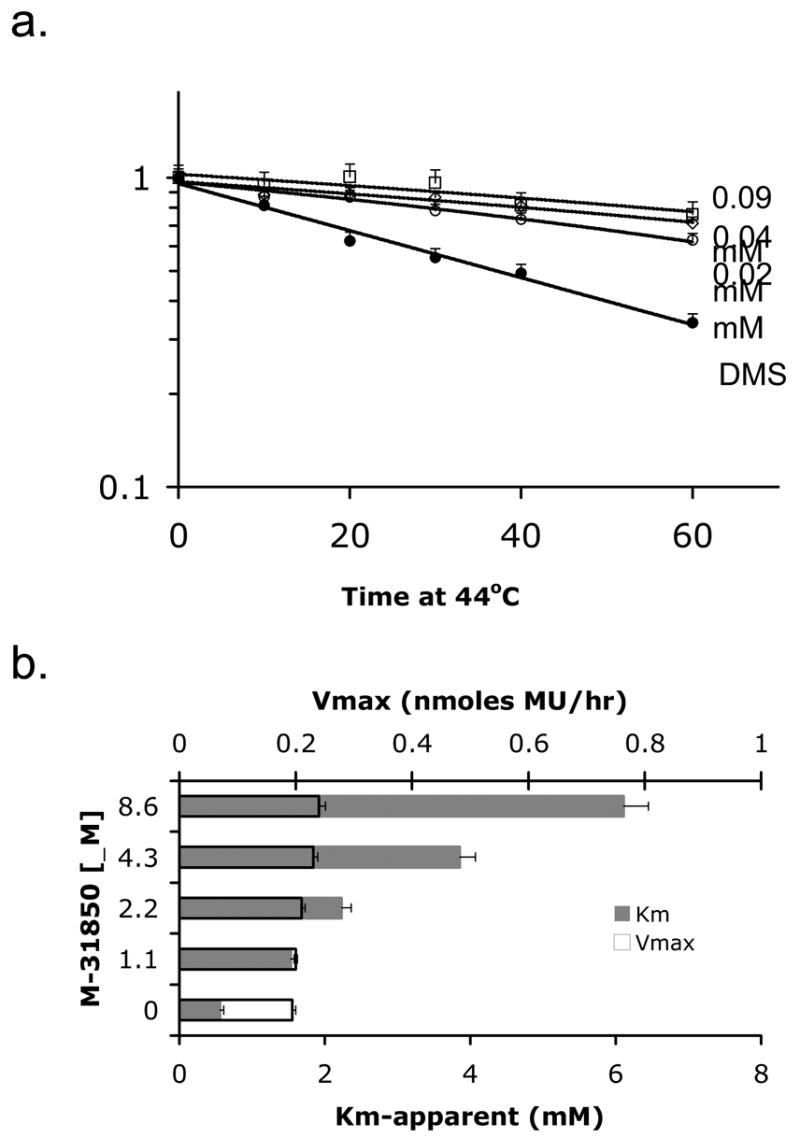
M-31850 attenuates thermal denaturation of Hex A, αGly269Ser mutant at 44°C and it is a competitive inhibitor. Panel a. Partially purified mutant Hex A from ATSD fibroblasts was heated at 44°C in the presence of DMSO (mock- treated, filled symbols), or different concentrations of M-31850 (open symbols) for 10, 20, 30, 40 or 60 minutes. “Fraction remaining MUGS activity,” (y-axis) was calculated using the formula (MU fluorescence of inhibitor-treated sample heated for a given time)/(MU fluorescence of inhibitor-treated sample left at 4°C (time = 0 min.), n=3), i.e. 1= no change. Panel b. The change in Km (grey bars) and Vmax (bars outlined in black) versus inhibitor concentration is shown for M-31850 (n=3) using MUG substrate. Michaelis-Menten plots were used to determine Km and Vmax in the absence and presence of increasing concentrations of M-31850 (dissolved in water).
These results validate the approach for identifying novel PC by first performing a HTS for Hex inhibitory compounds. The strongest inhibitors identified, M-22971, M-31850 and M-45373, are novel and structurally distinct from the known human Hex inhibitors, the majority of which are azasugars, e.g. [44, 45], and iminocyclitol derivatives [46]. These HTS derived inhibitors can serve as new, more drug-like, frameworks that can be further optimized using high throughput combinatorial chemistry [46]. This approach could be applied to the naphthalimide derivatives as they can be readily synthesized via a straightforward single step scheme [47].
Examination of the mechanism by which compound M-31850 acts as a PC for Hex A
Bisnaphthalimide compound M-31850 was examined in greater detail in terms of its mechanism of binding, because; 1) it was active in cells at the lowest concentration of the HTS compounds examined, 2) several naphthalimide derivatives, Elinafide (LU79533) and alrestatin had been evaluated or approved for use in humans, and 3) and other naphthalimide congeners can be easily synthesized. To confirm that M31850 functions as a PC in a manner similar to NGT, i.e. it binds at (or near) the active site and is capable of increasing the stability of the enzyme [8], the effects of the compound on enzyme kinetics and thermal denaturation were analyzed. M-31850 increased the half-life of the mutant Hex A from ATSD cells more than two-fold at 44° C, relative to the enzyme heated in the presence of DMSO (Fig. 4a). It acts as a classic competitive inhibitor of Hex (Km increases and Vmax is unaffected by increasing amounts of M-31850), with a Ki of 0.8 ± 0.1 μM (Fig. 4b).
All of the mono-naphthalimide derivatives, M-31860, M-31862 and M-31867 (Table 1 and Table 3) from the secondary screen had IC50 values that were at least 100fold higher than the bis-naphthalimide, M-31850 (Table 3). All of these derivatives contained a small hydrophobic group N-linked to the underlying naphthalimide by a short alkyl chain that lacked a secondary amine. Similarly, the decreased inhibitory activity of the mono-naphthalimides, BTB12933 and 5141402, further underline the importance of the second naphthalimide moiety.
Table 3.
Inhibitory activity of naphthalimide derivatives against purified human HexA.
| ID | Substituent (-R) | IC50 (mM) |
|---|---|---|
| Parent1 |
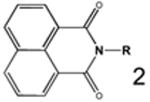
|
|
| PHG 00876 |
|
NI (200) |
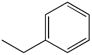
| M-31860 |
|
138 ± 1113 |
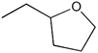
| M-31862 |
|
108 ± 53 |
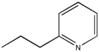
| M-31867 |
|
31 ± 15 |
| BTB12933 | -H | 18 ± 1.5 |
| 5141402 |
|
16 ± 1.5 |
| M-31850 |

|
0.6 ± 0.3 |
| LU79953 |
|
4.6 ± 1.9 |
| 7916963 |

|
NI (200) |
General structure naphthalimide derivatives evaluated.
Structures of N-linked R-groups from naphthalimide derivatives.
IC50 values based on 1.6 mM MUG (endpoint).
The critical role of the secondary amine group in the linker bridging the two naphthalimide moieties was underscored by the loss of all inhibitory activity when it was replaced by an ether linkage in the analogous position in compound 7916963. The behavior of this compound suggests that the secondary amine in M31850 may be involved in the formation of a hydrogen bond with an unidentified residue in Hex A. Although reduced more than ten-fold, the inhibitory activity of compound 5141402 suggests that an analogously positioned hydroxyl group could provide the necessary hydrogen atom. The importance of the N-alkylamine linker for inhibitory activity of both the mono- and bis-naphthalimide derivatives, 5141402 and M-31850, is reminiscent of recently described iminocyclitol based Hex inhibitors bearing an N-alkylamine moiety [48].
The attenuated inhibitory activity of compound 5141402, lacking the second naphthalimide, suggests that it may provide additional hydrophobic contacts important for high affinity binding. It is surprising that there is an approximately 8-fold decrease in the inhibitory activity of LU79553, as it bears two naphthalimide groups and the position of the amine in the linker relative to the naphthalimide group is identical to that in M31850. This suggests that the two naphthalimide groups must be spaced appropriately in order to effectively bind Hex A. The second active site in Hex A is not a candidate for the site that binds the second naphthamide group in M-31850, as the measured distance between the two active sites is more that 2.5 times the size of the alkylamine linker in M-31850. However the proposed hydrophobic aglycon pockets just outside either active site of Hex A [20, 48] remain possible candidates.
On the basis of the inhibitory activity of the naphthalimide derivatives in Table 2, and the fact that M-31850 is a competitive inhibitor, the following binding model is proposed. One of the naphthalimide groups binds directly in the substrate binding pocket of Hex A, while the second naphthalimide moiety binds to a secondary hydrophobic patch, e.g. an aglycon binding site. An appropriately sized linker between the two naphthalimide groups is needed to span the two sites and must also be able to form a stabilizing hydrogen bond with an acidic acceptor residue on the surface of Hex A.
Compound M-31850 was evaluated in greater detail in part due to its close similarity to LU79553 (Elinafide) which previously had been evaluated in phase I clinical trials as a solid tumor chemotherapeutic due its DNA intercalating ability and inhibitory activity against topoisomerase I [47]. Although, Elinafide did not proceed further in clinical trials [49, 50] due to its toxicity at therapeutic doses, these results demonstrate the utility of using the inhibitory compounds as seeds to identify in-trial or FDA approved drugs. Since the goals of the HTS screen was to identify more drug-like PCs, compounds such as Elinafide would be expected to expedite the development of therapeutics for the treatment of ATSD [51].
Significance
In Tay-Sachs and Sandhoff diseases, inadequate levels of Hex A (αβ-dimer) in the lysosome results in the intra-lysosomal accumulation of GM2 ganglioside in neuronal cells. Although the more common infantile forms have no residual enzyme activity, the late-onset, adult and juvenile forms have 2–8% of normal activity. To date carbohydrate-based inhibitors of Hex, e.g. NGT, act as pharmacological chaperones, enhancing lysosomal Hex enzyme levels in late-onset patients cells above the ~10% critical threshold.
We have developed a high throughput screening (HTS) method to identify novel drug-like inhibitors of Hex utilizing a fluorescence-based real-time enzyme assay. The simplicity and efficiency of this strategy to identify inhibitors utilizing existing methylumbelliferone-based substrates can be exploited to quickly identify candidate drug-like PC for other LSD where there is a non-existent or limited panel of inhibitory compounds available for the deficient enzyme.
Three structurally distinct single digit micromolar Hex inhibitors were identified that did not inhibit other lysosomal enzymes or neutral, cytosolic O-GlcNAcase. Unlike the majority of Hex inhibitors that are azasugar derivatives, these compounds consist of novel frameworks found in FDA approved drugs such as Alrestatin and Indacrinone. All of the HTS derived inhibitors were shown to function as PCs, since they enhanced the levels and activity of Hex S and mutant Hex A in ISD and ATSD patient fibroblasts, respectively. Importantly, the approximately three-fold increase in protein and enzyme activity levels in the lysosomes from M-32850 treated ATSD cells would be expected to raise Hex A levels above the critical 10% threshold. These results validate the approach of using HTS for inhibitory compounds to identify candidate drug-like PCs.
Experimental Procedures
Chemicals and Reagents
A total of 50,000 drug-like compounds from the Maybridge collection (Maybridge PLC, UK) were used in the initial screen. Compounds evaluated in the secondary screen and their derivatives were re-ordered from Maybridge PLC (UK) or Chembridge (USA) and solubilized using DMSO or water. Fluorogenic substrates purchased from SIGMA (USA) included, 4-methylumbelliferyl-β-D-glucopyranoside (MUbGlc), 4-methylumbelliferyl-β-D-galactosamine (MUbGal), 4-methylumbelliferyl phosphate (MUP), MUG, and MUGS. The colorimetric substrate p-nitrophenol-β-D-N-acetyl glucosaminide (SIGMA, USA) was also used to monitor Hex A activity. Placental Human Hex A was prepared as described [52]. NGT was synthesized and purified as previously described [53] [45].
Cell lines
The following cell lines were used: 1766 (ATSD) was from a ~40 year old female patient diagnosed with the chronic (adult) form of TSD (kindly provided by Dr. J.R. Donat, University of Saskatchewan, Kinsmen Children’s Centre, Saskatoon, Saskatchewan, Canada) homozygous for the αG269S mutation (Molecular Diagnostics Laboratory, SickKids, Toronto, Ont., Canada); 294 (ISD) was from an infantile Sandhoff disease, homozygous for the 16kb HEXB deletion mutation [54, 55]. All cell lines were grown in α-minimal essential media (α-MEM) (Invitrogen, USA) supplemented with 10% fetal calf serum (FCS) (Sigma, USA), and antibiotics Penicillin/Streptomycin (Invitrogen, USA) at 37°C in a humidified CO2 incubator.
Primary Screening
The statistical test, Z′ factor [31] measured the variability of the rate values for Hex in the presence (Low control) and absence (high control) of the Hex inhibitor NGT. To control for plate-to-plate variability, enzyme activity was expressed according to the equation:
(where “RA” = Residual Activity, “r“ = rate in the presence of the library compound; “μL” = mean low control rate, i.e. activity in the presence of NGT and DMSO; “μH“ = mean high control rate, i.e. activity in the presence of DMSO only. Reactions were setup in a black-walled 384 well plates using SAGIAN Core System (Beckman Coulter, Inc. Fullerton, CA, USA) equipped with an ORCA arm for labware transportation, a Biomek FX with a 96-channel head for liquid handling, an Analyst HT (Molecular Devices Corp., Sunnyvale, CA, USA) for fluorescence detection. All reactions were performed in duplicate (separate plates). High controls (64 wells) consisted of Hex A (100 ng/ml ) in Citrate Phosphate (CP) buffer containing 0.0025% human serum albumin and 0.5% DMSO. Low controls (64 wells) additionally contained NGT at 360 nM. Test wells contained 10 μM of each library compound. To take into account the effects of DMSO on enzyme activity, the expressed residual activity for the two replicates in Figure 1 was normalized to 100% (100 * raw activity/mean activity).
Secondary Screening
The dose-response curves of the 64 hits from the primary screen were determined by the continuous MUG Hex assay, in the presence of seven concentrations (0.1–100 μM) of the putative inhibitor diluted in DMSO. Data were fit to the equation:
using either Grafit (v.4.0.010, Erithacus Software, Surrey, UK) or Kaleidagraph (Synergy Software, PA, USA), (where “v” = background corrected reaction rate, “a” is the rate in the absence of the inhibitor, [I] is the concentration of the inhibitor and “s” is the slope factor) to calculate IC50 values. Compounds exhibiting sigmoidal dose response curves, were selected as bona fide inhibitors.
Hex Activity Assays
Hex A/B/S activity was measured by release of 4-Methylumbelliferyl fluorophore from MUG. Total Assay volume was 50 μL, and contained 10 mM Citrate–Phosphate buffer, pH 4.3, 0.025% Human Serum Albumin (HSA). For enzyme activity monitored continuously, reactions were initiated with 75 μM MUG at room temperature and monitored for 7 min using 330 nm and 450 nm excitation and emission filters, respectively. For the endpoint assay, the reaction at 37°C was terminated by raising the pH to 10.5, above the pKa of 4-MU, using 0.1M 2-amino 2-methyl 1-propanol. The increase in fluorescence was measured using a Spectramax Gemini EM MAX (Molecular Devices Corp, Sunnyvale, CA) fluorometer and detected at excitation and emission wavelengths set to 365 nm and 450nm, respectively.
Activity of Jack Bean Hex (Vector Labs, USA) and Streptomyces plicatus Hex (New England Biolabs, USA) in presence of DMSO, NGT and the HTS compounds were evaluated identically to procedures used for human Hex. In the case of Serratia marcesens chitobiase (glucosaminidase) (kindly provided by C. Vorgias [22, 56]), activity was monitored at pH 7.5 using otherwise identical conditions. The activity of human O-GlcNAcase was performed using 0.5 mM, 4-Nitrophenyl N-acetyl- β-D-glucosaminide substrate and reaction conditions as described in Macauley et al [35].
For primary and secondary screening of the compounds, enzyme activity was monitored continuously on the Analyst HT (Molecular Devices Corp, Sunnyvale, CA) fluorometer as describe above. Steady-state kinetic parameters were established using a range of MUG substrate (1.6 mM – 0.003 mM) and enzyme concentrations (1 – 0.01 mg/ml). The inhibitory activity of compounds which were fluorescent (M-31850 and derivatives) or quenched (M-22971) near the emission maxima of MU were also confirmed using the colorimetric substrate pNP-GlcNAc and conditions described for the MUG endpoint assay, except that absorbance was measured at 405 nm.
Kinetic parameters (Km, Vmax and Ki) were determined by fitting the non-linear least square data to the Michaelis-Menten equation with Kaleidagraph (Synergy Software) according to Kakkar et al. [57].
Heat inactivation assay
Heat inactivation experiments were performed using Hex A from ATSD fibroblasts treated with NGT (1mM) for 5 days as previously described [8], with Hex B removed by ion exchange chromatography. For heat inactivation experiments equal amounts of total protein (0.1 – 0.5 μg) from enriched mutant Hex A fractions were diluted in CP buffer pH 4.2 containing 0.5% HSA. Diluted samples of the enzyme containing Hex inhibitors or DMSO, were split into two aliquots, one was left on ice, and the other heat-treated at 44°C. The heat-treated enzyme was cooled on ice until completion of the time series. For assaying enzyme activity, samples were pre-equilibrated to 37°C for 10min, followed by addition of MUGS substrate and incubated at 37°C for a further 30 min.
Evaluating Chaperoning Activity of Compounds in Cell Culture
ISD fibroblasts were seeded onto 96 well tissue culture plates (Costar) at 10–50,000 cells per plate (ca. 50% confluence). The following day, the medium was replaced with fresh α-MEM-FCS with or without a test compound to be evaluated (1/100 dilution). Test compounds were either dissolved in water (NGT), DMSO. Mock or compound-treated cells were evaluated in triplicate and grown for 5 days at 37°C in a CO2 humidified incubator.
To measure Hex S activity in treated ISD fibroblasts, media was removed, cells were washed twice with PBS and subsequently lysed by the addition of 10 mM Citrate-phosphate buffer pH 4.8 containing 0.1% HSA and 0.1% Triton X-100. One aliquot (25 μL) of the lysate was mixed with an equal volume of MUG and assayed for total Hex activity. To control for variability in cell numbers between replicate wells, the remaining aliquot of the lysate, was used to assay for lysosomal β-galactosidase, with the substrate MUbGal [8] using the endpoint assay described above.
Western Blotting
Lysates prepared from ATSD cells were subjected to SDS-PAGE on a 10% bis-acrylamide gel, and the separated proteins were transferred to nitrocellulose. A rabbit polyclonal Ab against human Hex A was used as previously described [58]. Blots were developed using chemiluminescent substrate according to the manufacturers protocol (Amersham, Bioscences, UK). Bands were visualized and optical density quantitated using a high sensitivity gel documentation system (Fluorchem 8000) consisting of a cooled CCD camera coupled with Alpha Innotech software (Alpha Innotech Corp., USA).
Purification of Iron-dextran-labeled Lysosomes
Lysosomal fractions were prepared from ATSD fibroblasts treated with DMSO or M-31850 (1 mM) for five days, followed by labelling with Iron-dextran colloid and subsequent purification by magnetic chromatography as previously described [8]. Lysosomal, acid phosphosphase and Hex A was monitored fluorometrically using the substrate MUP and MUGS, respectively, as described [8].
Mass Spectrometry
The mass of selected secondary hits was confirmed by the Advanced Proteomic Centre at Sickkids (Toronto, ON, CANADA) using a QToF mass spectrometer (Waters/Micromass, Manchester, UK)
Supplementary Material
Acknowledgments
We acknowledge the excellent technical assistance of J. Buttner, A. Leung, D. Benedict and M. Skomorowski. Thanks to R. Bagshaw, J. T. R. Clarke, J. Callahan, J. Cechetto, M. Rigathan, R. Pomes, M. Ciufolini and B. Rigat for helpful discussions and suggestions. Special thanks to M. Brana for providing Elinafide and derivatives thereof, C.E, Vorgias for generously providing a sample of SmCHB, G. Maegawa for image analysis and quantitation, K. Stubbs and D. Vocadlo for evaluation of inhibitory activity HTS compounds against O-GlcNAcase. This project was supported in part by grants from the Protein Engineering Network of Centres of Excellence of CANADA (PENCE) (E.B., S.G.W and D. M.), and a bequest from the Uger Estate (M.B.T. and D.M.) and an NIH Grant (5R21NS051214-02) (M.B.T. and D.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004;15:222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Fan JQ. A contradictory treatment for lysosomal storage disorders: inhibitors enhance mutant enzyme activity. Trends Pharmacol Sci. 2003;24:355–360. doi: 10.1016/S0165-6147(03)00158-5. [DOI] [PubMed] [Google Scholar]
- 3.Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic. 2004;5:821–837. doi: 10.1111/j.1600-0854.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A. Intracellular Protein Degradation: From a Vague Idea, through the Lysosome and the Ubiquitin-Proteasome System, and onto Human Diseases and Drug Targeting (Nobel Lecture) Angew Chem Int Ed Engl. 2005;44:5944–5967. doi: 10.1002/anie.200501428. [DOI] [PubMed] [Google Scholar]
- 5.Yam GH, Zuber C, Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. Faseb J. 2005;19:12–18. doi: 10.1096/fj.04-2375com. [DOI] [PubMed] [Google Scholar]
- 6.Friedler A, Hansson LO, Veprintsev DB, Freund SM, Rippin TM, Nikolova PV, Proctor MR, Rudiger S, Fersht AR. A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc Natl Acad Sci U S A. 2002;99:937–942. doi: 10.1073/pnas.241629998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernier V, Bichet DG, Bouvier M. Pharmacological chaperone action on G-protein-coupled receptors. Curr Opin Pharmacol. 2004;4:528–533. doi: 10.1016/j.coph.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D. Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff Patients. J Biol Chem. 2004;279:13478–13487. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med. 1999;5:112–115. doi: 10.1038/4801. [DOI] [PubMed] [Google Scholar]
- 10.Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher disease. Proc Natl Acad Sci U S A. 2002;99:15428–15433. doi: 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuda J, Suzuki O, Oshima A, Yamamoto Y, Noguchi A, Takimoto K, Itoh M, Matsuzaki Y, Yasuda Y, Ogawa S, Sakata Y, Nanba E, Higaki K, Ogawa Y, Tominaga L, Ohno K, Iwasaki H, Watanabe H, Brady RO, Suzuki Y. Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis. Proc Natl Acad Sci U S A. 2003;100:15912–15917. doi: 10.1073/pnas.2536657100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gravel RA, Clarke JTR, Kaback MM, Mahuran D, Sandhoff K, Suzuki K. The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 7. Vol. 2. New York: McGraw-Hill; 1995. pp. 2839–2879. [Google Scholar]
- 13.Mark BL, Mahuran DJ, Cherney MM, Zhao D, Knapp S, James MN. Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay-Sachs disease. J Mol Biol. 2003;327:1093–1109. doi: 10.1016/s0022-2836(03)00216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou Y, Tse R, Mahuran DJ. The Direct Determination of the Substrate Specificity of the α-Active site in Heterodimeric β-Hexosaminidase A. Biochemistry. 1996;35:3963–3969. doi: 10.1021/bi9524575. [DOI] [PubMed] [Google Scholar]
- 15.Conzelmann E, Sandhoff K. Biochemical basis of late-onset neurolipidoses. Dev Neurosci. 1991;13:197–204. doi: 10.1159/000112160. [DOI] [PubMed] [Google Scholar]
- 16.Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MN. Crystallographic Structure of Human beta-Hexosaminidase A: Interpretation of Tay-Sachs Mutations and Loss of G(M2) Ganglioside Hydrolysis. J Mol Biol. 2006;359:913–929. doi: 10.1016/j.jmb.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahuran DJ. The Biochemistry of HEXA and HEXB Gene Mutations Causing GM2 Gangliosidosis. Biochim Biophys Acta. 1991;1096:87–94. doi: 10.1016/0925-4439(91)90044-a. [DOI] [PubMed] [Google Scholar]
- 18.Knapp S, Vocadlo D, Gao Z, Kirk B, Lou J, Withers SG. NAG-thiazoline, an N-Acetyl-beta-hexosaminidase inhibitor that implicates acetamido participation. J Am Chem Soc. 1996;118:6804–6805. [Google Scholar]
- 19.Henrissat B, Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol. 1997;7:637–644. doi: 10.1016/s0959-440x(97)80072-3. [DOI] [PubMed] [Google Scholar]
- 20.Maier T, Strater N, Schuette CG, Klingenstein R, Sandhoff K, Saenger W. The X-ray crystal structure of human beta-hexosaminidase B provides new insights into Sandhoff disease. J Mol Biol. 2003;328:669–681. doi: 10.1016/s0022-2836(03)00311-5. [DOI] [PubMed] [Google Scholar]
- 21.Mark BL, Vocadlo DJ, Knapp S, Triggs-Raine BL, Withers SG, James MNG. Crystallographic evidence for substrate-assisted catalysis in a bacterial beta-hexosaminidase. J Biol Chem. 2001;276:10330–10337. doi: 10.1074/jbc.M011067200. [DOI] [PubMed] [Google Scholar]
- 22.Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilson KS, Vorgias CE. Baterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay-Sachs disease. Nature Struc Biol. 1996;3:638–648. doi: 10.1038/nsb0796-638. [DOI] [PubMed] [Google Scholar]
- 23.Sawkar AR, Adamski-Werner SL, Cheng WC, Wong CH, Beutler E, Zimmer KP, Kelly JW. Gaucher disease-associated glucocerebrosidases show mutation-dependent chemical chaperoning profiles. Chem Biol. 2005;12:1235–1244. doi: 10.1016/j.chembiol.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Asano N, Ishii S, Kizu H, Ikeda K, Yasuda K, Kato A, Martin OR, Fan JQ. In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur J Biochem. 2000;267:4179–4186. doi: 10.1046/j.1432-1327.2000.01457.x. [DOI] [PubMed] [Google Scholar]
- 25.Zolli-Juran M, Cechetto JD, Hartlen R, Daigle DM, Brown ED. High throughput screening identifies novel inhibitors of Escherichia coli dihydrofolate reductase that are competitive with dihydrofolate. Bioorg Med Chem Lett. 2003;13:2493–2496. doi: 10.1016/s0960-894x(03)00480-3. [DOI] [PubMed] [Google Scholar]
- 26.Blanchard JE, Elowe NH, Huitema C, Fortin PD, Cechetto JD, Eltis LD, Brown ED. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du K, Sharma M, Lukacs GL. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12:17–25. doi: 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- 28.Bayleran J, Hechtman P, Kolodny E, Kaback M. Tay-Sachs disease with hexosaminidase A: Characterization of the defective enzyme in two patients. Am J Hum Genet. 1987;41:532–548. [PMC free article] [PubMed] [Google Scholar]
- 29.Tecan T. Shifts in emission and excitation spectra due to pH changes. Application Note SAFIRE. 2001;5:1–7. [Google Scholar]
- 30.Shulman ML, Kulshin VA, Khorlin AY. A continuous fluorimetric assay for glycosidase activity: human N-acetyl-beta-D-hexosaminidase. Anal Biochem. 1980;101:342–348. doi: 10.1016/0003-2697(80)90198-0. [DOI] [PubMed] [Google Scholar]
- 31.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 32.Shoemaker RH, Scudiero DA, Melillo G, Currens MJ, Monks AP, Rabow AA, Covell DG, Sausville EA. Application of high-throughput, molecular-targeted screening to anticancer drug discovery. Curr Top Med Chem. 2002;2:229–246. doi: 10.2174/1568026023394317. [DOI] [PubMed] [Google Scholar]
- 33.Tsai SC, Burnakis TG. Aldose reductase inhibitors: an update. Ann Pharmacother. 1993;27:751–754. doi: 10.1177/106002809302700616. [DOI] [PubMed] [Google Scholar]
- 34.Hutcheon DE, Martinez JC. A decade of developments in diuretic drug therapy. J Clin Pharmacol. 1986;26:567–579. doi: 10.1002/j.1552-4604.1986.tb02951.x. [DOI] [PubMed] [Google Scholar]
- 35.Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. O-GlcNAcase uses substrate-assisted catalysis: kinetic analysis and development of highly selective mechanism-inspired inhibitors. J Biol Chem. 2005;280:25313–25322. doi: 10.1074/jbc.M413819200. [DOI] [PubMed] [Google Scholar]
- 36.Brana MF, Castellano JM, Moran M, Perez de Vega MJ, Romerdahl CR, Qian XD, Bousquet P, Emling F, Schlick E, Keilhauer G. Bis-naphthalimides: a new class of antitumor agents. Anticancer Drug Des. 1993;8:257–268. [PubMed] [Google Scholar]
- 37.Robben JH, Sze M, Knoers NV, Deen PM. Rescue of vasopressin V2 receptor mutants by chemical chaperones: specificity and mechanism. Mol Biol Cell. 2006;17:379–386. doi: 10.1091/mbc.E05-06-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xing L, Remick DG. Mechanisms of dimethyl sulfoxideaugmentation of IL-1 beta production. J Immunol. 2005;174:6195–6202. doi: 10.4049/jimmunol.174.10.6195. [DOI] [PubMed] [Google Scholar]
- 39.Hasilik A, Neufeld EF. Biosynthesis of lysosomal enzymes in fibroblasts: synthesis as precursors of higher molecular weight. J Biol Chem. 1980;255:4937–4945. [PubMed] [Google Scholar]
- 40.Hasilik A, Neufeld EF. Biosynthesis of lysosomal enzymes in fibroblasts: phosphorylation of mannose residues. J Biol Chem. 1980;255:4946–4960. [PubMed] [Google Scholar]
- 41.d’Azzo A, Proia RL, Kolodny EH, Kaback MM, Neufeld EF. Faulty association of α- and β-subunits in some forms of β-hexosaminidase A deficiency. J Biol Chem. 1984;259:11070–11074. [PubMed] [Google Scholar]
- 42.Diettrich O, Mills K, Johnson AW, Hasilik A, Winchester BG. Application of magnetic chromatography to the isolation of lysosomes from fibroblasts of patients with lysosomal storage disorders. FEBS Lett. 1998;441:369–372. doi: 10.1016/s0014-5793(98)01578-6. [DOI] [PubMed] [Google Scholar]
- 43.Callahan JW, Pinsky L, Wolfe LS. G M1 -gangliosidosis (Type II): studies on a fibroblast cell strain. Biochem Med. 1970;4:295–316. doi: 10.1016/0006-2944(70)90056-6. [DOI] [PubMed] [Google Scholar]
- 44.Rye CS, Withers SG. Glycosidase mechanisms. Curr Opin Chem Biol. 2000;4:573–580. doi: 10.1016/s1367-5931(00)00135-6. [DOI] [PubMed] [Google Scholar]
- 45.Knapp S, Vocadlo D, Gao ZN, Kirk B, Lou JP, Withers SG. NAG-thiazoline, an N-acetyl-beta-hexosaminidase inhibitor that implicates acetamido participation. J Am Chem Soc. 1996;118:6804–6805. [Google Scholar]
- 46.Liu J, Numa MM, Liu H, Huang SJ, Sears P, Shikhman AR, Wong CH. Synthesis and high-throughput screening of N-acetyl-beta-hexosaminidase inhibitor libraries targeting osteoarthritis. J Org Chem. 2004;69:6273–6283. doi: 10.1021/jo049355h. [DOI] [PubMed] [Google Scholar]
- 47.Brana MF, Ramos A. Naphthalimides as anti-cancer agents: synthesis and biological activity. Curr Med Chem Anti-Canc Agents. 2001;1:237–255. doi: 10.2174/1568011013354624. [DOI] [PubMed] [Google Scholar]
- 48.Liang PH, Cheng WC, Lee YL, Yu HP, Wu YT, Lin YL, Wong CH. Novel five-membered iminocyclitol derivatives as selective and potent glycosidase inhibitors: new structures for antivirals and osteoarthritis. Chembiochem. 2006;7:165–173. doi: 10.1002/cbic.200500321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Awada A, Thodtmann R, Piccart MJ, Wanders J, Schrijvers AH, Von Broen IM, Hanauske AR. An EORTC-ECSG phase I study of LU 79553 administered every 21 or 42 days in patients with solid tumours. Eur J Cancer. 2003;39:742–747. doi: 10.1016/s0959-8049(02)00776-1. [DOI] [PubMed] [Google Scholar]
- 50.Villalona-Calero MA, Eder JP, Toppmeyer DL, Allen LF, Fram R, Velagapudi R, Myers M, Amato A, Kagen-Hallet K, Razvillas B, Kufe DW, Von Hoff DD, Rowinsky EK. Phase I and pharmacokinetic study of LU79553, a DNA intercalating bisnaphthalimide, in patients with solid malignancies. J Clin Oncol. 2001;19:857–869. doi: 10.1200/JCO.2001.19.3.857. [DOI] [PubMed] [Google Scholar]
- 51.Wermuth CG. Selective optimization of side activities: another way for drug discovery. J Med Chem. 2004;47:1303–1314. doi: 10.1021/jm030480f. [DOI] [PubMed] [Google Scholar]
- 52.Brown CA, Mahuran DJ. β-hexosaminidase isozymes from cells co-transfected with α and β cDNA constructs: Analysis of α subunit missense mutation associated with the adult form of Tay-Sachs disease. Am J Hum Genet. 1993;53:497–508. [PMC free article] [PubMed] [Google Scholar]
- 53.Kappes E, Legler G. Synthesis and inhibitory properties of 2-acetamido-2-deoxynojirimycin (2-acetamido-5-amino-2,5-dideoxy-D-glucopyranose, 1) and 2-acetamido-1,2-dideoxynojirimycin ( 2-acetamido-1,5-imino-1,2,5-trideoxy-D-glucitol, 2) J Carbohydrate Chemistry. 1989;8:371–388. [Google Scholar]
- 54.O’Dowd B, Klavins M, Willard H, Lowden JA, Gravel RA, Mahuran D. Molecular Heterogeneity in O-Variant GM2 Gangliosidosis. In: Freysz L, Dreyfus H, Massarelli R, Gatt S, editors. Enzymes of Lipid Metabolism II. Plenum Publishing Co; 1986. pp. 779–784. [Google Scholar]
- 55.Neote K, Brown CA, Mahuran DJ, Gravel RA. Translation initiation in the HEXB gene encoding the β-subunit of human β-hexosaminidase. J Biol Chem. 1990;265:20799–20806. [PubMed] [Google Scholar]
- 56.Tews I, Vincentelli R, Vorgias CE. N-acetylglucosaminidase (chitobiase) from Serratia marcescens: Gene sequence, and protein production and purification in Escherichia coli. Gene. 1996;170:63–67. doi: 10.1016/0378-1119(95)00848-9. [DOI] [PubMed] [Google Scholar]
- 57.Kakkar T, Pak Y, Mayersohn M. Evaluation of a minimal experimental design for determination of enzyme kinetic parameters and inhibition mechanism. J Pharmacol Exp Ther. 2000;293:861–869. [PubMed] [Google Scholar]
- 58.Hou Y, McInnes B, Hinek A, Karpati G, Mahuran D. A Pro504Ser substitution in the β-subunit of β-hexosaminidase A inhibits α-subunit hydrolysis of GM2 ganglioside, resulting in chronic Sandhoff disease. J Biol Chem. 1998;273:21386–21392. doi: 10.1074/jbc.273.33.21386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.