

Abstract
A series of ab initio (density functional) calculations were carried out on side chains of a set of amino acids, plus water, from the (intracellular) gating region of the KcsA K+ channel. Their atomic coordinates, except hydrogen, are known from X-ray structures[1–3], as are the coordinates of some water oxygen atoms. The 1k4c structure is used for the starting coordinates. Quantum mechanical optimization, in spite of the starting configuration, places the atoms in positions much closer to the 1j95, more tightly closed, configuration. This state shows four water molecules forming a “basket” under the Q119 side chains, blocking the channel. When a hydrated K+ approaches this “basket”, the optimized system shows a strong set of hydrogen bonds with the K+ at defined positions, preventing further approach of the K+ to the basket. This optimized structure with hydrated K+ added shows an ice-like 12 molecule nanocrystal of water. If the water molecules exchange, unless they do it as a group, the channel will remain blocked. The “basket” itself appears to be very stable, although it is possible that the K+ with its hydrating water molecules may be more mobile, capable of withdrawing from the gate. It is also not surprising that water essentially freezes, or forms a kind of glue, in a nanometer space; this agrees with experimental results on a rather different, but similarly sized (nm dimensions) system[4] It also agrees qualitatively with simulations on channels[5, 6] and on featureless channel-like systems[7], in that it forms a boundary on water that is not obvious from the liquid state. The idea that a atructure is stable, even if individual molecules exchange, is well known, for example from the hydration shell of ions. We show that when charges are added in the form of protons to the domains (one proton per domain), the optimized structure is open. No stable water hydrogen bonds hold it together; an opening of 11.0 Å appears, measured diagonally between non-neighboring domains as glutamine 119 carbonyl O – O distance. This is comparable to the opening in the MthK potassium channel structure that is generally agreed to be open. The appearance of the opening is in rather good agreement with that found by Perozo and coworkers. In contrast, in the uncharged structure this diagonal distance is 6.5Å, and the water “basket” constricts the uncharged opening still further, with the ice-like structure that couples the K+ ion to the gating region freezing the entrance to the channel. Comparison with our earlier model for voltage gated channels suggests that a similar mechanism may apply in those channels.
Keywords: KcsA K+ channel, gating, protons, ab initio calculations
INTRODUCTION
Ion channels make it possible for ions to enter and leave cells, by allowing the ions through the hydrophobic cell membrane. The gating (opening) mechanism of voltage-gated ion channels has been a long-standing problem. Determination of X-ray structures has made it possible to define the problem more precisely, but has not solved it. Prior to publication of the structure of the voltage-gated channel Kv1.2[8], the proton-gated channel KcsA [1–3] showed how the pore region of a potassium channel is structured. However, the mechanism by which a proton gates this channel is not transparent from the static structures of closed or open states, and is still controversial. Recent proposals for voltage gated channels by Bezanilla and Roux and coworkers[9, 10], and by Mackinnon and coworkers[11], are not compatible with each other. We here present evidence for a gating mechanism for the proton-gated channel KcsA, which, however, is expected to have critical characteristics in common with the voltage-gated channels; the motion of the gating segment is likely to be generally similar to those channels. This in turn suggests a third alternative for gating mechanisms of voltage gated channels, in which we propose that a proton-operated gate may resemble the final step of the mechanism of gating of a voltage-gated channel. Recent simulations by Shrivastava and Bahar[12] suggest a common conformational change on opening; although our work does not consider the entire channel, it suggests how a local change can lead to a more global change, as in their simulation. Our mechanism could be applied to voltage-gated channels as well, and would not be compatible with a mechanism dependent on a strictly mechanical linkage between the voltage sensor and the gate, as are the two mechanisms cited above.
The voltage sensing domain (VSD) of a standard voltage-gated potassium channel has been placed on the KcsA channel, causing it to behave as does a voltage-gated channel; Lu and coworkers [13] observed that certain conserved residues are required for coupling the VSD to the KcsA channel, particularly in the S4-S5 linker, between the S4 transmembrane (TM) segment that is part of the VSD, and the S5 TM segment that is part of the pore, and includes a part of the gate,. They interpreted this in terms of a mechanical linkage. However, the result could be understood instead as meaning that the VSD acts as a voltage to proton current transducer, with linkage through a set of hydrogen bonds. In addition, it is now known that the voltage-gated proton channel, hitherto considered unrelated, is essentially the voltage-gated channel’s VSD[14, 15]; it is not known whether the protons in that channel actually move along the S4, although that is clearly a possibility. It is known that single mutations in S4 disrupt the proton current, or, in a voltage gated channel, a single R to H mutation permits proton current. Therefore, it is very likely that the a proton current partially across the S4 is possible, and actually happens. If so, it would suggest that protons may also move along S4 to produce the gating current in voltage gated channels, which should mean that change in state of protonation is responsible for voltage gating far more generally than in the KcsA channel; the latter opens when the pH drops to approximately 4, showing that the channel is gated by addition of a proton (there may also be a form of gating at the selectivity filter, but that does not concern this discussion). Evidence concerning the nature of the linkage of S4 to the gating mechanism in voltage gated channels must be sought independently. We have argued that the final step in potassium (and, presumably, sodium) channel gating is related to the disposition of protons, and to the effect on the structure of the water hydrogen bonded to the gating region, a proposal we have previously made [16–18]. The example we present here shows how this could happen in a simpler channel in which the gating is known to be pH dependent, and thus almost certainly proton dependent. We must therefore determine the ways in which addition of proton(s) to the gating region of the KcsA channel could alter the hydrogen bonding network of water there. This should tell us whether protons weaken the hydrogen bonds that provide the force holding the domains together, allowing domain separation and thus channel opening. It may be that a similar effect exists if protons move along the VSD (presumably along S4) in a voltage gated channel, making charge alterations in the gating region of that channel analogous to those in the KcsA channel on change of pH. The protons that we believe gate a voltage-gated channel could move in either direction, extracellularly in the depolarization-gated channels, intracellularly in HCN (hyperpolarization-activated cyclic nucleotide gated) or other hyperpolarization activated (or partially activated, allowing for the ligand) channels. The point is to understand what happens to a channel when its charge changes; the change may in principle be of either sign; in KcsA, positive charges open the channel. The KcsA channel, where the structure is clearer than the Kv1.2 channel at the relevant residues, has several hydrogen bonding residues there: the most important are a glutamate, a glutamine, and an arginine, as these point toward each other. Other nearby residues include one glutamate and two arginines, but the latter point away from the center of the pore; perhaps they are salt bridged, although this is not obvious. The glutamine is not charged, although it contributes key hydrogen bonds; there are two sets of coordinates, 1k4c and 1j95, that differ. The 1k4c structure may even be partially open, although that is not likely; the 1j95 structure is closed. We compare the size of the opening for a K+ ion for the KcsA channel open structure to our results; they agree with this assignment within reasonable limits. Our model, described below, places a proton on the side chain of R122 at the intracellular end of the channel, where our calculation suggests gating occurs. This is a reasonable location to look at, as there are a number of cases known where protons move onto proteins along paths determined by arginines and carboxylate containing amino acids; we do have neighboring glutamates here. These include cytochrome C oxidase (cyt C)[19] and bacteriorhodopsin (bR)[20]. Other evidence comes from enzymes in which arginine is involved in enzymes in which proton abstraction is required[20]. It is not surprising to find that arginine is a key residue in the gating region of an enzyme in which protons are known to be responsible for the gating. The other possible key residue (not included in our model at this point, although a more detailed version could include this residue’s contribution; see discussion below) is a histidine, H124. This too has a pK in the appropriate range, and histidines have been suggested as part of a proton wire[21]. A number of biological systems depend on proton transport; several proteins have known paths along which to move protons, in addition to the examples given earlier: cytochrome C oxidase (cyt C) and bacteriorhodopsin (bR), and the proton channel (Hv) [15, 22] that is essentially the VSD of certain K+ and Na+ voltage gated ion channels with a salt bridge deleted.
Arginine is also involved in enzymes in which proton abstraction is required[23], when it is solvent accessible and adjacent to carboxylate groups, in spite of its pK of ≈ 12.5 in solution. Both of these conditions are met in the voltage sensing domain of ion channels, and therefore presumably in Hv; the key mutation in Hv is R133I, and secondarily D62S, removing a salt bridge, thus breaking the water column in the VSD[24]. Several enzymes have been reviewed by Brandsburg-Zabary et al[25], from the point of view of proton transport internal to the protein. These authors note that the diffusion constant of the proton in the protein is about half that in bulk water (or in a phospholipid bilayer, although that is less relevant to this work). The proton moves more slowly as the water is restricted,
All these results suggest that we can reasonably expect that a proton could reach the arginine that we are concerned with. Once it does the consequences are those that we calculate.
In the 1k4c structure side chains of three residues either point to the center of the channel (Q119), or form a salt bridge that appears to be crucial to the structure of that section of the channel (R122 of one domain with E120 of the neighboring domain). Because the channel structure grows wider above this region, and then narrows again, there seems no reason to have such a tightly fitted structure unless it has a significant role in holding the domains together in the closed channel. In other words, it is a good candidate for the main gating region; we must show that adding protons will cause it to open, and that it will hold together when it is not protonated. Cortes et al[26] suggested that the cluster of charged residues in this region was a prime candidate for the pH sensor; we have made this suggestion specific, and demonstrate how such a sensor might work. Data from Liu et al[27] show a pivot at residues 107–108 for an opening below (intracellular to) this level. The domains are necessarily held together below that level, and the most obvious location must be where the domains join. Therefore, this is the region we calculate.
These residues can form chains of hydrogen bonds with the water that must be present to fill the space available in the X-ray structure, although the water molecules in the X-ray structure tend to have positions different than those we find, with the water in the calculation often 3Å or so from the closest water in the 1k4c X-ray structure. However, we find the closed state is closer to 1j95, so this is not surprising; the 1j95 structure does not have relevant water positions. Nevertheless, the key “basket” water appears to be very robust. The energies calculated can be used to compare to kBT (kB=Boltzmann’s constant, T=temperature (K)) at room temperature, and distortions of the structure turn out to be very expensive energetically. We present here calculations on parts of the gating section of the channel (as defined above) that suggest the types of changes that must occur when protons are added to the channel. This helps make possible understanding the ways water may block a channel, and ways for the channel to open, releasing it from being held by hydrogen bonds. There have been numerous molecular dynamics simulations of the KcsA channel, of varying types [28–34]. The selectivity filter can now be reasonably well understood, but the gating region is more difficult; we believe this is related to subtleties of hydrogen bonding; in addition, the selectivity filter is more rigid. These hydrogen bonds may be difficult to reproduce by standard water potentials used in MD simulations[35, 36]. However, the qualitative behavior may be essentially correct, and the recent work by Shrivastava and Bahar[12] suggests, as previously noted, that the conformational change accompanying gating of all K+ channels may be similar; even if the details of that calculation are in some need of modification, the general conclusion is reasonable.
There have been a much smaller number of ab initio studies of the channel. The selectivity filter was investigated by Compoint et al [37], giving the charges on the selectivity filter. There has also been a density functional study of the selectivity filter [38]. A study of three sections of the channel, all in or near the selectivity filter, by Bliznyuk and Rendell, showed the effect of polarization in the calculation, even at a distance [39]. So far, it does not appear that the gating region has been treated by ab initio, or density functional, methods, nor with enough water to see its effect. In this work, we test the strength of the water intermolecular interactions with the key amino acids, as well as with other water molecules.
The calculations must show whether the channel is directly blocked by the water. A second alternative might show that the channel is blocked with protein held together by chains of hydrogen bonds that include water, and a third alternative would be that water does not play a major role. The importance of the intracellular amino acids studied here has been demonstrated by Perozo and coworkers [40, 41]; those results are also in reasonably good agreement with our open state structure. The neighboring amino acids may be indirectly important, contributing to the charge or the electric field, and thus affecting the pKa of the protonated group; however, they are distant from the key residues, and we are not here considering the pKa values. Another reason for looking at Q119, in particular, is the fact that the four domains come together closely enough for the side chains of this amino acid to be hydrogen bonded to each other. One more interdomain linkage involves E120 and R122 of neighboring domains; these two are at positions that allow the formation of an interdomain salt bridge. Taken together, these are the most probable amino acids to be critical for gating. The fact that gating occurs with lowered pH implies that addition of protons is a key step in gating. We need a relatively high-level computation of the side chains of those amino acids directly affected by this addition of protons. The channel’s “bundle crossing”, which is about 10 amino acids above the gate may play a role in the rotation that opens the channel, but does not itself appear to be the part that holds the gate closed. Instead, it is the pivot upon which the lower section of the channel rotates[42]. The gate itself has the ability to change bonding and thus actually open the channel. Mutations in the bundle crossing still affect gating, as they alter the pivot of the opening transition.
METHODS
Most of the calculations were carried out using the supercomputer facility at the Pacific Northwest National Laboratory, with NWChem software [43]. The work presented here followed the completion of preliminary studies, consisting principally of optimizing the hydrogens for the “monomer”, that is, one repeat of the side chains of three amino acids, Q119 and R122 amino acids from domains A and D, plus the E120 from domains D and C. The E120 of domain D and the neighboring domain A R122 are bridged. The side chains in the calculation began with the carbon two positions up the side chain from the end, converted to a methyl group (for glutamate, CA (alpha), for glutamine CB, for arginine CG), which is frozen. By freezing these atoms in place, at least most of the net forces exerted by the remainder of the protein on the backbone are included, although any effects of vibrations on side chains are omitted.
The single domain (“monomer”) from the 1k4c X-ray structure, plus added hydrogens, with the symmetry operations that are required to make the complete X-ray structure from the monomer, to construct a starting configuration for the final optimization. This provided the charged (open) and uncharged (closed) starting positions, which were identical save for the extra proton per domain in the open state. Four water molecule oxygen positions were also taken from the X-ray structure, with H added as for the amino acids. While the key closed calculation has all four domains, the open calculation was first done on a two domain construct; the complete four domain system is then constructed by symmetry. However (see below) the final calculation was done on the four domain structure. The extra proton representing the open state charge was added to the R122 guanidinium at the salt bridge, one per salt bridge, and then allowed to optimize. The initial position of the proton does not represent the actual structure; the optimized position does, and is the key to the structure. Placing the proton initially at R122 does not mean that we expect it to actually be there when an energy minimum is found, only that it would not surprise us to find it associated with the opening of the salt bridge. Thus it makes sense to start in the neighborhood of the salt bridge.
We noted in the introduction that it was reasonable to expect protons to move along a path containing arginine and a carboxylic acid (here, glutamate). The arginine in question appears to be a reasonable location for the proton to stop, thus destroying the salt bridge. The consequences of this choice are the subject of this work; the result shows that this leads to a reasonable gating mechanism, in which the closed state is bound by a combination of salt bridge and hydrogen bonding, and that this fails (i.e., the channel opens) when a proton destroys the salt bridge. It is conceivable that an additional proton could be added in a second location (involving H124), thus breaking additional hydrogen bonds not shown in the present calculation (see the discussion of a possible second basket of water, below); however, this does not alter the fundamental idea of this mechanism. The details may require modification, but not the mechanism.
The final position of the proton was determined by the calculation; there is no assumption that the guanidinium is uncharged in the closed state. Instead we have the salt bridge in the closed state, and the proton causes a redistribution of charge that destroys the salt bridge in the open state. Starting from the 1j95 coordinates was impossible, as R122 was missing.
Open state details
A two-domain density functional calculation using B3LYP/6-311++G** converged for the open (charged) state, starting from coordinates prepared as described above. (The B3LYP method was compared with the MP2 method by Kar and Scheiner[44], with the 6-31+G* basis set, to investigate water chains, as well as C-H…O hydrogen bonds, and found to be little different; the two methods produced similar trends in water chains. They found B3LYP to be satisfactory, and we use it here.) The system was optimized to determine the position of the side chains and the water molecules, save for the frozen end methyl groups. Eighteen water molecules were included from their oxygens in the X-ray structure for the dimer (hence, 36 for the tetramer created from the dimer), from which positions they were also optimized. The space is shown to be large enough to hold enough molecules that they would behave like bulk water. In this calculation, the two missing domains were added by symmetry. However, in this system, two out of four anchor points for the “basket” were absent. Providing a fair test of the possibility that structured water of some sort was going to form in any case required repeating the calculation with all four domains present; it was, at HF/6-31G* level. The water looked like bulk water, with the channel opening to almost the same extent as in the previous model. We conclude that the structured water can only form when the protons have not been added, hence the channel is closed, and the structure depends on the ability to hydrogen bond to the protein. When the protons are added so that the salt bridges break and the domains separate, the water cannot rescue the closed structure.
To check the adequacy of the assumptions, single point calculations at B3LYP/6-311++G** level were done around the minimum for one angle of rotation of the entire Q119 group, together with associated water molecules (single point calculations, unlike optimizations, are feasible at this level). The energy (units of Hartrees, H) of the tetrameric open state, with the structure of two domains determined by symmetry operations, was −6116.8016 H; with the Q119 plus associated water rotated +/− 5° in the plane of the amide carbons, the energy was (+5°) −6116.7922 H, (−5°) −6116.7950 H. Clearly this is a moderately steep minimum with respect to this rotation, approximately 6 to 9 kBT. With only the end atoms rotated 5o inwards, the energy was −6116.8011, a small change, and in the appropriate direction. However, a better minimum was found by taking one of the two domains of the optimized dimer and imposing C4 symmetry to create a tetrameric structure, single point energy −6016.8118. Therefore, the two minima are fairly close. This would be more accurate than the four domain calculation, as the higher level and basis set are needed for an accurate energy calculation; the geometry was so similar that we could use any of the geometries at its own minimum, to get the steepness of the minimum. Another check consisted of taking the optimization of the dimer discussed above, and comparing certain distances across opposite domains. If asymmetry were introduced by the optimization, these would differ (note: one of the diagonals is the same as what one gets by taking one domain, and applying C4 symmetry to create the tetramer). Comparing distances across opposite domains, we get the results in Table I:
The agreement between Column 2 and Column 3 is clearly excellent; the optimization of the dimer produces a symmetric tetramer. The four domain structure at HF/6-31G* level produced results within 0.1Å of the structures in Table I, with the key O – O distance (last row) 11.0Å.
Closed state details
No protons were added, so the charge was less by one charge per domain, but the same methyl groups were frozen. Because it was not possible to optimize the twelve amino acids plus associated waters of the full structure at the B3LYP/6-311++G** level, the following approaches were used for this part of the problem: 1) A system of four Q119 (one per domain) plus eight water molecules (starting positions again from the 1k4c structure for the oxygens) was optimized; unlike all else reported here, this optimization used Gaussian [45], was done on a Linux cluster, and used B3LYP/6-311+G**. 2) An HF/6-31G** (HF=Hartree-Fock) optimization (with NWChem) of the full 12 amino acid (four-domain) system with 36 molecules of water was possible; it confirmed the geometry, and showed the final positions of the side chains and the water. For the total system, there were 276 atoms, 2400 basis functions (without hydrated K+). (The same “basket” of water formed in the Gaussian optimization with only Q119 as in the NWChem optimization with the additional two amino acids, and a different method. This was tested further by displacing the “basket” of water vertically (i.e., along the central symmetry axis), and doing B3LYP/6-311++G** single point calculations to insure that the HF optimization corresponded also to the high level energy minimum. There was a very slight decrease in energy with 0.1Å displacement of the “basket” vertically toward the amino acids, but otherwise the minimum held (Energy = −6115.2800 H at the HF minimum, compared to −6115.2803 H with a 0.1Å displacement, for a difference of 0.0003 H, or about 1/3 kBT (0.001H ≈ kBT) — a further vertical 0.1Å displacement gave energy appreciably above the HF minimum, as did 0.1Å in the opposite direction). The “basket” was also rotated +/− 5°; the energy (H) was: −5°: −6115.2780; 0°: −6115.2795; +5°: −6115.2788. The minimum therefore comes at the same orientation as in the HF calculation. The 0° and HF minimum values as redone using B3LYP/6-311++G** differ by almost 0.0005H because the procedure used to do the rotation involved a coordinate transformation that allowed slight round-off error. The accuracy of the HF calculation, relative to a high level density functional calculation, is thus approximately 0.1 Å in distance and less than 5° in angle. This is of the order of a vibration, and thus adequate for our purposes. The absolute values of the energy cannot be compared to those of the open state, as that state has four additional protons. The fact that the tightly closed state was reached from the less closed 1k4c starting configuration demonstrates that sufficient length of side chains was included to show the change from 1k4c to 1j95 structures. The fact that the final atomic positions reproduced the X-ray structure reasonably closely suggests that the calculation is valid, and the omission of other amino acids did not distort the structure.
Finally, we did the calculation with an added hydrated K+. This tests the possibility that K+ would destroy the “basket”. It does not. In the four-domain state as optimized first at HF/6-31G* level, with potassium hydrated by eight waters, four waters again form a “basket”, in which the hydrogen bonds to a pair of neighbors stabilize the position. The hydrated potassium remains below the “basket”, without any significant change in its structure. The next optimization was done at B3LYP/6-31G** level at the EMSL facility supercomputer (technical reasons—the absence of a K+ basis set with diffuse functions in NWChem—prevented the use of diffuse basis functions, as in 6-31+G**). This was done with the same starting structure as before, this time with 32 water molecules total in the calculation; the system had a total of 2323 basis functions. Here we find the “basket” holds, together with additional water including part of the hydration shell of the K+ ion. The structure that forms looks like a nanocrystal of ice, blocking the path of the ion towards the channel. The 6-31G** basis set is large, although not as large as the 6-311++G** that might have been preferable, but the optimized geometry is still reliable—we have already seen that the structure does not seem to greatly depend on the calculation method. B3LYP includes at least some of the correlation energy of the electrons. Overall this is a rather accurate calculation, and reinforces the geometric result found earlier at HF/6-31G** level (i.e., with no correlation energy). It is also much more reliable than the MM potentials used in molecular dynamics simulations. Energy differences with position can also be considered reasonably reliable, although absolute energy values would be lower with a larger basis set. Single point calculations were done on the final structure to test the depth of the minimum.
The one modification of the model that appears to still be possible comes through the inclusion of H124 in the model; this residue may form a secondary basket of water, adding to the basket in the first calculation. Further calculations to study this question are difficult because of the size of the total system, but very preliminary indications, based in part on modeling, and on very approximate calculations, suggest that such a basket could exist. It appears that it would be a little too distant to hydrogen bond to the original basket as well (which remains, whether the additional basket exists or not). However, the key point of the model is the existence of a basket (a set of four hydrogen bonded molecules) of water that blocks the entrance to the channel, and is hydrogen bonded to the protein. If the histidines are involved at all, it reinforces the model, and adds detail, but does not contradict the calculation done so far. If the second basket does exist, the position at which the K+ ion would presumably be held is below the H124, but the “nanocrystal” would be expected to again form at that position, as the second anchoring basket would have the same structure as the first.. However, this remains to be confirmed by actual calculation, as does the existence of the second basket.
Charges
The charges shown (Table III) are fitted using Ahlrich’s Auxiliary basis sets protocol [46] in NWChem, which is considered much more accurate than other methods when doing DFT calculations. They are clearly better than the Mulliken charges, in particular. Charges are calculated with 6-31G** plus the Ahlrich’s Auxiliary Basis Set. Graphics: These are prepared using the program GOpenMol [47, 48]; this program shows hydrogen bonding, with bonds defined by these default criteria: donor-acceptor length < 3.9 Å, hydrogen acceptor < 2.5 Å, donor-hydrogen acceptor > 90°, hydrogen-acceptor-atom bonded to acceptor > 90°, donor-acceptor-atom bonded to acceptor > 90°. Therefore some weak bonds could be included. The distances for the critical bonds are shown in the figure and the tables, so that it is clear that the actual bonds in these figures are not very weak.
TABLE III.
Charges on important atoms in the calculation, with designations as given in text.
| ATOM | Charge (closed) | Scatter (RMS) | Charge (open) | Scatter (RMS) | Location |
|---|---|---|---|---|---|
| K | 0.70 | --- | --- | -- | Potassium |
| OA | −0.81 | 0.02 | -- | -- | Basket O |
| HAE | 0.38 | 0.01 | --- | -- | Basket H (E) |
| HAA | 0.40 | 0.02 | --- | --- | Basket H (A) |
| OB | −0.74 | 0.02 | --- | --- | Intermediate O |
| HBE | 0.38 | 0.01 | --- | --- | Intermediate H(E) |
| HBA | 0.41 | 0.01 | --- | --- | Intermediate H(A) |
| OC | −0.77 | 0.01 | --- | --- | K+ Hydration shell |
| HBE | 0.37 | 0.01 | --- | --- | K+ Hydration shell |
| HCA | 0.42 | 0.01 | --- | --- | K+ Hydration shell |
| OW | --- | --- | −0.86 | 0.01 | Water, not structured |
| HW1 | ---- | ---- | 0.47 | 0.04 | Water, not structured |
| HW2 | ---- | --- | 0.37 | 0.03 | Water, not structured |
| HCE | --- | --- | 0.45 | 0.45 | Hydrogen in O –HO hydrogen bond E120 carboxyls to water |
| O | −0.66 | 0.01 | −0.62 | 0.03 | Q119 |
| N | −0.91 | 0.01 | −0.78 | 0.04 | Q119 |
| HNB | 0.44 | 0.01 | 0.39 | 0.02 | Q119 (H-bonded) |
| HNF | 0.40 | 0.00 | 0.36 | 0.02 | Q119(no H-bond) |
| OSB | −0.78 | 0.01 | −.0.63 | 0.02 | E120 carboxyl (H-bond) |
| OSF | −0.61 | 0.00 | −0.59 | 0.01 | E120 carboxyl(no H-bond) |
| HS1 | 0.45 | 0.01 | 0.43 | 0.01 | R122, guanidinium, H-bond |
| HS2 | 0.46 | 0.00 | 0.41 | 0.02 | R122, guandinium, H-bond |
| NB | −0.93 | 0.02 | −0.91 | 0.01 | R122, guanidinium, H-bond |
| NF | −0.88 | 0.02 | −0.92 | 0.02 | R122, guanidinium, no H-bond |
| H1 | 0.45 | 0.01 | 0.43 | 0.01 | R122, guanidinum, no H-bond |
| H2 | 0.38 | 0.00 | 0.49 | 0.03 | R122, guanidinium, no H-bond |
RESULTS AND DISCUSSION
The structures of the optimized gating region, open and closed, are shown in Fig. 1. Fig. 1A has the charged, open, configuration. The distance from oxygen to oxygen of the glutamine carbonyl group across the center is 11.0 Å, compared to 8.2 Å for the 1k4c structure, the latter probably still too small for an open channel. The 11.0Å agrees very well, as noted above, with the 10.96 Å and 11.14 Å values given in Table I, for the calculations that used two domains plus symmetry. It appears that the open state structure is robust, and the geometry not dependent on the details of the calculation. For the KcsA channel, the appropriate dimensions of the open state are given by Perozo and coworkers[27]. It is interesting to note that this corresponds approximately to the channel opening of a voltage gated channel. For comparison, Long et al[8] have given the opening of the Kv1.2 channel as about 12 Å. The 1k4c opening is about 3 Å smaller than the calculated value, and about 4 Å smaller than the Kv1.2 structure. In the calculation, the water molecules have the appearance of unstructured bulk water. The conditions are not such as to hold the channel in a tightly fixed position, nor such as to block hydrated K+ from approaching.
Fig. 1.
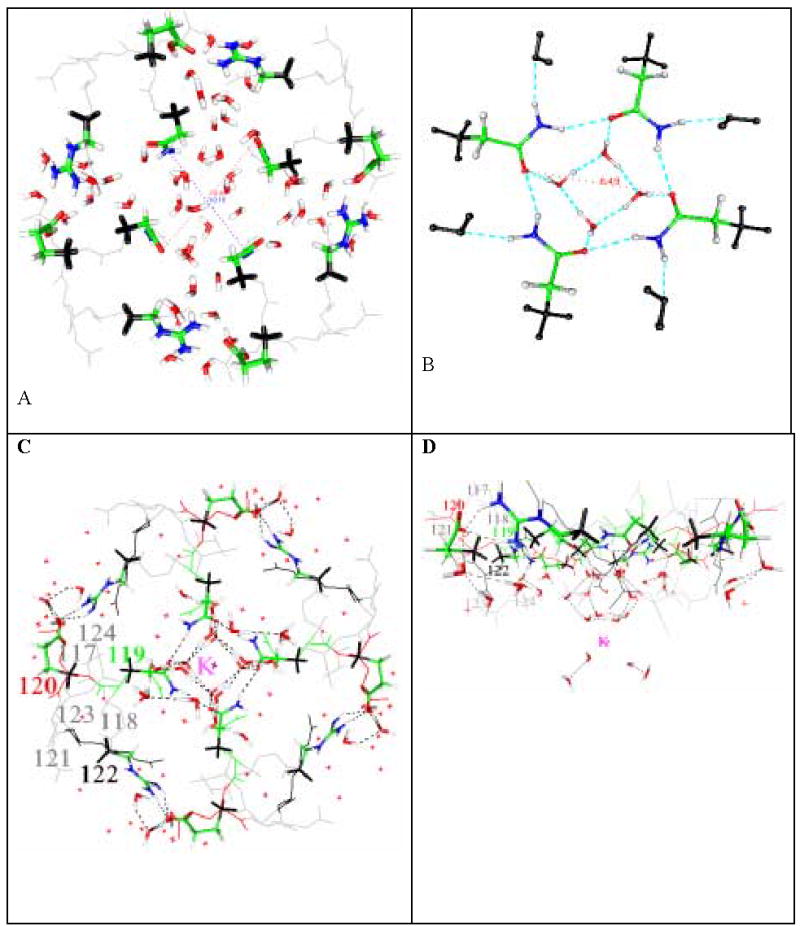
A) Open (protonated) state of the KcsA channel, showing results of calculations on three amino acids from all four domains, plus water, optimized at HF/6-31G** level. Outer methyl groups are frozen. The distance between the two oxygen atoms of Q119 is 10.9 Å on one diagonal, 10.2 on the other. Color code: see end of caption. Outer methyl groups are frozen. The central region of the set of amino acids opens, and the distance between two oxygen atoms of Q119 is 11.0 Å: B) The closed state, Q119 only, plus the water: this calculation is done at B3LYP/6-311++G** level. Instead of 11Å, the distance across is now only approximately 6Å, not enough for a hydrated K+ ion, even if the water molecules could exchange instead of blocking the channel. C,D) the uncharged (closed) state of all three amino acids (2 orientations), plus the remainder of the structure, not present in the computation: The system consisting of the Q119, E120, R122 amino acids (all four domains) plus 32 water molecules and a K+ is optimized using B3LYP/6-31G**. These are shown as heavy lines (tubes). The amino acids adjacent to these are not included in the calculation, and shown as thin gray lines; all amino acids are labeled in one domain. The small red x’s are 58 water molecules (oxygen atoms) from the 1k4c X-ray structure. The heavy water molecules are the result of the optimization. As in B) there is a “basket” of water molecules hydrogen bonded to Q119 that blocks the channel, and also holds the domains together: the set of 12 hydrogen bonded water molecules form a “nanocrystal”, shown at the center of Fig. 1C surrounding the K+, and at the bottom of Fig. 1D, is a strong barrier to the approach of K+ toward the channel. Hydrogen bonds are shown in Fig. 1A, B as dashed lines (as inserted by GOpenMol—criteria in text, but in Fig. 1C,D the key bonds are normat). Large amino acid labels are shown in one domain, but the corresponding amino acids are present in all four domains. Atoms: black = frozen; optimized atoms, green= carbon, blue = nitrogen, red = oxygen, white = hydrogen. See Table II for the relevant distances.
Fig. 1B shows the uncharged, hence presumably closed, configuration of the four domains, at B3LYP/6-31++G** level, for only the four glutamines (Q119), plus water. The “basket” of water molecules is clear, with a strong hydrogen bonded structure holding the water molecules in place. Fig. 1C and 1D show the twelve amino acids, plus the “basket” of water formed in the structure optimized using B3LYP/6-31G**, in two orientations, plus a hydrated K+ ion. The hydrogen bonds among the “basket” waters have oxygen-oxygen distance = 2.70Å, and for the bonds connecting the “basket” to the amino acid, the donor-acceptor distance is 2.67 Å. We observe that it is a plausible closed structure. (O – O distance is in the range 2.66 to 2.82 Å, for all relevant bonds, including those in the glutamines). The K+ to nanocrystal water oxygen bond length is 2.85Å, compared to 3.23 Å for the corresponding distance in an isolated hydrated K+ ion. The difference is apparently a consequence of a difference in charge on the O atom of the water hydrating the ion. Using B3LYP/6-31**, the oxygen charge is −0.69 q, where q is the electronic charge, for the nanocrystal case; −0.64 q for the hydrated ion (for comparison, in the latter case, the charge computed using B3LYP/6-311++G** in a single point calculation is −0.62 q; the difference in basis set is therefore not very important).
Sixteen water molecules out of the 32 in the computation are required for the hydration of the K+ and for the basket, including four that mediate between the two, with hydrogen bonds. Four of these constitute the basket, eight the water of hydration of the K+. The upper 12 of the molecules (the “basket”, the mediating four, and the upper four of the K+ water of hydration) form the nanocrystal with an ice-like structure that we discussed above.
The difference between the charged and uncharged states can easily be seen, in that water is structured, as a part of the protein structure, in the center of the figure with the uncharged system, showing a system of hydrogen bonds. In the charged (open) case, those waters are essentially as in bulk.
In short, Fig. 1A has the result of calculation on the four domain configuration for the open state. Figs 1 B, C, D give the closed state, C and D showing how the water blocks the hydrated K+ ion. Fig. 1 thus shows the essential result of this work: when the channel is charged, the domains separate; when the channel is not charged, the domains do not separate, but are held by hydrogen bonding among the amino acids and water molecules that are in the gating region, and a “basket” of water blocks the channel. The remaining amino acids that are omitted here will influence the details of the mechanism, but they will produce second order effects. A complete calculation would show how the gating pH of KcsA can be adjusted by neighboring amino acids, and would give the effects of mutation of these amino acids on the pKa of the residues in the gating region. That calculation awaits more powerful computers, or better methods of treating hydrogen bonds, so that they can be calculated to equal accuracy, but much more quickly. (The better minimum (by ≈ 0.01 H) referred to in the methods section is indistinguishable in geometry from the figure, at the scale shown, and within less than 0.2 Å in interatomic distances in the center of the channel.) Hydrogen bond lengths were discussed earlier, and are quite reasonable. Also the short K+ - water distance (2.85 Å and 2.86 Å (two different bonds) vs. 3.23 Å in normal hydrated K+) suggests a structure that is stabilized by its surroundings.
Indeed, the structure appears to be extremely stable. This was tested by distorting the structure slightly and recomputing the energy. The energy of the system, as optimized, is −6407.1586 H (H=Hartree, and 0.001H ≈ kBT). Displacing the K+ and its 8 waters of hydration upward (i.e., in the extracellular direction) by 1 Å gives a single point energy of −6406.9236 H, more than 200 kBT higher, thus totally out of reach. Displacing the 4 mediating water molecules 0.5 Å upward produces −6407.0835 H, raising the energy >75 kBT. A lateral displacement of the upper water of hydration of the K+ by 1 Å, 2 adjacent water in one direction, 2 in the opposite direction, gave −6407.0587 H (K+ stationary) or −6407.0585 (K+ displaced upward into the space left by moving the water molecules by 1Å). While it is possible that the K+ might move away from the gate (a possibility of less interest to us, and thus not tested), we have found no path by which the K+ could plausibly approach closer to the gate. In that direction, a displacement (even without moving the “basket” attached to the Q119 groups) is energetically extremely costly. This leaves the question of why the K+ does not appear in the X-ray structure, and in fact some of the water that should be there is at least hard to see. However, the X-ray structure does not extend down to the level of the K+, which may account for its absence. Whether the “basket” is composed of water molecules that are close enough in the X-ray structure (there are some water molecules roughly 2 Å away) to be within experimental uncertainty remains to be determined. Fig 2 shows the energetic cost of removing one molecule from the “basket”, and the cost of moving the entire “basket” outward.
Fig. 2.


Two views, at 90°angles comparing the calculated and 1k4c and 1j95 X-ray structures: Carbons from the1k4c structure are red-orange, from the calculation green, from 1j95 black. Nitrogen is blue, hydrogen not shown, and carbons frozen in the calculation that are used as methyl groups are circled in Fig 2A ( the 1k4c and calculated positions of those atoms are forced to coincide). Water oxygens (only calculated shown) are pale blue, and can be seen to be in locations that outline the nanocrystal. R122 is missing in the 1j95 X-ray structure. One instance of each amino acid is labeled. The two oxygen atoms of the E120 carboxyl to the neighboring domain R122 guanidinium C atom have optimized distances of 3.09 and 4.12 Å, compared to 4.19 and 6.17 Å in the 1k4c X-ray structure. The 3.09 Å distance is marked on the figure.
Table II shows the distances in the converged structures, compared with the 1k4c and 1j95 structures. The final coordinates of the closed state are in approximate agreement with the 1j95 structure, having moved during the optimization. The charged state opened, making a considerable difference with 1k4c.
TABLE IIA.
Distances (Å) of side chain amide Q119 atom pairs in calculated open (protonated) and closed (unprotonated) states (calculated = B3LYP/6-31G** for closed four-domain values, two-domain B3LYP/6-31++G** values for open; distances are shown for atoms in opposite domains); comparison to X-ray structures 1j95 and 1k4c.
| ATOMS | UNPROTONATED | 1j95 | PROTONATED | 1k4c |
|---|---|---|---|---|
| C – C | 7.47 | 6.61 | 10.28 | 8.91 |
| N – N | 7.09 | 4.28 | 10.42 | 9.43 |
| O – O | 6.51 | 7.04 | 10.96* | 8.18 |
Of Table IIB distances, only N—O is available from the 1j95 X-ray structure, and it is 2.84 Å. The water oxygens are those in the “basket” waters (which are the upper four water molecules of the nanocrystal). This agreement is satisfactory, considering the inaccuracies left in the calculation, the error limits of the X-ray structure, and the 1k4c starting point. The open MthK channel has an opening that is only about 1Å larger than we find for the glutamine oxygen to diagonally opposite glutamine oxygen distance, which agrees within the uncertainties of the calculation and the structure (also, it is a different channel, but that may not be as important). Jiang et al also give a 12 Å opening[3]. In addition, the agreement in Table IA within 1 Å between two out of three cases for the closed to 1j95 comparison suggests that the calculation is reasonable, needing only some fine tuning, which might have been obtained had a larger basis function, or additional amino acids, been possible. The amide groups of Q119 become coplanar in the calculation, and this agrees with the 1j95 structure. In the open state these groups rotate, with the oxygen pointing down (i.e., intracellularly), the nitrogen up. Fig. 3 makes the point even more clearly.
TABLE IIB.
Calculated H-bond donor-acceptor distances (Å) for three types of hydrogen bonds in the closed state: (HF/6-31G**: Ow = water oxygen; O = carbonyl oxygen, N=nitrogen, Q119 amide)
| ATOMS | CLOSED |
|---|---|
| N—O | 3.16 |
| Ow—O | 2.67 to 2.68 |
| Ow—Ow | 2.70 |
Fig. 3.
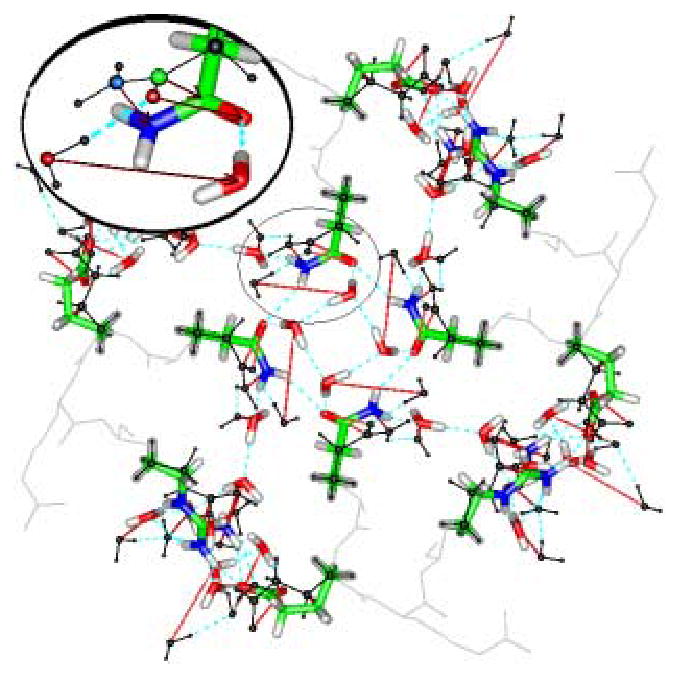
This figure shows how much the quantum optimization for KcsA moves atoms from the starting 1k4c positions; initial positions for all atoms in the calculation are shown in black. All thin red lines show motion of >1Å; absence of thin red lines for atoms shown in color shows that the motion is < 1Å; these atoms essentially remained at the X-ray structure positions; most of the protein atoms are in this group. Gray atoms were not included in the calculation; they are shown in their X-ray positions. Some water moved more than 1Å; the guanidinium nitrogen moved about 2 Å, and the water hydrogen bonded to it moves about 4 Å. The inset enlarges the key step: the glutamine amine nitrogen rotates about the single bond, and the hydrogen bonded water accompanies it, rotating to form the “basket”. For clarity, the water molecule to the left is removed from the inset. All four domains show the same move. Atom colors: C, green; O, red; N, blue; H (added, not from X-ray structure) white. The water O-H bond length is close to 1Å, so that the dimensions of the figure can be seen by using water as a scale bar. Dashed blue lines are hydrogen bonds, thin black lines, initial positions. The “basket” is in the center, and only exists after the optimization
The small rotation of the side chains produces better agreement with the 1j95 than the 1k4c structure, in spite of the fact that the backbone ends of the side chains were frozen in the 1k4c position. The motion suggests that the 1j95 positions may be more stable. Possibly, the 1k4c coordinates may be a form of intermediate, partially open state. Near agreement of the inner part of the uncharged case with 1j95, in spite of starting with 1k4c (see also Table II), shows that the system we are studying is large enough to tell the difference between the two structures, as well as between open and closed..
The distribution of the charges when protons are added is of some interest also. We have calculated the charges on the atoms, and present the charges on the key atoms in Table III. Because of the near four fold symmetry, there is very little difference in the charges on the corresponding atoms of different domains. The charges are therefore reported as averages over corresponding atoms, with the scatter given as well; generally the scatter is no greater than 0.02 charges. Atoms that are reported in Table III are those of the water molecules in the gate, the atoms to which they are hydrogen bonded, and those involved most directly in the E120-R122 salt bridge. The atoms can be listed with their Table III designations as follows: the water molecules, most clearly seen in Fig. 1D, where there are three layers of interest in the closed state: the water basket, which is hydrogen bonded to the Q119 residues (OA, HAE (equatorial), HAA(axial), where the axial hydrogen bonds are to the next layer), the next layer toward the K+, intermediate between the basket and the hydration shell (OB, HBE, HBA), and finally the water of the hydration shell (OC, OCE, OCA). Other important atoms are the Q119 O and N involved in hydrogen bonding to the basket (O) and to each other (O and N), and the two hydrogens attached to the nitrogen, one hydrogen bonded (HNB) to the next O, one not (HNF). In the salt bridge, there are eight atoms of interest: the glutamate carboxylate oxygens, one hydrogen bonded (OSB), one not (OSF), two hydrogen-bonded hydrogens on the guanidinium group of R122 (HS1, HS2), two hydrogens that are not hydrogen bonded (H1, H2), and two guanidinium group nitrogens, again, one hydrogen bonded (NB), one not (NF).
In the open state, there are four additional protons; E120 carboxyl groups each show an proton absent in the closed state. The water molecules are no longer structured; the three layers described above disappear. Table III has charges on water molecules that are in the center of the system, but these are hydrogen bonded to other water molecules, much as in bulk water. The key difference appears to be the protons on the glutamates, however, designated HEC in the Table..
Note that the water molecules have a small net charge. Other than this, there appear to be no real surprises. The charges are a little less than textbook values, suggesting some charge delocalization, but the charges are close to those one would expect. Neither the salt bridge nor the basket, nor the key amino acid atoms, appear to have charges that would cause us to look for special explanations. The carboxyl oxygens are less negative in the open state, as expected. A hydrogen in a hydrogen bond that exists only in the open state (HCE) has more than half of the charge difference between open and closed states. The total charges do not add to the total difference in charge (3 charges more positive in the open state, as the K+ is not present in the open state), because some charge is on atoms not shown.
We have also done a very limited calculation of the effects of a Q119C mutation. In this, only the single amino acid was present, and the backbone carbon was frozen; only one rotation of the terminal C – S – H group was allowed; several water molecules (as in Fig. 1b) were present. Here, no “basket” or other ordered water structure formed. The –S – H pointed toward the center of the channel, with the S forming an almost exactly 5Å square, the hydrogens pointing in and forming an approximately 4 Å square. The channel would have been blocked (a little weakly), but not by the mechanism suggested by the “basket” of water in the WT channel. It is likely that such a channel would still require protons to gate, to break salt bridges. We have not done a Q119A calculation, but that may allow the passage of ions, making a channel that would, from our results, be constitutively open (the salt bridges would still remain, but the water and the potassium should behave completely differently). However, it is possible that a double mutation (Q119A/H124A) would be required for a fully open channel. This is a question that can be tested experimentally.
One final question can be considered. The calculation finds the energy minimum. However, the channel operates near 300K. Is the minimum deep enough to make the basket stable at that temperature? Considering only the basket, we can assume there is a loss of entropy associated with the effective freezing of four water molecules. If these are as frozen as in ice, a reasonable analogy, there should be a loss of entropy of 22 J K−1 mol−1, corresponding to the ΔH of 6.01 kJ mol−1, or 2.4 kBT at 300K (or room temperature); for four molecules, this becomes 9.6 kBT, or 24 kJ for four moles of water. This is a small fraction of the energy to move a molecule 0.5 Å (>60 kJ mol−1-- see also Fig. 2). Even if the analogy to ice is less than perfect the net ΔG for formation of the basket remains favorable.. Fig. 4 shows how large the energy change is when the system is distorted.
Fig. 4.
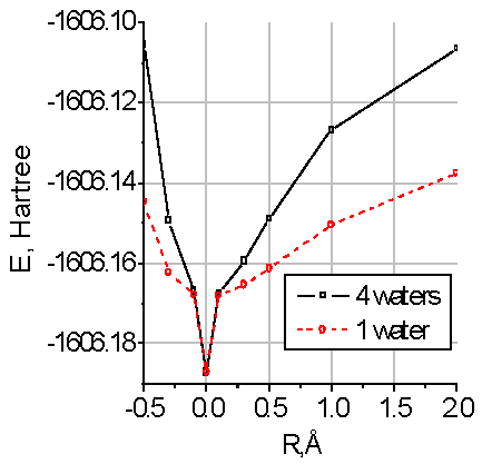
The cost of moving one water molecule, and of moving all four water molecules in the plane of the basket, determined from the basket alone plus Q119. The unit is again Hartrees, so that a 0.5Å distortion costs approximately 25 kBT for one molecule, over 30 kBT for the entire basket; to move the entire basket 1.0 Å costs approximately 60kBT, while even a single molecule costs about 35 kBT. The values are from single point calculations using B3LYP/6-311+G**, starting from the previously optimized structures. Note that these results are for the basket alone, together with only the Q119 residues, not the entire nanocrystal with 12 amino acids. These energy values are about 1/3 those for the full nanocrystal.
All this said, does the KcsA result also tell us anything about voltage gated channels? We believe that it does. We have, for some time, argued for a three-step mechanism for voltage gating [17, 18]: 1) proton tunneling, leading to 2) a proton cascade, producing gating current that 3) alters local charge in the gating region, breaking hydrogen bonds and opening the channel. Much earlier, Green argued [49] that channel block depended on water existing in an essentially frozen configuration, and the three planes of four water molecules each (the “ice nanocrystal”) in Fig 1C,D has as strong a resemblance to this idea as seems possible. The finding that the proton channel (Hv) is very similar to the voltage sensing domain (or an oligomer of it)[14, 15] is at least consistent with step 2) in the voltage gating model. In KcsA, protons are added directly, presumably from the intracellular medium, making the charge in the gating region positive. In voltage gated channels, it appears that the sign of the effect should be reversed, but the principle that change of charge alters hydrogen bonding may be conserved. If the depolarization of the membrane containing the voltage-gated channel moves protons, thus altering charge, it too could disrupt the pattern of hydrogen bonds holding the channel closed, producing a gating mechanism similar to that proposed here for KcsA, although details will be different. As there are also hyperpolarization gated channels, with apparently somewhat similar structure, it is possible that proton transport leading to the same sign of charge on the gating region as in KcsA may be found in nature.
SUMMARY
A calculation of open and closed states of the KcsA channel leads to a proposed gating mechanism, showing how proton addition to the gating region of the channel disrupts hydrogen bonds, producing channel opening. Omission of the main chain structure still allows the calculated positions of the side chains to close the channel, but leads to conservation of the X-ray structure in the closed state.. A “basket” of water molecules blocks the channel in the closed state, and disappears in the open state. In the closed state the basket attaches to the waters of hydration of K+, forming a robust nanocrystal of ice. This shows that water plays a key role in keeping the closed state closed. It is suggested that a similar mechanism may apply to voltage-gating.
Table I.
Distance between atoms, opposite domains. Column 2, one of two diameters with dimer plus C2 symmetry (equivalently, from one monomer plus C4 symmetry), and. Column 3, the second diameter with C2 symmetry. (Distances in Å)
| C (carboxyl), E120 | 23.71 | 23.65 |
| C (guanidinium), R122 | 20.32 | 20.34 |
| O (amide) Q119 | 11.14 | 10.96 |
Acknowledgments
This research was performed in part using the MSCF supercomputer facility in the William R. Wiley Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the U.S. DOE, Office of Basic Energy Research, and located at PNNL. Support has been provided by the NIH through a SCORE grant to City College (individual subgrant to MEG), and a PSC-CUNY grant to MEG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 2.MacKinnon R, Cohen SL, Kuo A, Lee A, Chait BT. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 1998;280:106–109. doi: 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- 3.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature. 2001;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 4.Jinesh KB, Frenken JWM. Capillary condensation in atomic scale friction: how water acts like a glue. Phys Rev Letters. 2006;96:166103/1–4. doi: 10.1103/PhysRevLett.96.166103. [DOI] [PubMed] [Google Scholar]
- 5.Anishkin A, Sukharev S. Water dynamics and dewetting transitions in the small mechanosensitive channel. MscS Biophys, J. 2004;86:2883–2895. doi: 10.1016/S0006-3495(04)74340-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beckstein O, Sansom MSP. Liquid-vapor oscillations of water in hydrophobic nanopores. Proc Natl Acad Sci USA. 2003;100:7063–7068. doi: 10.1073/pnas.1136844100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu J, Green ME. Simulation of water in a pore with charges: application to a gating mechanism for ion channels Progress in Colloid and Polymer. Science. 1997;103:121–129. [Google Scholar]
- 8.Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 9.Chanda B, Asamoah OK, Blunck R, Roux B, Bezanilla F. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature. 2005;436:852–856. doi: 10.1038/nature03888. [DOI] [PubMed] [Google Scholar]
- 10.Posson EJ, Ge P, Miller C, Bezanilla F, Selvin PR. Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature. 2005;436:848–851. doi: 10.1038/nature03819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 12.Shrivastava IH, Bahar I. Common mechanism of pore opening shared by five different potassium channels. Biophys, J. 2006;90:3929–3940. doi: 10.1529/biophysj.105.080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Z, Klem AM, Ramu Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J Gen’l Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller C. Lonely voltage sensor seeks protons for permeation. Science. 2006;312:534–535. doi: 10.1126/science.1127186. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki M, Takagi M, Okamura Y. A voltage-sensor-domain protein is a voltage-gated proton channel. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 16.Sapronova AV, Bystrov VS, Green ME. Water,proton transfer, and hydrogen bonding ion channel gating. Frontiers in Bioscience. 2003;8:s1356–s1370. doi: 10.2741/1179. [DOI] [PubMed] [Google Scholar]
- 17.Lu J, Yin J, Green ME. A model for ion channel voltage gating with static S4 segments. Ferroelectrics. 1999;220:249–271. [Google Scholar]
- 18.Green ME. Water as a structural element of an ion channel: Gating in the KcsA channel, and implications for voltage-gated ion channels. JBiomolec Struct Dynamics. 2002;19:725–730. doi: 10.1080/07391102.2002.10506779. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Voth GA. Computer simulation of explicit proton translocation in cytochrome c oxidase: The D-pathway. Proc Natl Acad Sci USA. 2005;102:6795–6800. doi: 10.1073/pnas.0408117102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murata K, Fujii Y, Enamoto N, Hata M, Hoshino T, Tsuda M. A study on the mechanism of the proton transport in bacteriorhodopsin: the importance of the water molecule. Biophys, J. 2000;79:982–991. doi: 10.1016/S0006-3495(00)76352-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemukhin AV, Grigorenko BL, Topol IA, Burt SK. Quantum Chemical Simulations of the Proton Transfer in Water Wires Attached to Molecular Wells. J Phys Chem B. 2003;107:2958–2965. [Google Scholar]
- 22.Ramsey IS, Moran MM, Chong JA, Clapham DE. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guillen YV, Schlippe Hedstrom L. A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch Biochem Biophys. 2005;433:266–278. doi: 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 24.Freites JA, Tobias DJ, White SH. A Voltage-sensor water pore. Biophys J: Biophys Letters. 2006;91:L90–L92. doi: 10.1529/biophysj.106.096065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandsburg-Zabary S, Fried O, Marantz Y, Nachliel E, Gutmann M. Biophysical aspects of intra-protein transfer. Biochim Biophys Acta. 2000;1458:120–134. doi: 10.1016/s0005-2728(00)00063-3. [DOI] [PubMed] [Google Scholar]
- 26.Cortes DM, Cuello LG, Perozo E. Molecular architecture of full-length KcsA: role of cytoplasmic domains in ion permeation and activation gating. J Gen’l Physiol. 2001;117:165–180. doi: 10.1085/jgp.117.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu YS, Sompornpisut A, Perozo E. Structure of hte KcsA channel intracellular gate in the open state. Nat Struct Biol. 2001;8:883–887. doi: 10.1038/nsb1001-883. [DOI] [PubMed] [Google Scholar]
- 28.Luzhkov VB, Aqvist J. A computational study of ion binding and protonation states in the KcsA potassium channel. BiochemBiophysActa. 2000;1481:360–370. doi: 10.1016/s0167-4838(00)00183-7. [DOI] [PubMed] [Google Scholar]
- 29.Guidoni L, Torre V, Carloni P. Water and potassium dynamics inside the KcsA K+ channel. FEBS Letters. 2000;477:37–42. doi: 10.1016/s0014-5793(00)01712-9. [DOI] [PubMed] [Google Scholar]
- 30.Allen TW, Kuyucak S, Chung SH. Molecular Dynamics Estimates of ion diffusion in model hydrophobic and KcsA potassium channels. Biophys Chem. 2000;86:1–14. doi: 10.1016/s0301-4622(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 31.Burykin A, Schutz CN, Villa J, Warshel A. Simulations of ion current in realistic models of ion channels: the KcsA potassium channel Proteins-Struct. Func and Genetics. 2002;47:265–280. doi: 10.1002/prot.10106. [DOI] [PubMed] [Google Scholar]
- 32.Garofoli S, Jordan PC. Modeling permeation energetics in the KcsA potassium channel. BiophysJ. 2003;84:2814–2830. doi: 10.1016/S0006-3495(03)70011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Domene C, Sansom MSP. Potassium channel, ions, and water: simulation studies based on the high resolution X-ray structure of KcsA. Biophys J. 2003;85:2787–2800. doi: 10.1016/S0006-3495(03)74702-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tikhonov DB, Zhorov BS. In silico activation of KcsA K+ channel by lateral forces applied to the C-terminal of inner helices. Biophys, J. 2004;87:1526–1536. doi: 10.1529/biophysj.103.037770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Znamenskiy VS, Green ME. Topological changes of hydrogen bonding of water with Acetic Acid: AIM and NBO studies. J Phys Chem A. 2004;108:6543–6553. [Google Scholar]
- 36.Znamenskiy VS, Green ME. Quantum calculations on hydrogen bonds in certain water clusters show cooperative effects. J Chem Theory and Computation ASAP article. 2006 doi: 10.1021/ct600139d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Compoint M, Ramseyer C, Huetz P. Ab initio investigation of the atomic charges in the KcsA channel selectivity filter. Chem PhysLetters. 2004;397:510–515. [Google Scholar]
- 38.Guidoni L, Carloni P. Potassium permeation through the KcsA channel: a density functional study. BiochemBiophysActa. 2002;1563:1–6. doi: 10.1016/s0005-2736(02)00349-8. [DOI] [PubMed] [Google Scholar]
- 39.Bliznyuk A, Rendell AP. Electronic effects in biomolecular simulations: investigation of the KcsA potassium channel. JPhys Chem B. 2004;108:13866–13873. [Google Scholar]
- 40.Cuello LG, Sompornpisut A, Perozo E. Molecular characterization of the pH sensor in KcsA. Biophys J. 2001;80:A839. [Google Scholar]
- 41.Perozo E, Marien Cortes D, Cuello L. Structural Rearrangements underlying K+-channel gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- 42.Perozo E, Cortes DM, Sompornpisut P, Kloda A, Martinac B. Open channel structure of MscL, and the gating mechanism of mechanosensitive channels. Nature. 2002;418:942–948. doi: 10.1038/nature00992. [DOI] [PubMed] [Google Scholar]
- 43.Straatsma 1EATP, Windus TL, Dupuis M, Bylaska EJ, de Jong SHW, Smith DMA, Hackler MT, Pollack L, Harrison JNRJ, Tipparaju V, Krishnan M, Auer AA, Brown GCE, Fann GI, Fruchtl H, Garza J, Hirao K, Kendall JANR, Tsemekhman K, Valiev M, Wolinski JAK, Bernholdt D, Borowski P, Clark T, Clerc HDD, Deegan M, Dyall K, Elwood D, Glendening MGE, Hess A, Jaffe J, Johnson B, Ju J, Kobayashi RKR, Lin Z, Littlefield R, Long X, Meng B, Nakajima SNT, Rosing M, Sandrone G, Stave M, Taylor H, Thomas JvLG, Wong A, Zhang Z. comprehensive computational chemistry package. 4.6. Pacific Northwest National Laboratory; Richland, Washington: 2004. Secondary, NWChem, A Computational Chemistry Package for Parallel Computers, Version 4.6. [Google Scholar]
- 44.Kar T, Scheiner S. Comparison of cooperativity in CH...O and OH...O hydrogen bonds. JPhys Chem A. 2004;108:9161–9168. [Google Scholar]
- 45.Frisch 1GWTMJ, Schlegel HB, Scuseria GE, Robb JRCMA, Montgomery JA, Jr, Vreven T, Kudin JCBKN, Millam JM, Iyengar SS, Tomasi J, Barone BMV, Cossi M, Scalmani G, Rega N, Petersson HNGA, Hada M, Ehara M, Toyota K, Fukuda JHR, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai MKH, Li X, Knox JE, Hratchian HP, Cross JB, Adamo JJC, Gomperts R, Stratmann RE, Yazyev O, Austin RCAJ, Pomelli C, Ochterski JW, Ayala PY, Morokuma GAVK, Salvador P, Dannenberg JJ, Zakrzewski SDVG, Daniels AD, Strain MC, Farkas DKMO, Rabuck AD, Raghavachari K, Foresman JVOJB, Cui Q, Baboul AG, Clifford S, Cioslowski BBSJ, Liu G, Liashenko A, Piskorz P, Komaromi RLMI, Fox DJ, Keith T, Al-Laham MA, Peng ANCY, Challacombe M, Gill PMW, Johnson WCB, Wong MW, Gonzalez C, Pople JA, Gaussian I, PA Pittsburgh. Secondary, Gaussian 03. Revision B.03. Gaussian Inc; Pittsburgh, PA: 2003. 2003. [Google Scholar]
- 46.Skylaris CK, Gagliardi L, Handy NC, Ioannou AG, Spencer SS, Willets A. On the resolution of identity Coulomb energy approximation in density functional theory. J Molec Struct(THEOCHEM) 2000;501–502:229–239. [Google Scholar]
- 47.Bergman DL, Laaksonen L, Laaksonen A. Visualization of solvation structures in liquid mixtures. J Molec Graph Model. 1997;15:301–306. doi: 10.1016/s1093-3263(98)00003-5. [DOI] [PubMed] [Google Scholar]
- 48.Laaksonen A. A graphics program for the analysis and display of molecular dynamics trajectories. J Mol Graph. 1992;10:33–34. doi: 10.1016/0263-7855(92)80007-z. [DOI] [PubMed] [Google Scholar]
- 49.Green ME. Electrorheological effects and gating of membrane channels. JTheor Biol. 1989;138:413–428. doi: 10.1016/s0022-5193(89)80042-6. [DOI] [PubMed] [Google Scholar]